REVIEW: Glucocorticoids Orchestrate Adult Hippocampal Plasticity: Growth Points and Translational Aspects*
Natalia V. Gulyaeva1,2
1Institute of Higher Nervous Activity and Neurophysiology, Russian Academy of Sciences, 117485 Moscow, Russia2Research and Clinical Center for Neuropsychiatry of Moscow Healthcare Department, 115419 Moscow, Russia
*The article is published as a part of the Special Issue “Biochemical Aspects of Different Levels of Neuroplasticity: Molecules, Genes, Synapses, Cells, Cognitive Processes” (Vol. 88, Nos. 3-4).
Received March 27, 2023; Revised April 10, 2023; Accepted April 10, 2023
The review analyzes modern concepts about the control of various mechanisms of the hippocampal neuroplasticity in adult mammals and humans by glucocorticoids. Glucocorticoid hormones ensure the coordinated functioning of key components and mechanisms of hippocampal plasticity: neurogenesis, glutamatergic neurotransmission, microglia and astrocytes, systems of neurotrophic factors, neuroinflammation, proteases, metabolic hormones, neurosteroids. Regulatory mechanisms are diverse; along with the direct action of glucocorticoids through their receptors, there are conciliated glucocorticoid-dependent effects, as well as numerous interactions between various systems and components. Despite the fact that many connections in this complex regulatory scheme have not yet been established, the study of the factors and mechanisms considered in the work forms growth points in the field of glucocorticoid-regulated processes in the brain and primarily in the hippocampus. These studies are fundamentally important for the translation into the clinic and the potential treatment/prevention of common diseases of the emotional and cognitive spheres and respective comorbid conditions.
KEY WORDS: neuroplasticity, hippocampus, glucocorticoids, hypothalamic-pituitary-adrenal axis, synaptic plasticity, stress, neurogenesis, neuroinflammation, glutamatergic transmission, proteases, BDNF, insulin resistance, depression, Alzheimer’s disease, agingDOI: 10.1134/S0006297923050012
Abbreviations: AD, Alzheimer’s disease; ACTH, adrenocorticotropic hormone; AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors; APP, amyloid precursor protein; BACE1, β-secretase 1; BDNF, brain-derived neurotrophic factor; CRH, corticotropin-releasing hormone; GCs, glucocorticoids; GR, glucocorticoid receptors; HPAA, hypothalamic-pituitary-adrenal axis; MAPK, mitogen-activated protein kinase; mGluR, metabotropic glutamate receptors; MMP, metalloproteinases; MR, mineralocorticoid receptors; NMDAR, N-methyl-D-aspartate receptors; TrkB, tropomyosin tyrosine kinase receptor of BDNF.
INTRODUCTION. THE HIPPOCAMPUS IS A TARGET OF GLUCOCORTICOIDS IN
NORMAL AND PATHOLOGICAL CONDITIONS
The plasticity of neurons and glial cells plays a vital role in the transmission and integration of signals in the central nervous system. Neuroplasticity, adaptive change of the nervous system in response to changes in external signals, covers a variety of processes and mechanisms of their implementation, from birth, survival, migration and integration of new neurons, growth of neurites, synaptogenesis and modulation of mature synapses to the formation and transformation of neural networks. The fundamental mechanism of the adult brain plasticity is the activity-dependent reorganization of the pre-existing structure, and it is the plasticity of the adult brain that allows human beings and animals to learn throughout the life. Studies of recent decades have made it possible to characterize not only plastic structural and functional rearrangements of the brain, but also several forms of synaptic plasticity, defining them as key processes that allow the brain to work dynamically and carry out learning, memorization, and memory use [1]. Neuroplasticity is the basis of the brain’s adaptation to changing conditions of the external and internal environment, and aberrant changes in plasticity are associated with pathological conditions.
Glucocorticoids (GCs), acting in concert with catecholamines, control behavioral adaptation to stress and improve the preservation of meaningful emotional information; they dynamically regulate synaptic function and synaptic plasticity underlying the formation of emotional memory [2]. The formation and use of memory is a complex process involving several brain structures, such as the hippocampus, amygdala and adjacent areas of the cortex, usually defined as the medial structures of the temporal lobe. It is believed that after learning, memory is initially encoded in the hippocampus, but subsequently stabilizes and is stored for a long time in other areas of the brain, such as the neocortex (this process is known as consolidation of systemic memory). Synaptic plasticity is the main cellular mechanism underlying learning and memory, and is therefore considered key in this process [3]. In the adult hippocampus, the limbic structure responsible for both cognitive functions and emotions, synaptic plasticity is important for information processing, learning, and memory encoding. The dentate gyrus of the adult hippocampus constantly generates cohorts of neurons, some of which survive, mature and integrate into existing neural circuits, and this process is regulated by both global and local neural activity, providing a unique cellular and synaptic plasticity of the hippocampus. Apparently, the emergence of new hippocampal neurons throughout life makes it possible to constantly “rejuvenate” the brain of adult mammals, including humans, and maintain its adaptive plastic properties [4].
Stress as an adaptive response to the demands of the environment is necessary for the survival of the organism. GCs, steroid “stress hormones” secreted by the zona fasciculata of the adrenal cortex are crucial for successful adaptation to stressors, and in the implementation of this important function of GCs for the survival of the body, the key place belongs to the ability of these hormones to regulate the rapid and long-term plasticity of the brain. The impact of stress causes activation of the key neurohumoral system of the body, the hypothalamic-pituitary-adrenal axis (HPAA) and related neurochemical reactions after the release of GCs from the adrenal glands, which underlies rapid physiological reactions. HPAA stimulation leads to activation of certain areas of the brain, including the hippocampus, amygdala and prefrontal cortex, in which the density of corticosteroid receptors is high [5].
Acting through specific intracellular receptors in the brain and on the periphery, GCs regulate behavior, as well as metabolic, cardiovascular, immune and neuroendocrine activity. GCs bind to two subtypes of receptors, mineralocorticoid receptors (MR) and glucocorticoid receptors (GR), which differ in their affinity for GCs. Both MR and GR can be localized intracellularly or on the membrane. MRs and GRs are activated by the action of GCs and mediate their effects, including influence on synaptic plasticity, on both fast and slow time scales. GRs are present in every cell of the nervous system, but the level of their expression varies, so different cell types react differently to the activation of GRs [6]. GCs exert their influence on the brain through “classical” genomic mechanisms, including direct binding of intracellular MRs and GRs to DNA, as well as through non-genomic mechanisms implemented through membrane MRs and GRs [7] (Fig. 1).
Fig. 1. Glucocorticoid receptors in the glutamatergic synapse (the scheme based on the data presented in recently published papers [7-11]). Corticosterone (in rodents) or cortisol (in humans) are released from the adrenal glands into the bloodstream. As lipid-soluble molecules, glucocorticoids (GCs) can freely penetrate the cell membrane. When GCs bind to cytosolic glucocorticoid receptors (GR) and mineralocorticoid receptors (MR), this leads to the release of regulatory complexes such as HSP90, FKBP5 and BAG1, followed by dimerization of the receptors and translocation into the nucleus. The binding of dimerized GR (MR) to the putative glucocorticoid response elements (GREs) localized in the promoter regions of DNA induces the activation of transcription factors. The sex steroid hormones estradiol (E2) and testosterone (T) can modulate the expression of GR-dependent genes (E2R, TR – receptors of estradiol and testosterone, respectively). GC-regulated genes control the expression of ionotropic glutamate receptors (NMDAR, AMPAR), trophic factor systems (brain-derived neurotrophic factor, BDNF), including the synthesis of pro-BDNF, its proteolytic transformation into mature BDNF (mBDNF), and the synthesis of the BDNF receptor TrkB. The expression of GC-dependent genes controls the functioning of the hypothalamic-pituitary-adrenal axis (HPAA), neuroplasticity, behavior, immune system and metabolism. Unlike classical cytosolic/nuclear receptors, membrane GR and MR realize rapid effects of GCs, modulating the release of glutamate in the presynapse, and in the postsynaptic membrane adjusting the activity of the ionotropic γ-aminobutyric acid receptor (GABAAR), cation channels: several types of calcium channels (CaChs) and potassium channel KCh(IA), as well as rapid changes in dendritic spines. Estradiol (E2) competes with corticosterone to inhibit GR signaling.
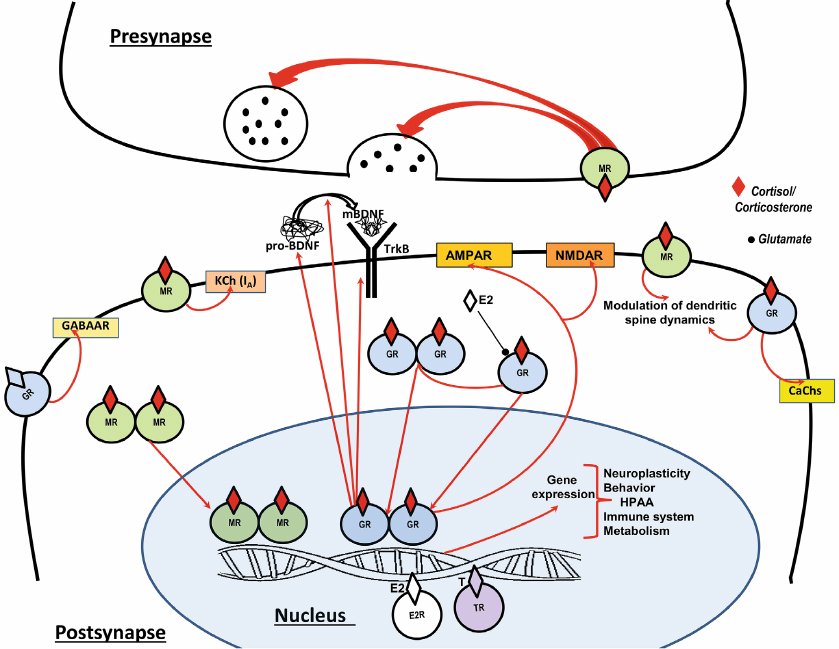
The effects of GCs significantly vary depending on the type of receptor, as well as on the brain region, cell type, and physiological context. These differences ultimately depend on the differential interactions of MR and GR with other proteins that determine ligand binding, nuclear translocation, and transcriptional activity. MR and GR can mediate very different and sometimes opposite effects in the brain. MR expression in the brain is much more limited as compared to GR. MR is very common in the hippocampus of rodents and humans, where MR expression equals or even exceeds GR expression in CA3 pyramidal cells [8]. It is known that MRs and GRs move between cytoplasmic and nuclear compartments, and the intracellular distribution of MR and GR depends on the equilibrium between nuclear import and export. This equilibrium seems to depend on the type of cells [9, 10].
GCs interact both genomically and non-genomically with neurotransmitters, neurotrophic factors, sex hormones and other stress mediators, forming present and future responses of the organism to stress challenges. In the hippocampus and other stress-reactive areas of the brain, allostatic overload resulting from chronic stress also can change the functioning of the HPAA through epigenetic modification of genes [7]. In basic conditions with low GCs secretion, MRs are close to saturation. When GCs rise during stress or at the peak of the circadian cycle of these hormones, MRs become fully occupied and GCs begin to bind to GRs. The important role of GCs in neuroplasticity was postulated several decades ago [12], but the specific mechanisms of the regulatory function of GCs have not been fully decoded. The wide prevalence of GRs in various populations of neuronal and glial cells, also outside the classical brain regions responsible for the execution of stress reactions, confirms the idea that in the central nervous system, GCs can act as a conductor, organizing and controlling a brain orchestra consisting of different musicians – various cells included in specific networks. The pleiotropy of GCs effects is directly related to the multiple mechanisms they trigger and/or control at all levels from molecular to network and organismic.
Stress through GCs induces the structural plasticity of neurons, Schwann cells, microglia, oligodendrocytes and astrocytes, and also affects neurotransmission by altering the release and reuptake of glutamate. Exposure to stressors causes a spectrum of reactions that range from potentially adaptive to maladaptive consequences at the structural, cellular, and physiological levels. These responses are particularly evident in the hippocampus, where they also affect hippocampus-dependent cognitive function and emotionality [13]. Unlike chronically elevated levels, circadian and acute stress-induced periods of augmented GCs are necessary for the survival of hippocampal neurons, the acquisition and consolidation of memory, the facilitation of glutamatergic neurotransmission, the formation of excitatory synapses, the induction of immediate early genes and the formation of dendritic spines. Negative feedback from the GCs includes several mechanisms leading to limiting the activation of HPAA and preventing the harmful effects of excessive generation of GCs. Adequate secretion of GCs is regulated by the nervous system that controls the secretion of hypothalamic corticotropin-releasing hormone (CRH) and vasopressin, the main regulators of pituitary adrenocorticotropic hormone (ACTH). Rapid feedback mechanisms involving non-genomic actions of GCs mediate immediate inhibition of hypothalamic secretion of CRH and ACTH, and slower genome-mediated mechanisms include modulation of limbic networks and peripheral metabolic messengers [14].
An excess of GCs can have negative effects, especially in the hippocampus, in which the density of MRs and GRs is high [15]. These effects include impaired synaptic plasticity, atrophy of dendrites, impaired ability of neurons to survive influences of various damaging factors and, ultimately, the death of neurons [16]. The selective vulnerability of the hippocampus to stress, mediated by the receptors of GCs secreted under stress, is the price of high functional plasticity and pleiotropy of this limbic structure [17]. Common molecular and cellular mechanisms of hippocampal plasticity disorders include dysfunction of GRs, neurotransmitters and neurotrophic factors, the development of neuroinflammation leading to neurodegeneration and death of hippocampal neurons, as well as disturbances of neurogenesis in the subgranular neurogenic niche and the formation of aberrant neural networks.
Normal plasticity of the nervous system is necessary for adaptation, learning and memory, and plasticity caused by stress is often inadequate and contributes to the development of neuropsychiatric disorders and other brain pathologies [18]. The structural plasticity of the hippocampus plays a key role in the etiopathogenesis of neurodegenerative diseases [19]. The effect of GCs on the brain is crucial for maintaining homeostasis, therefore, these hormones are also involved in the aging process, which is defined as a period of ontogenesis with a reduced ability to maintain homeostasis, increased lability of HPAA after stress and impaired behavioral adaptation [20]. Apparently, the dysfunction of GCs-dependent processes is associated with almost all pathologies of the brain, thus, an incomplete understanding of the relevant mechanisms prevents us from revealing the full potential possibilities of preventing and treating brain diseases.
This review analyzes the current understanding of the key mechanisms of hippocampal neuroplasticity, which are controlled by GCs. These are mechanisms forming growth points in the field of research of processes regulated by GCs in the brain. These mechanisms are fundamentally important for translation into the clinic and potential treatment/prevention of common diseases of the emotional and cognitive spheres, including depressive disorders and Alzheimer’s disease (AD).
NEUROGENESIS AS A FORM OF GLUCOCORTICOID-REGULATED HIPPOCAMPAL
PLASTICITY
The nervous system of an adult organism is not static; it undergoes morphological and physiological changes at various levels. This plastic mechanism ensures that the behavioral regulation of the nervous system adapts to various environmental stimuli. It is generally believed that in the mammalian hippocampus, the process of formation and development of functionally integrated neurons occurs throughout life making a significant contribution to the highly plastic nature of the mature central nervous system. The dentate gyrus of the hippocampus is one of the main neurogenic niches in the adult organism, containing newborn neurons, some of which mature and are included in neural networks [4]. Neurogenesis in the adult hippocampus is a dynamic process intimately associated with cognitive functions such as learning and memory. Importantly, a number of researchers believe that neurogenesis is not a mechanism for replacing cells in the adult brain, but instead supports the plastic neural network of the hippocampus due to the continuous addition of immature, new neurons. These new neurons have unique properties and structural plasticity and induce changes in the plasticity of mature neurons [21].
It is generally believed that in humans and other mammals, memories of events are encoded by ensembles of neurons (engrams) in the hippocampus. The mnemonic information stored in these engrams can then be used to control future behavior, including predicting and decision-making in a dynamic environment. Some engrams of the hippocampus can be stored permanently, others change over time, and this suggests that the memories presented can also be transformed. It is most likely that neurogenesis in the adult hippocampus is one of the processes that constantly rebuilds the neural networks of the hippocampus, presumably including stored engrams [22]. It is believed that the neural circuits of the dentate gyrus – the CA3 region of the adult hippocampus are constantly modified due to the integration of dentate granular cells born as a result of neurogenesis. These cells undergo a long maturation period, while they demonstrate increased synaptic plasticity, effective electrophysiological properties and connectivity. It is also assumed that neurogenesis of the adult hippocampus allows generating a library of events/new experiences, each of which is registered in the physiological properties and connectivity of mature granular cells of the dentate gyrus [23]. Apparently, neurogenesis provides the flexibility of the dentate gyrus, which allows to quickly generate a context-specific distributed representation of important sensory stimuli, such as spatial signals, which ultimately leads to their differentiation at the behavioral level [24]. However, most of the results underlying the concepts of the role of adult hippocampal neurogenesis in neuroplasticity were obtained in mammals, mainly rodents, and a number of methodological limitations cause ongoing debates about the intensity (and generally the occurrence) of neurogenesis in the adult hippocampus [25]. However, despite the methodological difficulties of studying neurogenesis in humans, most researchers accept that the dentate gyrus of the hippocampus is evolutionarily preserved as one of the few sites of neurogenesis in adult mammals, although it remains unclear whether new neurons integrate into existing hippocampal networks on a par with neurons born during development, or whether they represent a discrete cell population with unique functions [26]. Perhaps, as a result of neurogenesis, a specialized subpopulation of neurons is created in the adult hippocampus, which can play a key role in the hippocampal functions, such as episodic memory.
The addition of new neurons in adulthood occurs as part of a sequential multi-step process. Neurogenic stages include proliferation, differentiation, migration, maturation/survival, and integration of new neurons into an existing neural network. Most studies evaluating the influence of exogenous (e.g., GCs, stress) or endogenous (e.g., neurotrophins and their receptors) factors on neurogenesis in adults at different levels have focused on proliferation, survival and differentiation of neurons [27]. The interaction between external and internal factors plays a fundamental role in the regulation of neurogenesis. Over the past decades, several studies of “internal” pathways, including transcription factors, have revealed their fundamental role in regulating each stage of neurogenesis. However, it is likely that regulation of transcription is part of a more complex regulatory network that includes epigenetic modifications, non-coding RNAs, and metabolic pathways [28]. It is important that hormones, primarily steroid hormones, exert a multifaceted effect on all stages of neurogenesis in the adult hippocampus: there is evidence of hormonal stimulation (via gonadal and thyroid hormones), inhibition (via GCs) and neuroprotection (mediated by neurotrophins and neuromodulators) [29].
Neurogenesis in the adult hippocampus has been increasingly viewed in recent years through the prism of the brain’s response to stress. It is assumed that neurogenesis plays a key role in the formation of adaptation to environmental requirements, which underlies its role in the response to stress (or excess of GCs). The hippocampus, with its high convergence at the input and divergence at the output, is a kind of computing center ideally located in the brain for detecting signals and contexts related to past, current and predicted stress experiences, as well as for monitoring stress responses at the cognitive, affective, behavioral, and physiological levels. Neurogenesis in the adult hippocampus seems to promote contextual discrimination and cognitive flexibility, reducing the anticipation and generalization of stress experiences to a safe level. Thus, the underlying brain regions receive more accurate and contextual information, which allows them to respond more precisely to stress in definite contexts [30]. Nevertheless, the specific role of neurogenesis in the adult hippocampus in mediating the behavioral response to chronic stress remains not fully understood and the question of whether newborn neurons act as a kind of buffer or, conversely, increase susceptibility to stress-induced emotional maladaptation remains controversial [31].
In the early 2000s, it was confirmed that GCs/chronic stress/neuroinflammation are among the most important negative regulators of neurogenesis in adults. Although the effects of acute and moderate stress on neurogenesis are usually short-lived and can be quickly overcome, chronic exposure or more severe forms of stress can cause longer-term decreases in neurogenesis, which can only partially be overcome by subsequent exposure to exercise, adaptogenic drugs and some antidepressants [32]. There is no doubt that GCs-mediated disorders of neurogenesis in adults contribute to the occurrence of brain diseases, including cognitive and affective disorders, neurodegenerative diseases. In addition, exposure to stress, especially during critical periods of early childhood, disrupting the processes of neurogenesis for a long period, can reprogram the plasticity of the hippocampus, increasing the risk of cognitive impairment or anxiety symptoms common to a number of cerebral pathologies, such as dementia and depression, associated with chronic changes in the plasticity of the hippocampus. The review [33] analyzed in detail the effect of GCs on the mechanisms and physiological features of neurogenesis in the adult hippocampus and changes in neurogenesis in cerebral pathologies. Neuropsychiatric disorders are usually GCs-dependent, resulting from chronic stress or pain followed by (neuro)inflammation; all these conditions are associated with impaired neurogenesis and cognitive deficits. Susceptibility to stress and the ability to adapt to new conditions with the help of resistance mechanisms is directly related to specific features of neurogenesis in the adult hippocampus and its regulation with GCs [34].
The speed of the cell cycle and potential addition of new neurons to existing hippocampal neural circuits undoubtedly decrease with age. Nevertheless, neural stem cells/progenitor cells that persist in the hippocampus of an aging brain can be activated and produce a significant number of new functional neurons that demonstrate increased survival and integration under optimal conditions [35]. In the dentate gyrus, newborn neurons coexist with mature granular neurons that have arisen during development, and the connection between these two types of cells is regulated by both cooperative and competitive processes. It can be assumed that newborn neurons in the aging hippocampus have a noticeable potential for optimizing processes at the level of neural circuits and behavior, making neurogenesis a potential target for therapy. Both the hippocampus and the main region innervating this structure, the entorhinal cortex, demonstrate pronounced atrophy in patients with AD, the most common form of dementia in the elderly [25, 36]. It is important that, along with GCs, glycogen synthase kinase-3β (GSK-3β) and hyperphosphorylated tau protein, the two main molecules important for the pathogenesis of AD, are powerful negative regulators of adult hippocampal neurogenesis [37].
In states such as memory decline with age, neurodegeneration, and mental illness, mature neurons die or become defective, thus, stimulation of neurogenesis in adults is believed to have a potential for providing a therapeutic strategy to overcome these conditions. Hippocampus-dependent learning tasks, an enriched environment, physical exercises and activity-dependent synaptic plasticity powerfully activate the proliferation of nerve cell precursors in the hippocampus [21]. To explain the activation of neurogenesis by such influences, the mechanisms of strengthening neurotrophic and other influences that are normally positive regulators of neurogenesis (e.g., BDNF levels) are considered. Interestingly, exercise is associated with an increase in the level of GCs, but the absence of a negative effect of these hormones on neurogenesis is explained not by excessive and prolonged, but by their “moderate” and short-term release, so that their concentrations remain in the stimulating neurogenesis region. One of the new hypotheses examines the effect of lactate accumulated during exercise on the plasticity of neurons. Lactate, apparently, is one of the essential factors, since it participates in metabolism and signal transmission in most, if not all, cells of the central nervous system, including various types of cells in the neurogenic niche [38].
GLUTAMATERGIC TRANSMISSION AND HYPERGLUTAMATERGIC STATES ARE
GLUCOCORTICOID-DEPENDENT
The storage and processing of information at the synaptic level is provided by the ability of synapses to constantly change their effectiveness. It is believed that this phenomenon, a key event of synaptic plasticity, underlies multiple forms of long-term memory in mammals. It is the excitatory glutamatergic synapses that play a crucial role in synaptic transmission, synaptic plasticity and behavioral adaptation. Almost three decades ago, it was suggested that GCs and sex steroids regulate the neurochemical and structural plasticity of the hippocampus through cellular mechanisms mediated by N-methyl-D-aspartate receptors (NMDAR) of glutamate [39], and since then this has been confirmed by numerous experimental results. The synaptic plasticity of the hippocampus, which depends on glutamate receptors, is considered by many scientists to be the basis of learning processes and behavioral adaptation. Glutamate is known as the main excitatory neurotransmitter in the central nervous system, and the functioning of the glutamatergic system is provided by numerous receptors, including ionotropic and metabotropic subtypes. The first ensures the passage of cations through the postsynaptic membrane, while the metabotropic subtype activates signaling cascades through secondary messengers.
Ionotropic glutamate receptors mediate the synaptic and metabolic actions of glutamate. Along with NMDAR, ionotropic glutamate receptors include families of functional receptors of α-amino-3-hydroxy-5-methyl-4-isoxazolpropionic acid (AMPAR) and kainate receptors. The functioning of ionotropic receptors in the hippocampus depends on their subunit composition, the amino acid sequence of protein domains and scaffold proteins in synaptic membranes. These receptors are reactively plastic and rearranged by regulating subunits (quantitatively and qualitatively). It is the rearrangements of ionotropic receptors and changes in their subunit composition that turn neurons into “pathological” cells, determining the states of plasticity or pathology of the hippocampus [40]. For example, it is assumed that the GluN2B subunit may be particularly important for plasticity and memory formation, and its role is associated with interaction with calcium/calmodulin-dependent protein kinase II [41]. NMDARs are believed to provide a key trigger for the induction of long-term plasticity leading to changes in AMPAR expression. AMPAR, one of the fastest-acting neurotransmitter receptors, are located in excitatory synapses, are activated within hundreds of microseconds and deactivated within milliseconds due to their low affinity for glutamate, and are also capable of deep desensitization [42]. The third type of ionotropic receptors, kainate, contribute to critical postsynaptic signals, and also function as presynaptic autoreceptors [43]. Kainate receptors are considered as homeostatic modulators of neurotransmitter release, capable of bidirectionally regulating plasticity depending on the “functional history” of the synapse [44]. Metabotropic glutamate receptors (mGluR), a group of G-protein-coupled receptors, exert a wide range of modulating actions on excitatory synapses of the central nervous system. In the hippocampus, selective activation of various mGluRs modulates internal excitability, the strength of synaptic transmission, and induces multiple forms of long-term plasticity [45]. These receptors are critically needed both for stable forms of memory and for stable synaptic plasticity. Metabotropic glutamate receptors are divided into several separate groups based on their binding to G-protein and affinity for agonists and perform different functions in the regulation of hippocampus-dependent long-term plasticity and long-term memory [46].
As mentioned above, the hippocampus is one of the most important targets of steroid hormones in the brain. Steroid hormones and neurotransmitters function in concert, and this coherence is regulated by numerous mechanisms. The review [11] provides a detailed scheme of the currently known genomic and non-genomic mechanisms of direct modulation of various types of glutamate receptors carried out by GCs via MR and GR in glutamatergic presynapse and postsynapse. GCs quickly tune synaptic NMDARs through membrane dynamics and signal transmission via MR and cause long-term changes and preservation of “settings” through GR-mediated mechanisms. Thus, GCs modulate the transmission of glutamatergic signals and NMDAR-dependent synaptic plasticity of the hippocampus, contributing to the implementation of adequate behavioral responses to environmental factors. GCs specifically control the dynamics of GluN2B-NMDAR and synapse states by tuning NMDAR-dependent synaptic adaptation [47].
Along with the direct action on the glutamatergic synapse, the regulatory action of GCs is also carried out on other critical processes that ensure synaptic plasticity, including neurogenesis (described in the previous chapter). It is known that neurogenesis in the adult and developing dentate gyrus is “limited” by GCs, as well as by excitatory amino acids (primarily glutamate), and in both cases the mechanisms involve NMDAR. These results not only indicate a high degree of interdependence between some neurotransmitters and GCs, but also emphasize how important an aspect of the action of steroid hormones is structural plasticity [48]. New neurons are constantly generated from resident pools of neural stem cells and progenitor cells in the adult hippocampus, while NMDARs are involved in the regulation of progenitor cell proliferation. NMDAR agonists and antagonists modulate proliferation both in vivo and in vitro, and the presence of NMDAR is shown in the neural precursor cells.
Glutamate uptake is a process mediated by sodium-dependent glutamate transporters that prevents the leakage of glutamate from the synapse. As a rule, astrocytes are responsible for most of the glutamate uptake by expressing glutamate transporters; neurons can also express these transporters, albeit in smaller quantities. These transporters are actively involved in the realization of the most studied phenomena of long-term synaptic plasticity – long-term potentiation and long-term depression [49]. Insufficient reuptake of glutamate leads to hyperglutamatergic states [50] and excitotoxicity-mediated neuronal death. Corticosterone has been shown to inhibit the expression of the microglial glutamate transporter GLT-1 [51], modulate the expression of the astrocytic glutamate transporter GLT-1 [52, 53], and reduce the activity of the glutamate transporter type 3 (EAAC1 or EAAT3) [54].
Considering the importance of glutamatergic signaling in synaptic plasticity, learning and memory, and the realization of movements, it is believed that the state of glutamatergic neurotransmission is the key link in the plasticity of the hippocampus, and glutamate serves as a link and the basis for the balance between states of cognitive health and disease [55]. Glutamatergic transmission largely determines the functional properties of the hippocampus in specific situations, providing it with an appropriate position in the continuum of “plasticity–pathology” [56]. Excess extracellular glutamate causes excessive stimulation of ionotropic glutamate receptors, causing hyperglutamatergic states [50] with their characteristic excitotoxicity, oxidative stress, structural and functional damage to brain cells. These processes play a crucial role in the development of various brain pathologies associated with neuroplasticity disorders, such as strokes, epilepsy and neurodegenerative diseases [57].
The main cause of age-related cognitive decline has long been considered the loss of neurons, but now these changes are associated with gradual synaptic dysfunction caused by calcium dyshomeostasis and changes in ionotropic/metabotropic receptors. NMDARs play a central role in the mechanisms mediating synapse aging. Areas of the brain that are vulnerable to aging show the earliest pathological changes in AD. In the hippocampus, a region of the brain selectively vulnerable to adverse factors and aging, impaired synaptic function during aging is associated with shifts in Ca2+-dependent synaptic plasticity, which is believed to contribute to early cognitive deficits associated with the development of dementia, including Alzheimer’s type [58]. Changes in the intracellular redox state are accompanied by disturbances in the regulation of Ca2+, including NMDAR hypofunction and increased release of Ca2+ from intracellular stores, which alters synaptic plasticity. In AD, β-amyloid and mutated presenilin 1 can also impair the function of NMDAR, contributing to the release of Ca2+ from intracellular stores and increasing oxidative stress. The control of mGluR5-dependent NMDAR activation and subsequent Ca2+ dysfunction during aging is carried out by adenosine A2A receptors [59]. With aging, changes in the expression and functionality of other metabotropic glutamate receptors are also observed, including those in the synapses of mossy fibers – CA3 of the hippocampus [45].
Since GCs regulate the state of the glutamatergic system of the brain both directly, through receptors on glutamatergic synapses, and indirectly [11], disruption of the functioning of HPAA and its inability to optimally regulate glutamatergic synaptic plasticity leads to the development of neuropsychiatric diseases, in the pathogenesis of which hyperglutamatergic states play a key role. Violation of glucocorticoid control of glutamatergic processes underlies cognitive and emotional disorders, epilepsy and a number of other cerebral pathologies, being a common underlying mechanism for the development of many brain diseases and their comorbidities [50]. In this regard, the study of the mechanisms of interaction between HPAA and the glutamatergic system of the brain has a priority translational value.
GLUCOCORTICOIDS AND BDNF JOINTLY CONTROL HIPPOCAMPAL
PLASTICITY
A family of small secreted proteins called neurotrophins includes powerful molecular mediators of central synaptic plasticity. In particular, brain- neurotrophic factor (BDNF) and neurotrophin-3 (NT3) play a key role in the neurobiological mechanisms associated with learning and memory. The influence of BDNF and NT3 on synaptic plasticity may be permissive, establishing conditions for occurring plastic changes, or it may be instructive, directly modifying the communication and morphology of synapses. BDNF stands out among all neurotrophins with a high level of expression in the brain, in particular in the hippocampus, and a powerful effect on synapses [60]. BDNF has a pleiotropic effect on neuronal morphology and synaptic plasticity, which underlies the development of hippocampal neural circuits and cognitive function. BDNF contributes to the stabilization and maturation of existing synapses, as well as the generation of new synaptic contacts. Since BDNF modulates both the electrical properties and the structural organization of synapses, it is considered an important biological marker of learning and memory processes [61].
The function of BDNF is controlled and diversified by molecular and cellular mechanisms, including delivery and subcellular compartmentalization of various types of Bdnf mRNA, pre- and postsynaptic release of BDNF, control of BDNF signaling via TrkB receptors (tropomyosin B receptor kinase) and conversion of pro-BDNF into mature BDNF and BDNF-propeptide. The diverse effects of BDNF on excitatory synapses are determined by the activation of TrkB receptors and underlying signaling pathways, as well as the functions of its immature form (pro-BDNF) activating p75NTR receptors, which are opposite in comparison with mature BDNF. Violation of these regulatory mechanisms affects the formation of dendritic spines and the morphology of pyramidal neurons, as well as synaptic integration [62]. The most important aspects of BDNF biology, such as transcription, processing, and secretion, are regulated by synaptic activity. It was these observations that led to the idea that BDNF can regulate activity-dependent forms of synaptic plasticity, such as long-term potentiation (LTP), a steady increase in excitatory synaptic efficiency in the hippocampus, which is believed to underlie learning and memory [60]. BDNF is released from the presynaptic terminals of excitatory neurons (probably also from postsynaptic endings) and acts through the TrkB receptor on pre- and postsynaptic sites of excitatory, primarily glutamatergic neurons and inhibitory neurons [63]. BDNF is considered a central regulator of neuronal plasticity in the adult hippocampus, not only because it mediates morphological correlates of neuronal plasticity – changes at the level of dendritic spines, but, at least in the dentate gyrus of the adult hippocampus, BDNF controls plasticity at the level of neurogenesis. Specific changes in the dendritic spines, as well as in the neurogenesis of the adult hippocampus, can be observed in the context of several forms of learning and memory, on the other hand, depression is known to be accompanied by a decrease in the rate of neurogenesis and the density of spines [64].
The “behavioral decisions” made by the brain during stress depend on the signaling pathways of GCs and BDNF acting synchronously in mesolimbic (reward) and corticolimbic (emotions) neural networks. A violation of the control of BDNF and GR expression in the areas of the brain that evaluate external factors, including the hippocampus, may jeopardize the integration of signals. Phosphorylation of GRs during transmission of BDNF signals in neurons is one of the mechanisms underlying the integration of BDNF and GC signals, which, in case of imbalance, can serve as a basis for maladaptation to stress. The interaction between GCs and BDNF determines the stress response to a large extent: stress-induced remodeling of the hippocampus, prefrontal cortex and amygdala coincides with changes in BDNF levels [65]. Chronic stress is usually associated with a decrease in BDNF levels, although the effect of exposure is determined by the nature and severity of the stimulus, varies in different areas of the brain and changes over time during and after exposure to the stressor [66]. The effects of GCs and BDNF on the structural and cellular plasticity of the hippocampus, as a rule, have the opposite character [13, 66, 67]. The interaction between GCs and BDNF can also play a role in acute, rapid, and adaptive stress responses, and are also an important target for BDNF-mediated signaling [68]. BDNF induces phosphorylation of GR, which, providing coordinated/paired actions of BDNF and GCs, is apparently an essential link in the neuroplasticity response to stress. Thus, the interaction between GCs and BDNF against the background of the influence of other molecules can contribute to plasticity in the adaptive response to stress. Changing this interaction under extreme conditions may also be involved in maladaptive responses to stress, leading to cognitive impairments and disease states. Consistency between TrkB and GR signaling is a determining factor for adequate cellular responses to stress [60].
As noted earlier, chronic exposure to GCs causes damage to neurons and dendrite atrophy, reduces neurogenesis in the hippocampus and worsens synaptic plasticity. Since GCs also alter the expression and signaling of BDNF, which promotes neuroplasticity, increases cell survival, hippocampal neurogenesis and cellular excitability, it has been suggested that specific side effects of GCs may be mediated by reducing expression and signaling of BDNF [13]. Glucocorticoid regulation of BDNF occurs at several levels, from GC-induced changes in BDNF mRNA to changes in TrkB-mediated signaling. Bdnf transcription can be modulated by GCs either by direct binding to putative glucocorticoid response elements (GREs) present in promoter regions, or by interfering with the activity of other factors contributing to Bdnf transcription, such as activator protein-1 complex (AP-1) and transcription factor CREB (cAMP response element-binding protein) [69]. In addition to transcriptional regulation of Bdnf, GC can also potentially alter the translation of the Bdnf gene by modulating the activity of the translational mechanism. The transformation of secreted pro-BDNF into mature BDNF is mediated by a variety of intracellular and extracellular proteases, including furin and prohormone convertases inside the cell, as well as the plasmin–tissue plasminogen activator (tPA) system and matrix metalloproteinases outside the cells. GCs can modulate the levels or activity of intracellular and extracellular proteases and thus regulate the levels of available mature BDNF. Mature BDNF binds to intracellular chaperones that allow sorting of BDNF by regulated secretory pathway or constitutive pathway. Pro-BDNF and mature BDNF are packaged and transported to either dendrites or axons. BDNF is released in response to neuronal activity and postsynaptically interacts with its TrkB receptor or the low-affinity p75NTR receptor to activate various signaling cascades. By binding to TrkB, BDNF activates the pathways of mitogen-activated protein kinase (MAPK), phospholipase C (PLC) and phosphatidylinositol-3-kinase (PI3-kinase). Activation of these signaling pathways induces functional modulation of the underlying target molecules involved in synaptic plasticity, neuronal survival and cellular excitability. GCs potentially modulate the activity of these signaling pathways at several levels. GCs prevent the interaction of TrkB with specific adapter proteins such as Shp2, thus disrupting the activation of the MAPK pathway. In addition, they stimulate the transcription of the MAPK inhibitor protein, MAPK-1 phosphatase (MKP-1), which stops the transmission of MAPK signals. GCs also impair the interaction of TrkB and GRs, thereby weakening the activation of the phospholipase C pathway [69].
The role of BDNF as a trophic factor and a central regulator of synaptic plasticity is implemented in close interaction with other systems. First of all, the effect of BDNF depends on the coactivation of GCs and other factors as determinants of the final cellular response [66]. Excitatory neurotransmitter glutamate and BDNF are the most important “regulatory pair” for synaptic plasticity throughout the central nervous system [70]. The BDNF system and the glutamatergic system are closely related and actively interact in the formation of hippocampal plasticity. The connections between the two systems are numerous and bidirectional, and it is the complex and well-coordinated nature of these connections that ensures optimal synaptic and cellular plasticity [71]. Both systems are under the control of the GCs, which ensure their coordination and synchronized functioning. Both systems are associated with the pathogenesis of depression, and the disruption of close and well-balanced connections between them leads to adverse changes in neural plasticity underlying depressive disorders and other mental illnesses.
All neurotrophins are first synthesized as pro-neurotrophins, and then cleaved intracellularly and extracellularly. More and more evidence suggests that pro-neurotrophins and mature neurotrophins play opposite roles in the central nervous system. This idea concerns the participation of nerve growth factor (NGF), BDNF, neurotrophins 3 and 4 (NT3, NT4) and their corresponding forms in cellular processes related to learning and memory. Among the mechanisms of “maturation” of BDNF, a completely specific posttranslational mechanism has been revealed, namely, the transformation of the BDNF precursor into mature BDNF by proteolytic cleavage [72]. In addition to the actively studied role of BDNF, it turned out that specific biological roles in synaptic plasticity are also played by endogenously secreted by nerve cells BDNF precursor protein and BDNF prodomain, called BDNF propeptide [73]. Initially, it was believed that pro-neurotrophins are simple inactive precursors responsible only for ensuring the folding of the mature domain and for regulating the secretory pathway of neurotrophins. However, it turned out that pro-neurotrophins are biologically active due to the transmission of signals through specific receptors. Recent studies show that pro-neurotrophins can be secreted into the extracellular space, bind with high affinity to specific receptor complexes and induce activation of the apoptotic mechanism with subsequent cell death of various neuronal populations. In addition to obvious pathological situations, extracellular pro-neurotrophins also play a key role in many other cellular mechanisms in the nervous system [74]. It has been shown that pro-neurotrophins mediate synaptic plasticity phenomenon, namely long-term depression in hippocampal neurons, and are important for the development of axons. The transformation of proneurotrophins into the corresponding mature form is controlled by the activity of several enzymes (proteinases) and regulatory molecules. A failure in this regulation is currently considered one of the possible mechanisms responsible for the pathological cell death associated with pro-neurotrophins. According to modern concepts, neurobiological actions of three different subtypes of brain neurotrophic factor are distinguished and a multiligand model of growth factor signaling is formulated [75]. The effects of BDNF on the synaptic proteome are realized either by affecting the mechanism of protein synthesis, or by regulating protein degradation by calpains and, possibly, by the ubiquitin-proteasome system (UPS). This fine-tuned control of the synaptic proteome, rather than just activation of protein synthesis, may play a key role in BDNF-mediated synaptic potentiation [76].
Stress, which may remain a risk factor of the “lowest common denominator” for a number of mental and neurological diseases, affects the BDNF system in areas and circuits of the brain that are selectively vulnerable to external factors. This view is based on both experimental and clinical data. It is noted that BDNF plays a still underestimated multifactorial role as both a regulator and a target for the transmission of GCs stress signals in the brain [77]. One of the hypotheses explaining the occurrence and severity of mental and neurological disorders is the loss of trophic support [13]. Indeed, changes in BDNF levels and activity occur in numerous neurodegenerative and neuropsychiatric diseases. BDNF deficiency contributes to vulnerability, whereas enhanced function contributes to recovery by increasing survival, synapse formation, and synaptic plasticity. Normal levels of GCs support normal brain function, excessive secretion of GCs accelerates the development of affective disorders associated with stress. Another logic leading to the same conclusions is based on the fact that the synergistic interactions between neuronal activity and synaptic plasticity mediated by BDNF make it an ideal and important regulator of cellular processes underlying cognition and other complex behaviors, and the deficiency of BDNF signaling contributes to the pathogenesis of a number of brain diseases, such as Huntington’s disease, AD, and depression [78].
The idea that impaired BDNF signaling may be associated with affective disorders arose primarily from data on the opposite effects of antidepressants and stress on BDNF signaling. Antidepressants enhance BDNF signaling and synaptic plasticity, and harmful environmental factors, such as severe stress, suppress BDNF signaling, disrupt synaptic activity and reduce resistance to affective disorders [79]. Studies in humans with a single nucleotide polymorphism in the Bdnf gene, BDNF Val66Met, which affects the regulated release of BDNF, have shown a deep deficit in plasticity of the hippocampus and prefrontal cortex, as well as in cognitive function. BDNF regulates synaptic mechanisms responsible for various cognitive processes, including attenuation of aversive memories, which is a key process in the regulation of affective behaviors. Therefore, the unique role of BDNF in cognitive function and affective behavior suggests that cognitive deficits due to altered BDNF signaling may underlie affective disorders. Stress and depression are associated with neuronal atrophy and decreased synaptic connections in brain regions such as the hippocampus and prefrontal cortex, and these contribute to depressive behavior, while treatment with antidepressants can reverse this deficiency. Exposure to stress and depression reduces the expression of BDNF in these structures, and treatment with antidepressants can activate BDNF in the adult brain and neutralize the effects of stress. New data on the mechanisms of action of fast-acting antidepressants, in particular, the NMDAR antagonist ketamine shown that the observed rapid synaptic and antidepressive behavioral effects of ketamine are associated with activity-dependent release of BDNF [80].
BDNF acts as a paracrine and autocrine factor on both presynaptic and postsynaptic target sites. This is crucial for converting synaptic activity into long-term synaptic changes associated with memories. BDNF affects dendritic spines and, at least in the hippocampus, neurogenesis, namely, changes in the rate of neurogenesis and the density of spines can affect some forms of learning and memory on the one hand or contribute to depressive behavior on the other. With this in mind, it is not surprising that BDNF, one of the key molecules that modulate brain plasticity and affect cognitive deficits, is associated with aging and neurodegenerative diseases. Cognitive decline with age is a major risk factor for cognitive diseases, and changes in BDNF generation and secretion, as well as BDNF/TrkB signaling, have been found in various neurodegenerative diseases such as AD and Parkinson’s disease, as well as mood disorders such as depression, eating disorders, and schizophrenia [81, 82].
GLIAL MECHANISMS OF HIPPOCAMPAL PLASTICITY MANAGED BY
GLUCOCORTICOIDS: ASTROCYTES, MICROGLIA, NEUROINFLAMMATION
Astrocytes, the most common glial cells in the brain, play a key role in regulating the synaptic plasticity of the hippocampus. Previously, astrocytes were described as a homogeneous cell population, but now it has been shown that in the adult hippocampus, astrocytes are very heterogeneous and can react differently to changes in neuronal activity depending on the hippocampal subregion, actively modulating synaptic plasticity [83]. Changes in the local activity of neurons regulate interactions between astrocytes and synapses, either by modulating the secretion of gliotransmitters and synaptogenic proteins, or through signaling pathways triggered by direct intercellular contacts [84]. Such specific reactions induced in astrocytes mediate interactions between astrocytes and neurons, thereby forming synaptic communications in the adult hippocampus. Violation of the regulation of these interactions and signal transduction can cause dysfunction of the hippocampal neural networks in pathological conditions, leading to cognitive impairment and neurodegeneration. It has been shown that reactive astrocytes, in which the regulation of the transmission of signals potentially related to learning and memory is disrupted, are induced in the brains of patients with AD and in transgenic murine models of AD [85].
Along with neurons, astrocytes are stress-reactive cells, and the presence of astrocytic GRs determines the direct regulation by GCs of these glial cells, including in stressful situations. “Early life stress” (adversive experiences in early ontogenesis, one of the most significant risk factors for the development of mood disorders and anxiety disorders later in life) is currently one of the most commonly used and translationally valid GCs-dependent stress models in rodents. Specifically in the limbic system, stress at an early age causes long-term changes in neural networks, neurotransmitter systems, neural architectonics and plasticity, and these changes further significantly affect the processing of emotional and cognitive information. It has been shown that astrocytes, along with neurons, also change functions almost for life after early life stress [86]. As a component of the tripartite synapse, astrocytes interact with neurons in several ways, affecting the uptake and metabolism of neurotransmitters, secreting gliotransmitters and providing energy to neurons in local networks. Thus, astrocytes modulate the plasticity of neurons, mediating the long-term effects of stress triggered by GCs at an early age.
As already mentioned, neurogenesis in the adult hippocampus is one of the most remarkable forms of plasticity, and there is increasing evidence that this process is associated with both memory mechanisms and the development of cognitive and depressive disorders. Astrocytes are part of the neurogenic niche that provides a structural and molecular support for stem cell proliferation and differentiation as well as functional integration of new neurons [87]. Astrocytes make a significant contribution to the control of neurogenesis, and changes in the function of astrocytes can disrupt the regulation of neurogenesis in adults and contribute to cognitive impairment, including in the context of AD and emotional disorders.
The central nervous system was previously considered an immune-privileged part of the body with no immune cell responses, but this point of view has now been completely revised. Microglia are resident tissue macrophages, innate immune cells of the brain responsible for supporting the functioning of neurons and immune protection of the brain parenchyma. These are the primary immune effector cells in the central nervous system, which regulate the broad interaction between the nervous and immune systems in response to various immunological, physiological, and psychological stressors. Therefore, microglia contributes to normal brain function, but is also involved in various cerebral pathologies [88]. Microglia possesses high plasticity and plays an integral role in the formation of the brain structure, the improvement of neural circuits and synapses, actively contributing to the plasticity of neurons in a healthy brain. Recently, studies have revealed various features of microglia specific to certain brain regions and have shown that the maturation and function of individual neural circuits may be potentially related to the molecular identity of microglia in different brain structures [89]. Microglia can play a role in physiological and pathological conditions, regulating the growth of axons and dendrites, contributing to the formation, elimination and movement of synapses, modulating the functioning of excitatory synapses, participating in functional synaptic plasticity. Ultimately, depending on environmental conditions, microglia modulate the state of the hippocampus in different ways and affects memory function [90]. As mentioned above, new neurons are constantly being generated from stem cells and integrated into the adult hippocampus, contributing to the highest level of neuroplasticity, memory function. Data have been obtained that indicate the modulating participation of microglia both in the formation of new neurons and in the mechanisms governing their inclusion in the neuronal circuits associated with the realization of memory [90]. The hippocampal microglia interacts with local factors, such as BDNF, and external stimuli that promote neurogenesis. Microglia interacts with serotonin, a neurotransmitter that is definitely involved in neurogenesis in adults and is known for its role in antidepressant action [91].
Stressful events cause, among other things, a rapid increase in the level of brain and a slower increase in GCs. Microglia, a key regulator of neuronal function, contain adrenaline and GC receptors and can potentially be involved in modulating the effects of stress on neuronal function, learning, and memory [92]. Since microglia in the mature brain affects synaptic signaling, provides trophic support, and forms synaptic plasticity, these cells are, along with neurons, participants in the implementation of the regulatory effects of GCs on various forms of brain plasticity.
Hyperactivity of HPAA in chronic stress and a number of neuropsychiatric diseases is caused by reduced inhibition of GC secretion by feedback, due to a decrease in the transmission of HPAA signals and increased secretion of CRH from the hypothalamic paraventricular nucleus and extrahypothalamic neurons. During inhibition of systemic feedback caused by chronic stress, there is an increase in the level of pro-inflammatory cytokines secreted by both immune and non-immune cells, and the levels of cytosolic GRs in the hippocampus and prefrontal cortex change. Prolonged response to stress and an excess of cytokines disrupts the plasticity of neurons, and inflammatory reactions in the brain contribute to cell damage [93]. Stress, especially chronic, causes proliferation of microglia, as well as its shift towards a pro-inflammatory phenotype. As a result of significant stress effects, the interaction of microglia with neurons and the transmission of the glutamate signal is disrupted; the immune reactions of microglia after stress affect metabolism of tryptophan by activating the kynurenine pathway generating metabolites that disrupt glutamate transmission (kynurenic acid is an endogenous antagonist of NMDARs). All these effects may underlie memory disorders and synaptic plasticity alterations caused by severe or prolonged stress of different nature. For example, psychological stress can disrupt the function of microglia, which contributes to impaired plasticity of neurons and the development of changes in emotional behavior. Stress-induced microglial dysfunction may underlie neuroplasticity deficiency associated with many mental illnesses [88].
Activation of microglia is a distinctive feature of almost all known pathologies of the brain. Chronic activation of microglia can, in turn, cause damage to neurons due to the release of potentially cytotoxic molecules, such as pro-inflammatory cytokines, oxygen radicals, proteinases and complement proteins [94]. The acute inflammatory response of microglia to injury, stress or infection involves the release of cytokines and phagocytosis of damaged cells. Accumulating data indicate chronic microglia-mediated inflammation in almost all diseases of the central nervous system associated with the emotional and cognitive spheres, and its connection with the progression of diseases. The hippocampus is particularly vulnerable to neuroinflammation. One of the reasons is that chemokines and cytokines are involved in normal neurogenesis, cellular plasticity, learning and memory. Neuroimmune interactions and immune signaling molecules, especially chemokines, may be the main mechanism combining plasticity and vulnerability of the hippocampus and switching these states under the influence of external and internal factors [95].
Microglia regulates neuroimmune pathways that affect neuroplasticity and potentially lead to depressive disorders, the pathogenesis of which is directly related to excessive secretion of GCs and impaired regulation of HPAA. Several hypotheses have been proposed about the role of microglia in the onset of depression, but all of them somehow involve key molecular pathways mediating microglia-related neuroinflammation and degeneration of hippocampal neurons. Excess of GCs and associated changes in the expression of neurotrophic factor genes, as well as neuroactive substances secreted by the intestinal microbiota, affect the morphology and phenotype of microglia [96]. Neuropsychiatric disorders (e.g., mood disorders, schizophrenia) and inflammation are closely intertwined and possibly reinforce each other; for example, depression contributes to inflammatory reactions, and inflammation contributes to depression and other neuropsychiatric disorders. Patients with neuropsychiatric disorders show all the main signs of inflammation, including an increased level of circulating inflammatory inducers, activated targets and inflammatory mediators affecting all tissues. Inflammation can contribute to the pathophysiology and clinical progression of these disorders. It should be noted that an excess of pro-inflammatory cytokines negatively modulates emotional behavior and cognition, reducing the level of monoamines in the brain, activating neuroendocrine responses, contributing to excitotoxicity and disrupting brain plasticity. At the same time, changes in the regulation of HPAA act as an important trigger of inflammation [97].
It is believed that the inflammation in the central nervous system plays a key role in the processes leading to the death of neurons in a number of neurodegenerative diseases, including Parkinson’s disease, AD, prion diseases, multiple sclerosis and HIV dementia. Repeated exposure to stress, mediated by excessive secretion of GCs and impaired control of HPAA, increases the risk of neurodegenerative diseases, including sporadic AD. Microglia is causally associated with the accumulation of β-amyloid, tau pathology, neurodegeneration and loss of synapses in AD, although it plays a positive role, especially in the phagocytic elimination of an excess of amyloid peptides. The involvement of microglia altered as a result of chronic stress is associated, in particular, with the appearance of new microglia phenotypes, presumably related to neuroprotection in AD [98]. AD is considered as a kind of pathological aging, but microglia is a key cellular element in the mechanisms of normal aging. Older people often experience cognitive decline after stressful events (for example, infections or injuries) that trigger activation of the immune system. This is partly because aging increases the sensitivity of the microglial response to immune signals. In the aging brain, microglia respond to these signals by producing more cytokines and for a longer period. Although the presence of microglia is necessary for the realization of memory, overactivated by immune signals, microglia excessively produces inflammatory cytokines. This is unfavorable for memory function due to the powerful negative effect of cytokines on hippocampal synaptic plasticity. BNDF helps protect neurons from damage caused by infection or trauma, and plays a critical role in the same processes of memory and plasticity of the hippocampus, which are altered by a violation of the regulation of interleukins production by microglia [99]. The excessive inflammatory response of the brain that occurs during aging under the influence of a secondary immune challenge may weaken the ability to provide BDNF necessary for memory-related plasticity processes in hippocampal synapses. HPAA malfunction in aging is most likely the key cause of the multi-stage cascade, which is associated with impaired reactivity of microglia to stress factors.
GLUCOCORTICOIDS ARE INVOLVED IN PROTEASE-DEPENDENT PLASTICITY OF
THE HIPPOCAMPUS
Since all plastic rearrangements in the brain are somehow connected with the modification of the structure of protein molecules and the resulting changes in their functioning, the regulation of enzymes that catalyze posttranslational modifications of proteins is undoubtedly important for the realization of neuroplasticity phenomena. Learning and memory require changes in the number and strength of existing synaptic connections, and extracellular proteolysis in synapses plays a key role in synaptic plasticity, determining the structure, function and number of synapses. The early phase of the well-studied phenomena of synaptic plasticity, long-term synaptic potentiation and depression depends on posttranslational modifications of synaptic proteins. There is ample evidence of the role of various types of proteases in synaptic plasticity, which reflects the diversity of mechanisms involved in the regulation of intracellular and extracellular protein content [100]. The cleavage of extracellular proteins is associated with changes in postsynaptic intracellular mechanisms, and additional changes in this compartment are the result of protease-mediated cleavage of intracellular proteins. Both mechanisms contribute to the initiation of signaling cascades that control the downstream pathways associated with synaptic plasticity. The review [100] summarizes data on the role of extracellular and intracellular proteases with different specificity, localization, and regulation of these enzymes in synaptic plasticity. The combined actions of proteases and translational mechanisms ensure tight control of the synaptic proteome, which is important for long-term plasticity. Nevertheless, it can be recognized that the role of proteases in neuroplasticity is underestimated due to an insufficient number of studies.
Regulated extracellular proteolysis plays a key role in the structural and functional remodeling of synapses during brain development, learning and memory formation. Synapses of mossy fibers on pyramidal cells of the hippocampus CA3 subfield demonstrate several unique functional features, including short-term facilitation, presynaptic mechanisms of long-term potentiation independent of NMDAR activation, and NMDA-dependent metaplasticity. Functional and structural plasticity of mossy fiber synapses is mediated by extracellular proteolysis. It has been shown that among perisynaptic proteases, tissue plasminogen activator (tPA)/plasmin system, β-secretase (BACE1), an enzyme that cleaves the amyloid precursor protein (APP) and metalloproteinases play vital roles in the plastic changes [101]. Although it is generally recognized that it is the synaptic plasticity of the excitatory synapses of the hippocampus that plays a crucial role in the formation of memory traces, some components of neuroplasticity are associated with non-synaptic alterations. The activity of extracellular proteases can affect the processing of information in neuronal networks by affecting targets outside synapses. Interestingly, extracellular proteolysis can change the internal excitability of neurons and the balance of excitation/inhibition both in the short term (from minutes to hours) and in the long-term range. Moreover, it turns out that by cleaving the components of the extracellular matrix, proteases can modulate the function of ion channels or change inhibition and, consequently, facilitate the active participation of dendrites and initial axon segments in regulating the function of neurons. In general, both rapid and prolonged extracellular proteolysis can affect some aspects of information processing in neurons, such as initiation of action potential, adaptation of spike frequency, properties of action potential and back propagation in dendrites [102].
The regulatory effects of GCs on the activity of proteases important for neuroplasticity are also insufficiently studied; however, there are results indicating that this group of enzymes is under the control of GCs, although the specific mechanisms are not always described in detail. A number of data on changes in the activity of proteases important for neuroplasticity induced by stress, GCs or their receptor ligands point to the role of proteases as one of the main factors in the regulation of neuroplasticity coordinated by GCs.
Before considering the data on the GC-related proteolytic activity of brain proteases in the realization of neuroplasticity phenomena, it should be stated that the detrimental effect of chronically elevated levels of GCs on the structure and function of neurons up to their death has been repeatedly described and can be considered a common place. The pro-apoptotic effects of GCs, leading to the death of neurons, by definition include activation of apoptotic proteases, including caspases and calpains. Nevertheless, even such “destructive” enzymes are expressed in basal conditions and cleave proteins important for normal neuroplasticity. For example, earlier we showed that caspase-3 is necessary to maintain normal plasticity of the hippocampus, since there are many proteins (including cytoskeletal) among its substrates, limited proteolysis of which is necessary for the implementation of normal synaptic plasticity [103, 104]. Calpains are a family of soluble calcium-dependent proteases that are involved in many regulatory pathways and also play an essential role in neuroplasticity. Two isoforms of calpain, calpain-1 and calpain-2, execute opposite functions in the brain. Activation of calpain-1 is necessary for certain forms of synaptic plasticity and the corresponding types of learning and memory, while activation of calpain-2 limits the degree of plasticity and learning. Calpain-1 has a neuroprotective effect both during postnatal development and in adulthood, whereas calpain-2 has a neurodegenerative effect [105]. Unfortunately, there are little direct data on the mechanisms of regulation by GCs of “apoptotic” proteases, caspases and calpains, in the realization of normal plasticity. It was shown that in the hippocampus of adult Wistar rats, 10-day immobilization stress led to a significant decrease in the number of neuronal and astroglial cells in the CA1 and CA3 regions, an increase in the number of caspase-3-positive cells, an increase in GR mRNA levels and a decrease in MR mRNA levels (maximum in the dentate gyrus and CA3 region) [106]. In this experiment, the appearance of active caspase-3 was apparently associated with apoptotic cell death.
Based on the pleiotropic function of proteases, it can be assumed that there are many potential targets of proteases in the realization of synaptic plasticity in the hippocampus and that these processes are associated with GCs. For example, GCs can modulate the levels or activity of intracellular and extracellular proteases and thus regulate the levels of available mature BDNF [13]. Diffuse and structured extracellular matrix makes up about 20% of the brain volume and plays an important role in the development and plasticity of the adult brain. Perineuronal networks, specialized structures of the extracellular matrix, surround certain types of neurons in the brain. Stress affects the diffuse matrix, as well as perineuronal networks, and the effects of stress depend on age and brain region [107]. Metalloproteinases are a component of the extracellular matrix and targets of GCs/stress. The activity of metalloproteinase 9 (MMP9), a gelatinase involved in the processes of synaptic plasticity, learning and memory, was increased both in animal models with chronic stress and in peripheral blood samples of patients with depression. In a mouse model of depression/anxiety due to chronic corticosterone administration, MMP9 activity and protein levels were significantly increased, and levels of the MMP9 substrate nectin-3 were reduced in the hippocampus, mainly in the CA1 and CA3 regions [108]. MMP9 activity correlated with despair behavior in this depression model. Remodeling of the hippocampus under chronic stress is accompanied by overexpression of GRs, proteasomes and caspase-3. In cultured rat astrocytes, the addition of the glucocorticoid dexamethasone reduced the basal levels of MMP3 and MMP9 mRNA, however, pretreatment with dexamethasone reduced the endothelin-induced increase in mRNA of MMPs. The effects of endothelin-1 on the release of MMP3 and MMP9 proteins were reduced by pretreatment with dexamethasone. These results show that dexamethasone suppresses astrocytic endothelin receptors and reduces endothelin-induced MMP production [109]. In primary cultures of endothelial cells of rat brain microvessels, dexamethasone partially inhibited cytokine-induced activation of MMP9 [110], and in the mouse line of cerebral vascular endothelium cEND dexamethasone induced expression of the MMP inhibitor TIMP-1, which effectively suppresses the activity of MMP9 [111]. It has also been shown in Danio rerio fish that the expression of the Mmp13 gene in the brain is a target for GCs [112].
In most cases, the molecular mechanism underlying the pathogenesis of sporadic AD is unknown. Elevated basal cortisol levels in patients with AD suggest that GCs may contribute to the development and/or maintenance of AD. Amyloid plaques are a hallmark of AD, and it is believed that they play a role in the early stages of AD. However, little is known about how their formation is regulated by stress and GCs. It was shown in [113] that GCs stimulate the formation of astrocytic β-amyloid peptide by increasing the expression of APP and APP-cleaving enzyme BACE1, as well as reducing the expression of β-amyloid-degrading proteases. The accumulation of astrocytes is one of the earliest changes in AD brain. It has been shown that GCs increase the production of β-amyloid in primary astrocyte cultures by increasing the expression of the App gene and the BACE1 β-site. Noteworthy, the administration of GCs to normal middle-aged mice contributed to the expression of APP and BACE1 in astrocytes. GCs significantly reduced the degradation and clearance of β-amyloid by astrocytes in vitro, thereby limiting the neuroprotective capabilities of astrocytes. This could be due to a decrease in the activity of several proteases that degrade β-amyloid peptides, such as insulin-degrading enzyme and MMP9. These effects were mediated by activation of receptors of GCs. Thus, GCs can enhance the accumulation of β-amyloid, reduce its degradation in astrocytes, and thus form a molecular mechanism linking stress factors with the development of AD. In primary cultures of neocortical neurons, corticosterone (1 µm), depending on the duration of application, inhibited the production of the enzyme BACE1 (with a single application), which was accompanied by a decrease in amyloid-β(1-42) levels, or activated the expression of BACE1 and β-amyloid(1-42) (with prolonged use) [114]. Apparently, in this way, GCs are able to regulate the accumulation of β-amyloid fragment 1-42 in AD.
GLUCOCORTICOIDS ARE INVOLVED IN THE REGULATION OF HIPPOCAMPAL
PLASTICITY BY METABOLIC HORMONES
GCs, “stress hormones” coordinate the metabolism and energy status of the body, they are associated with the metabolism of nutrients. For example, the circadian rhythm of GCs secretion is regulated by the time of food consumption in both rodents and humans: GCs levels increase before food intake, and manipulations with the eating regime change the nature of GCs secretion [115]. At the same time, low energy reserves quickly stimulate the secretion of ACTH and GCs in situations of negative energy balance. Patients with an excess of GCs, either endogenous (Cushing syndrome) or exogenous (corticosteroid treatment) origin, are characterized by increased appetite and fat accumulation, while patients with a deficiency of GCs (Addison disease) – decreased appetite and weight loss; similar patterns were obtained in animal studies [115]. Indeed, the system regulating stress responses, HPAA, also regulates responses to food intake, since the neural circuits regulating food intake converge in the paraventricular nucleus, which includes CRH- and urocortin-containing neurons. Given the same anatomy, the systems that control food intake and stress response can influence each other. Complex mechanisms of such interaction include GCs levels (depending on the severity of the stressor), interaction between GCs and food intake-related neuropeptides, in particular neuropeptide Y, α-melanocyte-stimulating hormone, agouti-like protein, melanocortins and their receptors, CRH, urocortin and peripheral signals (leptin, insulin, ghrelin) [116]. Hormones regulating eating behavior regulate HPAA and are controlled by this axis.
At the periphery, insulin plays a critical role in regulating metabolic homeostasis by stimulating glucose uptake by peripheral organs. For decades, the brain has been mistakenly considered an insulin-insensitive organ, but recently research has begun on the mechanisms by which insulin contributes to critical brain functions such as metabolism, cognition and motivated behavior. There is no more doubt about the existence of insulin-mediated synaptic plasticity in the brain both in normal and pathological conditions [117]. In the central nervous system, insulin plays a crucial role in the formation of neural circuits and synaptic connections and contributes to the plasticity of the adult brain. A decrease in the activity of insulin receptors and the transmission of its signals in the brain (insulin resistance), as shown by clinical and preclinical studies, causes a deficit of neuroplasticity, leading to a decrease in cognitive function and an increase in the risk of neuropsychiatric disorders (in particular, this is observed in obesity and type 2 diabetes mellitus) [118]. The brain’s insulin resistance is directly related to the plasticity of the hippocampus, since the transduction of insulin signals affects the molecular cascades in the hippocampus underlying plasticity, learning and memory, and also modulates neurogenesis in the subgranular neurogenic niche [119]. It has also now become apparent that the metabolic hormone leptin performs many functions in the brain that go beyond its established role in hypothalamic control of energy balance. The hippocampus contains regions with a high density of leptin receptors, which, in particular, are localized on the synapses of the CA1 field. Leptin has a pro-cognitive effect, since it rapidly changes the synaptic efficiency in excitatory Shaffer collateral-CA1 and temporoammonic-CA1 synapses, increasing the efficiency of memory in tasks dependent on the hippocampus. The sensitivity of the hippocampus to leptin declines functionally with age, in particular, the modulatory effect of leptin in synapses of both types decreases during aging [120].
The hippocampus expresses high levels of both insulin and leptin receptors, as well as key components of respective signaling cascades, whereas both hormones modulate hippocampus-dependent cognitive functions [121]. Both leptin and insulin affect key cellular events in the hippocampus underlying learning and memory, including activity-dependent synaptic plasticity and transport of glutamate receptors to the synapses. The hippocampus is selectively sensitive to neurodegenerative processes, in particular in AD, in which the functions of leptin or insulin are impaired. Thus, the ability of metabolic hormones, leptin and insulin, to regulate the synaptic function of the hippocampus is important for normal brain function, as well as for the development of cognitive decline. The brain insulin resistance may be a key factor causing cognitive impairments observed in metabolic and neurodegenerative diseases. The critical role of insulin resistance in comorbidities of metabolic and neurodegenerative diseases is established. Type 2 diabetes mellitus is a risk factor for the development of cognitive deficits and AD (people with diabetes have a 2.5 times higher risk of developing dementia). Importantly, the effect on hippocampal neurogenesis is considered as one of the important targets of the pathogenic influence of insulin resistance [122].
The general neurobiological pathways characteristic of type 2 diabetes and depressive symptoms are currently being widely discussed, taking into account the morphological and neurocognitive data obtained recently. Clinical studies have shown frequent coexistence of depression and diabetes; both diseases are associated with similar changes in the brain and behavior. Some morphological and functional changes that occur in these diseases appear to be the result of an excess of pro-inflammatory cytokines or glutamate. Stress-induced GCs not only affect synaptic plasticity, but also disrupt glucose metabolism in the brain and reduce insulin sensitivity [123]. A similar decrease in neuroplasticity in stress-sensitive brain regions, primarily in the hippocampus, may be associated with both type 2 diabetes and depressive symptoms, diabetic patients showing a decrease in neuroplasticity, including morphological abnormalities and subsequent neurocognitive deficits, similar to those characteristics of patients with depressive symptoms [124]. Functional neuroimaging studies have demonstrated altered glucose metabolism in the brain of patients with depression. Changes in the number or activity of key metabolic enzymes and lower sensitivity of insulin receptors were found in the brains of animal models of both diseases. It is obvious that an excess of GCs can disturb the action of insulin and glucose metabolism and restrict energy supply for the proper functioning of neurons and, ultimately, lead to a violation of synaptic plasticity [123, 125].
HIPPOCAMPAL PLASTICITY IS SEX-DEPENDENT: INVOLVEMENT OF STEROID
HORMONES
Numerous studies have demonstrated differences between men and women in the structure, function, and plasticity of the hippocampus. Sex differences in the mechanisms of neuroplasticity are one of the most important, but obviously insufficiently studied problems. The importance of this research area is determined not only by obvious fundamental issues, but also by the fact that almost all diseases associated with neuroplasticity disorders (and these are all cerebral pathologies affecting both the cognitive and emotional spheres) specifically depend on gender. There is a lot of data on the different predisposition of men and women to diseases in which the hippocampus plays an important role, and it is obvious that sex differences in the function of the hippocampus are essential for understanding the mechanisms of cognitive and mental disorders depending on gender [126]. The insufficient number and quality of experimental studies in this area also has an objective reason – the obvious convenience and ease of working with males of small experimental animals. As a result, the predominant number of studies were performed on males, the minimum – on females, and comparative studies with the design including animals of both sexes, remain in the overwhelming minority. Nevertheless, in recent years, data have been obtained that allow us to get closer to understanding the steroid-associated mechanisms of the sex-dependent neuroplasticity.
Sex differences in hippocampal function are observed in many mammalian species. However, the magnitude, degree, and specificity of these differences are unclear, as they may depend on factors such as age, methodology used, and environmental factors [127]. Pyramidal cells of the hippocampal CA3 subfield of male and female rats differ in structure, function and plasticity, and these sex differences cannot be explained simply by the action of circulating gonadal hormones [128]. To date, sex differences have been established for various mechanisms of hippocampal plasticity, cognitive functions, as well as for a number of pathological states affecting hippocampal plasticity. For example, meta-analysis shows that males outperform females in tasks that depend on the hippocampus in rodents and humans; in addition, women are more likely to have a more pronounced decrease in cognitive functions in AD and depression, and both diseases are characterized by hippocampal dysfunction. The hippocampus is a very plastic structure, important for processing high order information and sensitive to environmental factors such as stress. The structure retains the ability to produce new neurons, and this process plays an important role in pattern separation, proactive behavior, and cognitive flexibility. Interestingly, in rodents, noticeable sex differences were found in the level of neurogenesis and activation of new neurons in response to cognitive tasks dependent on the hippocampus [126].
Previously, it was believed that sex steroid hormones are synthesized exclusively in the gonads (for example, estrogens in the ovaries) and cause transcriptional changes in the period from several hours to several days. However, estrogens are also locally synthesized in neural circuits (neuroestrogens), where they quickly (within a few minutes) modulate a number of behavioral responses, including spatial learning and contacts. More and more experimental data suggest that neuroactive sex steroids (neurosteroids) are necessary for memory formation. Neurosteroids have a profound effect on the function and structure of nerve circuits, and local synthesis of these hormones is necessary for the induction of both long-term potentiation and long-term depression of synaptic transmission, as well as for the formation of nerve processes in various areas of the nervous system. With a certain degree of simplification, it can be concluded that in the hippocampus 17β-estradiol (E2) is necessary for the induction of long-term potentiation, and 5α-dihydrotestosterone (DHT) is necessary for the induction of long-term depression. The rapid effect of sex neurosteroids on long-term synaptic plasticity, including memory formation, requires the maintenance of tonic or phasic local synthesis of steroids controlled by neural activity, but may also depend on circulating hormones, age and sex of the animal [129].
The influence of sex hormones (estrogens, androgens, and progesterone) and GCs is mediated by various types of steroid receptors involved (membrane or nuclear) and is accompanied by local metabolic transformations. Sex-dependent characteristics of hippocampal plasticity are described for different regions of this structure and relate to almost all systems that implement plasticity mechanisms. For example, changes in the structural plasticity of the hippocampus in response to estrogens in female rodents have been well studied [130]. In particular, the idea that estradiol can act as a local neuromodulator in the brain, rapidly affecting synaptic function, has been confirmed by studies conducted over the past 30 years. De novo estradiol synthesis in the brain has been demonstrated, as well as signaling mechanisms mediating responses to the hormone, along with morphological data indicating rapid changes in synaptic input after an increase in local estradiol levels [131]. Estrogens modulate and mediate the formation of spines and synapses, cellular and molecular reactions of the hippocampus, as well as neurogenesis. Dendritic spines, postsynaptic structures of synapses, are essential for synaptic plasticity and learning. The formation and modulation of dendritic spines are altered by both the rapid (probably non-genomic) and classical (genomic) action of estrogens, and it is assumed that these mechanisms play a role in the effects of estrogens on learning and memory. Rapid non-genomic modulation of dendritic spines in the hippocampus by neurosteroids and androgens, along with GCs, has been described [132]. Sex steroids (dihydrotestosterone, testosterone, estradiol, and progesterone) and GCs modulate hippocampal synapses due to kinase-dependent signaling mechanisms, providing rapid non-genomic modulation of dendritic spinogenesis. Synaptic (classical) sex steroid receptors are also involved in triggering these rapid synaptic modulations.
Studies of sexual dimorphism in synaptic coding processes underlying learning have been systematically conducted recently. Experiments on male rodents were carried out on a model of long-term potentiation, activity-dependent increase in synaptic strength, as a coding mechanism for identifying synaptic receptors and signaling activity that coordinate activity-dependent remodeling of the subsynaptic actin cytoskeleton, crucial for persistent potentiation and memory. In females, the same mechanisms are utilized for long-term potentiation and memory, but only in females, the use of both modulatory receptors, such as TrkB and synaptic signaling mediators, including Src and ERK1/2, require estrogen synthesized in neurons and signaling through the membrane-associated estrogen receptor α (ERα). Due to the additional involvement of ERα, females have a higher threshold for long-term hippocampal potentiation and spatial learning [133]. The evidence that estrogen promotes learning-related plasticity by modifying the synaptic cytoskeleton, and the acute stimulating effects of estrogen on glutamatergic transmission and long-term potentiation allow us to explain the significant effect of the steroid on behavior. Recent study has identified the mechanisms underlying these synaptic actions [134]. 17β-estradiol triggers actin polymerization in the dendritic spines of the hippocampus through a signaling cascade starting with GTPase RhoA and ending with inactivation of the cofilin, the protein that cuts the filament. Along with direct action, the hormone activates the synaptic receptors TrkB of BDNF, a secreted neurotrophin that stimulates the pathway from RhoA to cofilin. Therefore, it is possible that 17β-estradiol acts through transactivation of neighboring receptors, modifying the composition and structure of excitatory contacts.
Several decades ago, it was suggested that BDNF mediates some of the effects of estrogen in the hippocampus and that these interactions play a role both in the normal brain and in some diseases. For example, the interaction between BDNF and estrogen is shown in the synapses of the mossy fibers of the hippocampus in rodents: 17β-estradiol alters the function of the hippocampus, affecting the expression of BDNF in these synapses. Apparently, estrogen affects the hippocampus through mechanisms related not only to the mature form of BDNF acting on TrkB receptors, but also by regulating the precursor, pro-BDNF acting on the p75NTR receptor. It is assumed that interactions between BDNF and 17β-estradiol in mossy fibers are potentially important for normal hippocampal function and are important for sex differences in functions dependent on mossy fibers, and also in diseases in which plasticity of mossy fibers is assumed to play an important role (AD, epilepsy, and drug addiction) [135]. In general, a special threshold of long-term potentiation in females is associated with sex-specific information processing and the specific features of their learning and memory.
In the last decade, there has been a significant increase in research studying the role of fundamental neuroimmune processes as key mechanisms that can form a natural bridge between normal physiology and pathological outcomes. To date, results have been obtained that stressful events cause time-dependent and stressor-specific patterns of cytokine/chemokine expression in the brain, and inflammation-related genes demonstrate unique expression profiles in females and males depending on individual, cooperative or antagonistic interactions between steroid hormone receptors (primarily receptors of estrogen and GCs) [136]. Thus, neuroimmune stress mechanisms, largely associated with hippocampal plasticity, depend on gender, and this is important for the pharmacotherapy of stress-related diseases.
As mentioned above, neuroestradiol synthesized in the hippocampus plays an important role in neuroplasticity, regardless of the circulating estradiol that is secreted from the gonads. The reaction of the hypothalamic-pituitary regions to the synthesis of neuroestradiol in the hippocampus opens up prospects for a new scientific concept in cognitive neuroscience. The existence of a new regulatory axis hypothalamus-pituitary-hippocampus is postulated. It is assumed that the plasticity of the hippocampus is regulated by neuroblasts, the main cellular unit of functional neurogenesis in the adult hippocampus, and defects in differentiation, integration and survival of neuroblasts in the hippocampus, apparently, are the main cause of neurocognitive disorders. Gonadotropin receptors and steroidogenic enzymes have been found to be expressed in hippocampal neuroblasts. Based on the available data, the cellular basis of neuroestradiol synthesis, the potential relationship between neuroestradiol synthesis and neuroblastosis in the hippocampus, the possible involvement of aberrant neuroestradiol production in mitochondrial dysfunctions and dyslipidemia in menopause and in adults, the pathogenesis of neurodegenerative disorders is re-evaluated. These phenomena are discussed within the framework of the hypothesis of the functioning of the hypothalamic-pituitary-hippocampal axis in the adult brain. Ultimately, understanding the regulation of hippocampal neurogenesis by abnormal levels of neuroestradiol on the feedback principle can provide an additional basis for the development of neuroregenerative therapeutic treatment of emotional disorders, depression and cognitive decline in menopause, as well as neurocognitive disorders [137].
CONCLUSION. TRANSLATIONAL ASPECTS OF GLUCOCORTICOID-REGULATED
HIPPOCAMPAL PLASTICITY
It is simple to make things complex, but complex to make things simple
Meyer’s 3rd Law
Animal studies have shown that the hippocampus is a particularly sensitive and vulnerable area of the brain that reacts to stress and stress hormones. The hippocampus is an important part of the brain for the realization of working and spatial memory in animals and humans, as well as a vulnerable and plastic structure with respect to its sensitivity to epilepsy, ischemia, traumatic brain injury, stress, and aging. Understanding the detrimental effects of elevated levels of GCs on the functioning of brain structures selectively sensitive to these hormones, primarily the hippocampus, stimulated interest in studies of the effect of chronic hypercorticism on the brain. In patients with Cushing syndrome, memory disorders associated with the hippocampus are revealed, while functional disturbances of the hippocampus precede structural anomalies detected by neuroimaging methods. Patients with Cushing syndrome also have impaired executive functions (including decision-making) and other functions, such as visual-constructive skills, speech, motor functions, and information processing speed. Early diagnosis and rapid normalization of hypercorticism can stop and prevent the progression of hippocampal lesions and memory disorders. Among patients with Cushing syndrome, there is a high prevalence of psychopathology, primarily depression and anxiety, also associated with excessive secretion of GCs and changes in the hippocampus [138].
Treatment of critical illnesses usually focuses on short-term physical recovery of the patient, however, people who have survived a serious illness have a significantly increased risk of developing persistent cognitive impairment and mental disorders. The paper [139] discusses in detail the role of endogenous and exogenous GCs in the development of neuropsychiatric pathologies after critical illnesses. Such diseases are characterized by a sharp increase in the level of free cortisol and suppression of the level of ACTH, which usually normalize after recovery, but sometimes there may be a long-term violation of HPAA regulation. High levels of GCs can cause long-term changes in plasticity and structural integrity of the hippocampus and prefrontal cortex; this mechanism may probably contribute to impaired memory and cognitive abilities in critical condition survivors, although rigorous evidence for this assumption has not yet been obtained. Nevertheless, a variety of cerebral pathologies affecting the cognitive and emotional spheres are associated with elevated levels of GCs, from depressive disorders to epilepsy [140], stroke [141, 142], and traumatic brain injury [143, 144]. The general mechanisms of various brain pathologies confirm not only frequent comorbidities, but also the development of some pathologies on the basis of others. For example, depression is a risk factor and a component of AD, it can also be a trigger of the initial stage of AD [145, 146].
Excess of GCs disrupts the function and impairs the structure of the hippocampus, an area of the brain key to learning, memory, and emotions. The selective vulnerability of the hippocampus to stress, mediated by GCs secreted during stress, is the price for the high functional plasticity and pleiotropic functions of this limbic structure. Molecular and cellular mechanisms common to many cerebral pathologies include GR dysfunction, hyperglutamatergic states, disruption of neurotrophic factor systems, the development of neuroinflammation leading to neurodegeneration and loss of hippocampal neurons, as well as disorders of neurogenesis in the subgranular neurogenic niche and the formation of aberrant neural networks [17, 147]. These GC-dependent processes are associated with an altered response to stress and the development of chronic concomitant pathologies caused by stress.
Over the past 50 years, the concept of stress has changed significantly, and our understanding of the underlying neurobiology has expanded dramatically [148]. Stress biology is relevant not only in unusual and threatening conditions, but modern understanding defines it as a continuous adaptive process of assessing the environment, overcoming its factors and giving a person the opportunity to anticipate future problems and cope with them. The fundamental neural circuit underlying these processes is generally understood, the key molecular players have been identified, and their influence on neuroplasticity has been established. However, concepts about the mechanisms of pathogenesis of brain diseases are increasingly being revised, which took into account the role of any one individual system as a key one. This is clearly seen in the example of the evolution of understanding the pathogenesis of depression. There are several theories of depression that suggest, alternatively, violations of the HPAA regulation, modulation of monoaminergic neurotransmission, changes in neurotrophic factors and activation of neurogenesis in the hippocampus, but none of these theories sufficiently explains the etiopathology and approaches to the treatment of depression. Simultaneously, concepts based on violations of the BDNF system or neurogenesis emphasize the important role of neuroplasticity in depression and the fact that the hippocampus is an important anatomical area associated with depression. Studies have shown that some antidepressants can treat depression by changing the plasticity of the hippocampus, and the latest versions of the theory postulate that it is the synthesis of multiple mechanisms of brain plasticity, including changes in neurogenesis and the BDNF system, that can fuse numerous “monotheories” [149]. The second example is the recognition of the role of stress in the mechanism of the AD pathogenesis, a multifactorial neurodegenerative disease [150]. Accumulating clinical and experimental data indicate the key role of stress in the development of AD. Chronic stress and its accompanying high level of GCs secretion trigger, along with many others, two main pathomechanisms of AD: improper processing of APP and the formation of β-amyloid, as well as hyperphosphorylation and aggregation of tau protein, also associated with changes in neuroplasticity. Considering that depression is directly related to stress, and evidence that depression is a risk factor for AD, the neurobiological mechanisms common to these diseases are the changes described in this review that lead to impaired plasticity of the hippocampus.
The main data considered in this review are summarized in a fairly simple diagram (Fig. 2). It is shown how the cascade signaling of the HPAA by binding its final hormones, GCs, to the corticosteroid receptors of the hippocampus controls various targets – key components of a complex and multilevel system of plasticity of the hippocampus. On the one hand, this scheme shows potential key targets of neuroplasticity regulation by GCs in accordance with experimental and clinical data accumulated to date. On the other hand, this scheme does not contain information about dozens of mechanistic connections between different target systems of GCs, the influence of each of these systems on the effects of GCs, as well as the regulatory influence of GCs on the interaction between these systems. These data are given in the text, but their display in this figure turned out to be almost impossible, in full accordance with the famous statement of Paul Valéry: “Everything that is simple is theoretically false, everything that is complicated is pragmatically useless.” Thus, analyzing the scheme, it should be borne in mind that almost all targets regulated by GCs are densely interconnected by numerous processes at the molecular, subcellular, and cellular levels. Taking into account the third Meyer’s law, which states as an epigraph to this chapter that “it is simple to make things complex, but complex to make things simple”, at this stage of the consideration we leave a simple scheme that illustrates the role of the GCs as a “conductor” of a multicomponent orchestra supporting the neuroplasticity of the hippocampus, but we imply the presence of complex relationships between the orchestra members and their feedback with the conductor.
Fig. 2. Control of the adult hippocampal plasticity by glucocorticoids. The summary diagram shows the main ways of control by glucocorticoid hormones (GCs) of various plasticity mechanisms. The hypothalamic-pituitary-adrenal axis is shown on the left (HPAA; the names of its components are marked in red; the names of the main hormones in the framework: corticotropin-releasing hormone (CRH), adrenocorticotropic hormone (ACTH), cortisol (in humans) or corticosterone (in rodents). The remaining components of the limbic system of the brain, the hippocampus and the amygdala are also indicated. The left column shows the key systems of the hippocampus that determine its plasticity and are simultaneously targets of GCs (blue background). In the next column (green background), the main pathological effects on these systems of an excess of GCs resulting from HPAA dysfunction and/or stress (severe, chronic in adults or stress in the early period of ontogenesis) are noted. The last column (red background) presents diseases and conditions for the pathogenesis of which the key is the development of pathological consequences of GC-dependent alterations of hippocampal plasticity. It should be noted that the influence of GCs on the targets indicated in the blue column is not necessarily direct. In addition, one red arrow can include multiple mechanisms by which GCs act on a specific target. Detailed explanations are provided in the respective sections of the text.
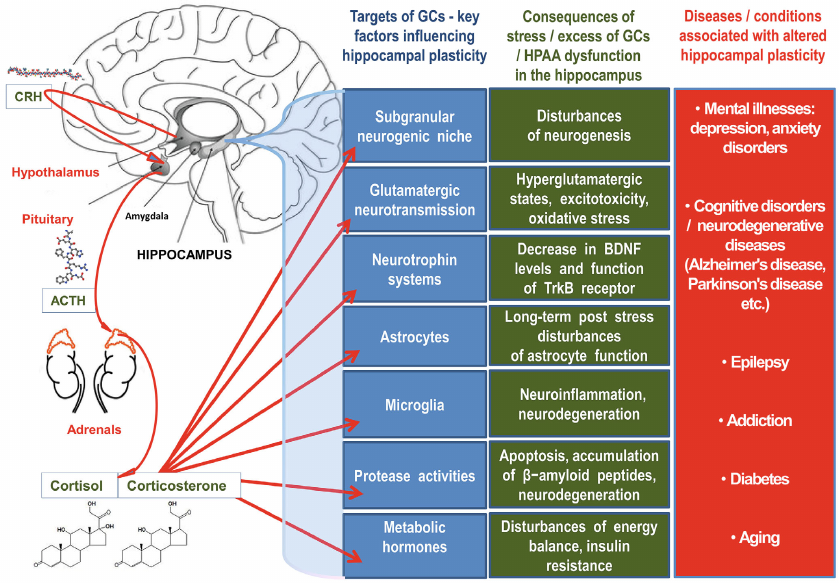
In various pathologies of the brain, along with violations of the HPAA control, secretion of GCs, expression and properties of their receptors, changes occur in each of the systems presented in the diagram. The importance of each of these systems is beyond doubt, therefore, the key components of each system are considered as targets for therapy and approaches (pharmacological and non-pharmacological) to the modulation of these targets are being developed. So far, such approaches have not led to outstanding success in the treatment of cerebral pathologies. If GCs really perform the conductor role of hippocampal neuroplasticity, perhaps more attention should be paid to the potential possibilities of correcting HPAA and the state of corticosteroid receptors? Logic suggests that if you manage to adjust the basic regulatory system, a positive effect will be exerted on the underlying components that are pathologically altered as a result of the central system dysfunction. It is obvious that it is extremely difficult to directly influence such a complex neurohumoral system as HPAA, but it is very likely that it is the achievement of control of this system that will allow maintaining an optimal level of neuroplasticity and thereby make the long-awaited breakthrough in the therapy of cognitive and emotional disorders, as well as the related comorbid states. The synthesis of new ligands of MR and GR, the results of their use in experiments on models of brain diseases and the preliminary results of their use in the clinic [151-157] allow us to hope that this approach will bring longed-for success.
Funding. The work was carried out within the framework of the state assignment of the Ministry of Science and Higher Education of the Russian Federation for 2021-2023.
Ethics declarations. The author declares no conflict of interest. This article does not deal with studies involving humans or animals as objects.
Open access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
REFERENCES
1.Gulyaeva, N. V. (2017) Molecular mechanisms of
neuroplasticity: an expanding universe, Biochemistry (Moscow),
82, 237-242, doi: 10.1134/S0006297917030014.
2.Xiong, H., and Krugers, H. J. (2015) Tuning
hippocampal synapses by stress-hormones: relevance for emotional memory
formation, Brain Res., 1621, 114-120, doi:
10.1016/j.brainres.2015.04.010.
3.Goto, A. (2022) Synaptic plasticity during systems
memory consolidation, Neurosci. Res., 183, 1-6, doi:
10.1016/j.neures.2022.05.008.
4.Toda, T., and Gage, F. H. (2018) Review: adult
neurogenesis contributes to hippocampal plasticity, Cell. Tissue
Res., 373, 693-709, doi: 10.1007/s00441-017-2735-4.
5.Deppermann, S., Storchak, H., Fallgatter, A. J.,
and Ehlis, A. C. (2014) Stress-induced neuroplasticity: (mal)adaptation
to adverse life events in patients with PTSD – a critical
overview, Neuroscience, 283, 166-177, doi:
10.1016/j.neuroscience.2014.08.037.
6.Den Boon, F. S., and Sarabdjitsingh, R. A. (2017)
Circadian and ultradian patterns of HPA-axis activity in rodents:
significance for brain functionality, Best Pract. Res. Clin.
Endocrinol. Metab., 31, 445-457, doi:
10.1016/j.beem.2017.09.001.
7.Gray, J. D., Kogan, J. F., Marrocco, J., and
McEwen, B. S. (2017) Genomic and epigenomic mechanisms of
glucocorticoids in the brain, Nat. Rev Endocrinol., 13,
661-673, doi: 10.1038/nrendo.2017.97.
8.Meijer, O. C., Buurstede, J. C., and Schaaf, M. J.
M. (2019) Corticosteroid receptors in the brain: transcriptional
mechanisms for specificity and context-dependent effects, Cell. Mol.
Neurobiol., 39, 539-549, doi: 10.1007/s10571-018-0625-2.
9.Koning, A. C. A. M., Buurstede, J. C., van Weert,
L. T. C. M., and Meijer, O. C. (2019) Glucocorticoid and
mineralocorticoid receptors in the brain: a transcriptional
perspective, J. Endocr. Soc., 3, 1917-1930, doi:
10.1210/js.2019-00158.
10.Meijer, O. C., Buurstede, J. C., Viho, E. M. G.,
Amaya, J. M., Koning, A. C. A. M., van der Meulen, M., van Weert, L. T.
C. M., Paul, S. N., Kroon, J., and Koorneef, L. L. (2023)
Transcriptional glucocorticoid effects in the brain: Finding the
relevant target genes, J. Neuroendocrinol., 35, e13213,
doi: 10.1111/jne.13213.
11.Gulyaeva, N. V. (2021) Glucocorticoid regulation
of the glutamatergic synapse: mechanisms of stress-dependent
neuroplasticity, J. Evol. Biochem. Phys., 57, 564-576,
doi: 10.1134/S0022093021030091.
12.Fuxe, K., Diaz, R., Cintra, A., Bhatnagar, M.,
Tinner, B., Gustafsson, J. A., Ogren, S. O., and Agnati, L. F. (1996)
On the role of glucocorticoid receptors in brain plasticity, Cell.
Mol. Neurobiol., 16, 239-258, doi: 10.1007/BF02088179.
13.Suri, D., and Vaidya, V. A. (2015) The adaptive
and maladaptive continuum of stress responses – a hippocampal
perspective, Rev. Neurosci., 26, 415-442, doi:
10.1515/revneuro-2014-0083.
14.Uchoa, E. T., Aguilera, G., Herman, J. P.,
Fiedler, J. L., Deak, T., and de Sousa, M. B. (2014) Novel aspects of
glucocorticoid actions, J. Neuroendocrinol., 26, 557-572,
doi: 10.1111/jne.12157.
15.Bolshakov, A. P., Tret’yakova, L. V.,
Kvichansky, A. A., and Gulyaeva, N. V. (2021) Glucocorticoids: Dr.
Jekyll and Mr. Hyde of hippocampal neuroinflammation, Biochemistry
(Moscow), 86, 156-167, doi: 10.1134/S0006297921020048.
16.McEwen, B. S. (2002) Sex, stress and the
hippocampus: allostasis, allostatic load and the aging process,
Neurobiol. Aging, 23, 921-939, doi:
10.1016/s0197-4580(02)00027-1.
17.Gulyaeva, N. V. (2021) Stress-associated
molecular and cellular hippocampal mechanisms common for epilepsy and
comorbid depressive disorders, Biochemistry(Moscow), 86,
641-656, doi: 10.1134/S0006297921060031.
18.Madalena, K. M., and Lerch, J. K. (2017) The
effect of glucocorticoid and glucocorticoid receptor interactions on
brain, spinal cord, and glial cell plasticity, Neural Plast.,
2017, 8640970, doi: 10.1155/2017/8640970.
19.Weerasinghe-Mudiyanselage, P. D. E., Ang, M. J.,
Kang, S., Kim, J. S., and Moon, C. (2022) Structural plasticity of the
hippocampus in neurodegenerative diseases, Int. J. Mol. Sci.,
23, 3349, doi: 10.3390/ijms23063349.
20.De Kloet, E. R., Sutanto, W., Rots, N., van
Haarst, A., van den Berg, D., Oitzl, M., van Eekelen, A., and Voorhuis,
D. (1991) Plasticity and function of brain corticosteroid receptors
during aging, Acta Endocrinol. (Copenh), 125 Suppl 1,
65-72.
21.Fares, J., Bou Diab, Z., Nabha, S., and Fares, Y.
(2019) Neurogenesis in the adult hippocampus: history, regulation, and
prospective roles, Int. J. Neurosci., 129, 598-611, doi:
10.1080/00207454.2018.1545771.
22.Ko, S. Y., and Frankland, P. W. (2021)
Neurogenesis-dependent transformation of hippocampal engrams,
Neurosci. Lett., 762, 136176, doi:
10.1016/j.neulet.2021.136176.
23.Miller, S. M., and Sahay, A. (2019) Functions of
adult-born neurons in hippocampal memory interference and indexing,
Nat. Neurosci., 22, 1565-1575, doi:
10.1038/s41593-019-0484-2.
24.Tuncdemir, S. N., Lacefield, C. O., and Hen, R.
(2019) Contributions of adult neurogenesis to dentate gyrus network
activity and computations, Behav. Brain Res., 374,
112112, doi: 10.1016/j.bbr.2019.112112.
25.Moreno-Jiménez, E. P., Terreros-Roncal,
J., Flor-García, M., Rábano, A., and
Llorens-Martín, M. (2021) Evidences for adult hippocampal
neurogenesis in humans, J. Neurosci., 41, 2541-2553, doi:
10.1523/JNEUROSCI.0675-20.2020.
26.Huckleberry, K. A., and Shansky, R. M. (2021) The
unique plasticity of hippocampal adult-bornneurons: contributing to a
heterogeneous dentate, Hippocampus, 31, 543-556, doi:
10.1002/hipo.23318.
27.Vilar, M., and Mira, H. (2016) Regulation of
neurogenesis by neurotrophins during adulthood: expected and unexpected
roles, Front. Neurosci., 10, 26, doi:
10.3389/fnins.2016.00026.
28.Niklison-Chirou, M. V., Agostini, M., Amelio, I.,
and Melino, G. (2020) Regulation of adult neurogenesis in mammalian
brain, Int. J. Mol. Sci., 21, 4869, doi:
10.3390/ijms21144869.
29.Jorgensen, C., and Wang, Z. (2020) Hormonal
regulation of mammalian adult neurogenesis: a multifaceted mechanism,
Biomolecules, 10, 1151, doi: 10.3390/biom10081151.
30.Surget, A., and Belzung, C. (2022) Adult
hippocampal neurogenesis shapes adaptation and improves stress
response: a mechanistic and integrative perspective, Mol.
Psychiatry, 27, 403-421, doi:
10.1038/s41380-021-01136-8.
31.Jones, K. L., Zhou, M., and Jhaveri, D. J. (2022)
Dissecting the role of adult hippocampal neurogenesis towards
resilience versus susceptibility to stress-related mood disorders,
NPJ Sci. Learn., 7, 16, doi:
10.1038/s41539-022-00133-y.
32.Lucassen, P. J., Oomen, C. A., Naninck, E. F.,
Fitzsimons, C. P., van Dam, A. M., Czeh, B., and Korosi, A. (2015)
Regulation of adult neurogenesis and plasticity by (early) stress,
glucocorticoids, and inflammation, Cold Spring Harb. Perspect.
Biol., 7, a021303, doi: 10.1101/cshperspect.a021303.
33.Podgorny, O. V., and Gulyaeva, N. V. (2021)
Glucocorticoid-mediated mechanisms of hippocampal damage: contribution
of subgranular neurogenesis, J. Neurochem., 157, 370-392,
doi: 10.1111/jnc.15265.
34.Vasic, V., and Schmidt, M. H. H. (2017)
Resilience and vulnerability to pain and inflammation in the
hippocampus, Int. J. Mol. Sci., 18, 739, doi:
10.3390/ijms18040739.
35.Kirschen, G. W., and Ge, S. (2019) Young at
heart: insights into hippocampal neurogenesis inthe aged brain,
Behav. Brain. Res., 369, 111934, doi:
10.1016/j.bbr.2019.111934.
36.Chen, P., Guo, Z., and Zhou, B. (2023) Insight
into the role of adult hippocampal neurogenesis in aging and
Alzheimer’s disease, Ageing Res. Rev., 84, 101828,
doi: 10.1016/j.arr.2022.101828.
37.Teixeira, C. M., Pallas-Bazarra, N.,
Bolós, M., Terreros-Roncal, J., Ávila, J., and
Llorens-Martín, M. (2018) Untold new beginnings: adult
hippocampal neurogenesis and Alzheimer’s disease, J.
Alzheimers Dis., 64 (s1), S497-S505, doi:
10.3233/JAD-179918.
38.Nicola, R., and Oku, E. (2021) Adult hippocampal
neurogenesis: one lactate to rule them all, Neuromol. Med.,
23, 445-448, doi: 10.1007/s12017-021-08658-y.
39.McEwen, B. S. (1996) Gonadal and adrenal steroids
regulate neurochemical and structural plasticity of the hippocampus via
cellular mechanisms involving NMDA receptors, Cell. Mol.
Neurobiol., 16, 103-116, doi: 10.1007/BF02088170.
40.Mihály, A. (2019) The reactive plasticity
of hippocampal ionotropic glutamate receptors in animal epilepsies,
Int. J. Mol. Sci., 20, 1030, doi:
10.3390/ijms20051030.
41.Shipton, O. A., and Paulsen, O. (2013) GluN2A and
GluN2B subunit-containing NMDA receptors in hippocampal plasticity,
Philos. Trans. R. Soc .Lond. B. Biol. Sci., 369,
20130163, doi: 10.1098/rstb.2013.0163.
42.Pampaloni, N. P., and Plested, A. J. R. (2022)
Slow excitatory synaptic currents generated by AMPA receptors, J.
Physiol., 600, 217-232, doi: 10.1113/JP280877.
43.Nair, J. D., Wilkinson, K. A., Henley, J. M., and
Mellor, J. R. (2021) Kainate receptors and synapticplasticity,
Neuropharmacology, 196, 108540, doi:
10.1016/j.neuropharm.2021.108540.
44.Valbuena, S., and Lerma, J. (2021) Kainate
receptors, homeostatic gatekeepers of synaptic plasticity,
Neuroscience, 456, 17-26, doi:
10.1016/j.neuroscience.2019.11.050.
45.Griego, E., and Galván, E. J. (2021)
Metabotropic glutamate receptors at the aged mossy fiber – CA3
synapse of the hippocampus, Neuroscience, 456, 95-105,
doi: 10.1016/j.neuroscience.2019.12.016.
46.Mukherjee, S., and Manahan-Vaughan, D. (2013)
Role of metabotropic glutamate receptors in persistent forms of
hippocampal plasticity and learning, Neuropharmacology,
66, 65-81, doi: 10.1016/j.neuropharm.2012.06.005.
47.Mikasova, L., Xiong, H., Kerkhofs, A., Bouchet,
D., Krugers, H. J., and Groc, L. (2017) Stress hormone rapidly tunes
synaptic NMDA receptor through membrane dynamics and mineralocorticoid
signalling, Sci. Rep., 7, 8053, doi:
10.1038/s41598-017-08695-3.
48.Gonçalves-Ribeiro, J., Pina, C. C.,
Sebastião, A. M., and Vaz, S. H. (2019) Glutamate transporters
in hippocampal LTD/LTP: not just prevention of excitotoxicity,
Front. Cell. Neurosci., 13, 357, doi:
10.3389/fncel.2019.00357.
49.Taylor, C. J., He, R., and Bartlett, P. F. (2014)
The role of the N-methyl-D-aspartate receptor in the proliferation of
adult hippocampal neural stem and precursor cells, Sci. China Life
Sci., 57, 403-411, doi: 10.1007/s11427-014-4637-y.
50.Gulyaeva, N. V. (2022) Neuroendocrine control of
hyperglutamatergic states in brain pathologies: the effects of
glucocorticoids, J. Evol. Biochem. Phys., 58, 1425-1438,
doi: 10.1134/S0022093022050131.
51.Jacobsson, J., Persson, M., Hansson, E., and
Rönnbäck, L. (2006) Corticosterone inhibits expression of the
microglial glutamate transporter GLT-1 in vitro, Neuroscience,
139, 475-483, doi: 10.1016/j.neuroscience.2005.12.046.
52.Zschocke, J., Bayatti, N., Clement, A. M., Witan,
H., Figiel, M., Engele, J., and Behl, C. (2005) Differential promotion
of glutamate transporter expression and function by glucocorticoids in
astrocytes from various brain regions, J. Biol. Chem.,
280, 34924-34932, doi: 10.1074/jbc.M502581200.
53.Chu, S. F., Zhang, Z., Zhou, X., He, W. B., Yang,
B., Cui, L. Y., He, H. Y., Wang, Z. Z., and Chen, N. H. (2021) Low
corticosterone levels attenuate late life depression and enhance
glutamatergic neurotransmission in female rats, Acta Pharmacol.
Sin., 42, 848-860, doi: 10.1038/s41401-020-00536-w.
54.Kang, M., Ryu, J., Kim, J. H., Na, H., Zuo, Z.,
and Do, S. H. (2010) Corticosterone decreases the activity of rat
glutamate transporter type 3 expressed in Xenopus
oocytes, Steroids, 75, 1113-1118, doi:
10.1016/j.steroids.2010.07.003.
55.Cox, M. F., Hascup, E. R., Bartke, A., and
Hascup, K. N. (2022) Friend or foe? Defining the role of glutamate in
aging and Alzheimer’s disease, Front. Aging, 3,
929474, doi: 10.3389/fragi.2022.929474.
56.Gulyaeva, N. V. (2022) Multi-level
plasticity-pathology continuum of the nervous system: functional
aspects, Neurochem. J., 16, 424-428, doi:
10.1134/S1819712422040092.
57.Bano, D., and Ankarcrona, M. (2018) Beyond the
critical point: an overview of excitotoxicity, calcium overload and the
downstream consequences, Neurosci. Lett., 663, 79-85,
doi: 10.1016/j.neulet.2017.08.048.
58.Foster, T. C., Kyritsopoulos, C., and Kumar, A.
(2017) Central role for NMDA receptors in redox mediated impairment of
synaptic function during aging and Alzheimer’s disease, Behav.
Brain Res., 322 (Pt B), 223-232, doi:
10.1016/j.bbr.2016.05.012.
59.Temido-Ferreira, M., Coelho, J. E., Pousinha, P.
A., and Lopes, L. V. (2019) Novel players in the aging synapse: impact
on cognition, J. Caffeine Adenosine Res., 9, 104-127,
doi: 10.1089/caff.2019.0013.
60.Leal, G., Bramham, C. R., and Duarte, C. B.
(2017) BDNF and hippocampal synaptic plasticity, Vitam. Horm.,
104, 153-195, doi: 10.1016/bs.vh.2016.10.004.
61.Gómez-Palacio-Schjetnan, A., and Escobar,
M. L. (2013) Neurotrophins and synaptic plasticity, Curr. Top.
Behav. Neurosci., 15, 117-136, doi:
10.1007/7854_2012_231.
62.De Vincenti, A. P., Ríos, A. S., Paratcha,
G., and Ledda, F. (2019) Mechanisms that modulate and diversify BDNF
functions: implications for hippocampal synaptic plasticity, Front.
Cell. Neurosci., 13, 135, doi: 10.3389/fncel.2019.00135.
63.Zagrebelsky, M., and Korte, M. (2014) Form
follows function: BDNF and its involvement in sculpting the function
and structure of synapses, Neuropharmacology, 76 Pt C,
628-638, doi: 10.1016/j.neuropharm.2013.05.029.
64.Von Bohlen Und Halbach, O., and von Bohlen Und
Halbach, V. (2018) BDNF effects on dendritic spine morphology and
hippocampal function, Cell. Tissue Res., 373, 729-741,
doi: 10.1007/s00441-017-2782-x.
65.Jeanneteau, F., Borie, A., Chao, M. V., and
Garabedian, M. J. (2019) Bridging the Gap between brain-derived
neurotrophic factor and glucocorticoid effects on brain networks,
Neuroendocrinology, 109, 277-284, doi:
10.1159/000496392.
66.Gray, J. D., Milner, T. A., and McEwen, B. S.
(2013) Dynamic plasticity: the role of glucocorticoids, brain-derived
neurotrophic factor and other trophic factors, Neuroscience,
239, 214-227, doi: 10.1016/j.neuroscience.2012.08.034.
67.Numakawa, T., and Odaka, H. (2022) The role of
neurotrophin signaling in age-related cognitive decline and cognitive
diseases, Int. J. Mol. Sci., 23, 7726, doi:
10.3390/ijms23147726.
68.Arango-Lievano, M., Lambert, W. M., and
Jeanneteau, F. (2015) Molecular biology of glucocorticoid signaling,
Adv. Exp. Med. Biol., 872, 33-57, doi:
10.1007/978-1-4939-2895-8_2.
69.Jeanneteau, F., and Chao, M. V. (2013) Are BDNF
and glucocorticoid activities calibrated? Neuroscience,
239, 173-195, doi: 10.1016/j.neuroscience.2012.09.017.
70.Rothman, S. M., and Mattson, M. P. (2013)
Activity-dependent, stress-responsive BDNF signaling and the quest for
optimal brain health and resilience throughout the lifespan,
Neuroscience, 239, 228-240, doi:
10.1016/j.neuroscience.2012.10.014.
71.Gulyaeva, N. V. (2017) Interplay between brain
BDNF and glutamatergic systems: a brief state of the evidence and
association with the pathogenesis of depression, Biochemistry
(Moscow), 82, 301-307, doi: 10.1134/S0006297917030087.
72.Gibon, J., Barker, P. A. (2017) Neurotrophins and
proneurotrophins: focus on synaptic activity and plasticity in the
brain, Neuroscientist, 23, 587-604, doi:
10.1177/1073858417697037.
73.Mizui, T., Ishikawa, Y., Kumanogoh, H., and
Kojima, M. (2016) Neurobiological actions by three distinct subtypes of
brain-derived neurotrophic factor: multi-ligand model of growth factor
signaling, Pharmacol. Res., 105, 93-98, doi:
10.1016/j.phrs.2015.12.019.
74.Kojima, M., and Mizui, T. (2017) BDNF propeptide:
a novel modulator of synaptic plasticity, Vitam. Horm.,
104, 19-28, doi: 10.1016/bs.vh.2016.11.006.
75.Costa, R. O., Perestrelo, T., and Almeida, R. D.
(2018) PROneurotrophins and CONSequences, Mol. Neurobiol.,
55, 2934-2951, doi: 10.1007/s12035-017-0505-7.
76.Leal, G., Afonso, P. M., Salazar, I. L., and
Duarte, C. B. (2015) Regulation of hippocampal synaptic plasticity by
BDNF, Brain Res., 1621, 82-101, doi:
10.1016/j.brainres.2014.10.019.
77.Notaras, M., and van den Buuse, M. (2020)
Neurobiology of BDNF in fear memory, sensitivity to stress, and
stress-related disorders, Mol. Psychiatry, 25, 2251-2274,
doi: 10.1038/s41380-019-0639-2.
78.Lu, B., Nagappan, G., and Lu, Y. (2014) BDNF and
synaptic plasticity, cognitive function, and dysfunction, Handb.
Exp. Pharmacol., 220, 223-250, doi:
10.1007/978-3-642-45106-5_9.
79.Ninan, I. (2014) Synaptic regulation of affective
behaviors; role of BDNF, Neuropharmacology, 76 Pt C,
684-695, doi: 10.1016/j.neuropharm.2013.04.011.
80.Duman, R. S., Deyama, S., and Fogaça, M.
V. (2021) Role of BDNF in the pathophysiology and treatment of
depression: activity-dependent effects distinguish rapid-acting
antidepressants, Eur. J. Neurosci., 53, 126-139, doi:
10.1111/ejn.14630.
81.Colucci-D’Amato, L., Speranza, L., and
Volpicelli, F. (2020) Neurotrophic factor BDNF, physiological functions
and therapeutic potential in depression,neurodegeneration and brain
cancer, Int. J. Mol. Sci., 21, 7777, doi:
10.3390/ijms21207777.
82.Numakawa, T., Adachi, N., Richards, M., Chiba,
S., and Kunugi, H. (2013) Brain-derived neurotrophic factor and
glucocorticoids: reciprocal influence on the central nervous system,
Neuroscience, 239, 157-172, doi:
10.1016/j.neuroscience.2012.09.073.
83.Jones, O. D. (2015) Astrocyte-mediated
metaplasticity in the hippocampus: help or hindrance?
Neuroscience, 309, 113-124, doi:
10.1016/j.neuroscience.2015.08.035.
84.Fuchsberger, T., and Paulsen, O. (2022)
Modulation of hippocampal plasticity in learning and memory, Curr.
Opin. Neurobiol., 75, 102558, doi:
10.1016/j.conb.2022.102558.
85.Wang, Y., Fu, A. K. Y., and Ip, N. Y. (2022)
Instructive roles of astrocytes in hippocampal synaptic plasticity:
neuronal activity-dependent regulatory mechanisms, FEBS J.,
289, 2202-2218, doi: 10.1111/febs.15878.
86.Çalışkan, G., Müller, A.,
and Albrecht, A. (2020) Long-term impact of early-life stress on
hippocampal plasticity: spotlight on astrocytes, Int. J. Mol.
Sci., 21, 4999, doi: 10.3390/ijms21144999.
87.Cassé, F., Richetin, K., and Toni, N.
(2018) Astrocytes’ contribution to adult neurogenesis in
physiology and Alzheimer’s disease, Front. Cell.
Neurosci., 12, 432, doi: 10.3389/fncel.2018.00432.
88.Delpech, J. C., Madore, C., Nadjar, A., Joffre,
C., Wohleb, E. S., and Layé, S. (2015) Microglia in neuronal
plasticity: Influence of stress, Neuropharmacology, 96 (Pt
A), 19-28, doi: 10.1016/j.neuropharm.2014.12.034.
89.Guedes, J. R., Ferreira, P. A., Costa, J. M.,
Cardoso, A. L., and Peça, J. (2022) Microglia-dependent
remodeling of neuronal circuits, J. Neurochem., 163,
74-93, doi: 10.1111/jnc.15689.
90.Rodríguez-Iglesias, N., Sierra, A., and
Valero, J. (2019) Rewiring of memory circuits: connecting adult newborn
neurons with the help of microglia, Front. Cell. Dev. Biol.,
7, 24, doi: 10.3389/fcell.2019.00024.
91.Turkin, A., Tuchina, O., and Klempin, F. (2021)
Microglia function on precursor cells in the adult hippocampus and
their responsiveness to serotonin signaling, Front. Cell. Dev.
Biol., 9, 665739, doi: 10.3389/fcell.2021.665739.
92.Sanguino-Gómez, J., Buurstede, J. C.,
Abiega, O., Fitzsimons, C. P., Lucassen, P. J., Eggen, B. J. L.,
Lesuis, S. L., Meijer, O. C., and Krugers, H. J. (2022) An emerging
role for microglia in stress-effects on memory, Eur. J.
Neurosci., 55, 2491-2518, doi: 10.1111/ejn.15188.
93.Gądek-Michalska, A., Tadeusz, J.,
Rachwalska, P., and Bugajski, J. (2013) Cytokines, prostaglandins and
nitric oxide in the regulation of stress-response systems,
Pharmacol. Rep., 65, 1655-1662, doi:
10.1016/s1734-1140(13)71527-5.
94.Dheen, S. T., Kaur, C., and Ling, E. A. (2007)
Microglial activation and its implications in the brain diseases,
Curr. Med. Chem., 14, 1189-1197, doi:
10.2174/092986707780597961.
95.Williamson, L. L., and Bilbo, S. D. (2013)
Chemokines and the hippocampus: a new perspective on hippocampal
plasticity and vulnerability, Brain. Behav. Immun., 30,
186-194, doi: 10.1016/j.bbi.2013.01.077.
96.Singhal, G., and Baune, B. T. (2017) Microglia:
an interface between the loss of neuroplasticity and depression,
Front. Cell. Neurosci., 11, 270, doi:
10.3389/fncel.2017.00270.
97.Bauer, M. E., and Teixeira, A. L. (2019)
Inflammation in psychiatric disorders: what comes first? Ann .N. Y.
Acad. Sci., 1437, 57-67, doi: 10.1111/nyas.13712.
98.Bisht, K., Sharma, K., and Tremblay, M. È.
(2018) Chronic stress as a risk factor for Alzheimer’s disease:
roles of microglia-mediated synaptic remodeling, inflammation, and
oxidative stress, Neurobiol. Stress, 9, 9-21, doi:
10.1016/j.ynstr.2018.05.003.
99.Patterson, S. L. (2015) Immune dysregulation and
cognitive vulnerability in the aging brain: Interactions of microglia,
IL-1β, BDNF and synaptic plasticity, Neuropharmacology,
96 (Pt A), 11-18, doi: 10.1016/j.neuropharm.2014.12.020.
100.Salazar, I. L., Caldeira, M. V., Curcio, M.,
and Duarte, C. B. (2016) The role of proteases in hippocampal synaptic
plasticity: putting together small pieces of a complex puzzle,
Neurochem. Res., 41, 156-182, doi:
10.1007/s11064-015-1752-5.
101.Wiera, G., and Mozrzymas, J. W. (2015)
Extracellular proteolysis in structural and functional plasticity of
mossy fiber synapses in hippocampus, Front. Cell. Neurosci.,
9, 427, doi: 10.3389/fncel.2015.00427.
102.Wójtowicz, T., Brzdąk, P., and
Mozrzymas, J. W. (2015) Diverse impact of acute and long-term
extracellular proteolytic activity on plasticity of neuronal
excitability, Front. Cell. Neurosci., 9, 313, doi:
10.3389/fncel.2015.00313.
103.Gulyaeva, N. V. (2003) Non-apoptotic functions
of caspase-3 in nervous tissue, Biochemistry (Moscow),
68, 1171-1180, doi: 10.1023/b:biry.0000009130.62944.35.
104.Yakovlev, A. A., and Gulyaeva, N. V. (2011)
Pleiotropic functions of brain proteinases: methodological
considerations and search for caspase substrates, Biochemistry
(Moscow), 76, 1079-1086, doi: 10.1134/S0006297911100014.
105.Wang, Y., Liu, Y., Bi, X., and Baudry, M.
(2020) Calpain-1 and calpain-2 in the brain: new evidence for a
critical role of calpain-2 in neuronal death, Cells, 9,
2698, doi: 10.3390/cells9122698.
106.Orlowski, R. Z. (1999) The role of the
ubiquitin-proteasome pathway in apoptosis, Cell Death Differ.,
6, 303-313, doi: 10.1038/sj.cdd.4400505.
107.Laham, B. J., and Gould, E. (2022) How stress
influences the dynamic plasticity of the brain’s extracellular
matrix, Front. Cell. Neurosci., 15, 814287, doi:
10.3389/fncel.2021.814287.
108.Breviario, S., Senserrich, J., Florensa-Zanuy,
E., Garro-Martínez, E., Díaz, Á., Castro, E.,
Pazos, Á., and Pilar-Cuéllar, F. (2023) Brain matrix
metalloproteinase-9 activity is altered in the corticosterone mouse
model of depression, Prog. Neuropsychopharmacol. Biol.
Psychiatry, 120, 110624, doi:
10.1016/j.pnpbp.2022.110624.
109.Koyama, Y. (2021) Endothelin ETB
receptor-mediated astrocytic activation: pathological roles in brain
disorders, Int. J. Mol. Sci., 22, 4333, doi:
10.3390/ijms22094333.
110.Harkness, K. A., Adamson, P., Sussman, J. D.,
Davies-Jones, G. A., Greenwood, J., and Woodroofe, M. N. (2000)
Dexamethasone regulation of matrix metalloproteinase expression in CNS
vascular endothelium, Brain, 123 (Pt 4), 698-709, doi:
10.1093/brain/123.4.698.
111.Förster, C., Kahles, T., Kietz, S., and
Drenckhahn, D. (2007) Dexamethasone induces the expression of
metalloproteinase inhibitor TIMP-1 in the murine cerebral vascular
endothelial cell line cEND, J. Physiol., 580 (Pt. 3),
937-949, doi: 10.1113/jphysiol.2007.129007.
112.Hillegass, J. M., Villano, C. M., Cooper, K.
R., and White, L. A. (2007) Matrix metalloproteinase-13 is required for
zebra fish (Danio rerio) development and is a target for
glucocorticoids, Toxicol. Sci., 100, 168-179, doi:
10.1093/toxsci/kfm192.
113.Wang, Y., Li, M., Tang, J., Song, M., Xu, X.,
Xiong, J., Li, J., and Bai, Y. (2011) Glucocorticoids facilitate
astrocytic amyloid-β peptide deposition by increasing the
expression of APP and BACE1 and decreasing the expression of
amyloid-β-degrading proteases, Endocrinology, 152,
2704-2715, doi: 10.1210/en.2011-0145.
114.Hou, Y., Luo, S., Zhang, Y., Jia, Y., Li, H.,
Xiao, C., Bao, H., and Du, J. (2019) Contrasting effects of acute and
long-term corticosterone treatment on amyloid-β, beta-secretase 1
expression, and nuclear factor kappa B nuclear translocation, J.
Integr. Neurosci., 18, 393-400, doi:
10.31083/j.jin.2019.04.1172.
115.Proulx, K., and Seeley, R. J. (2005) The
regulation of energy balance by the central nervous system,
Psychiatr. Clin. North Am., 28, 25-38, doi:
10.1016/j.psc.2004.09.005.
116.Maniam, J., and Morris, M. J. (2012) The link
between stress and feeding behaviour, Neuropharmacology,
63, 97-110, doi: 10.1016/j.neuropharm.2012.04.017.
117.Ferrario, C. R., and Reagan, L. P. (2018)
Insulin-mediated synaptic plasticity in the CNS: Anatomical, functional
and temporal contexts, Neuropharmacology, 136 (Pt B),
182-191, doi: 10.1016/j.neuropharm.2017.12.001.
118.Grillo, C. A., Woodruff, J. L., Macht, V. A.,
and Reagan, L. P. (2019) Insulin resistance and hippocampal
dysfunction: disentangling peripheral and brain causes from
consequences, Exp. Neurol., 318, 71-77, doi:
10.1016/j.expneurol.2019.04.012.
119.Spinelli, M., Fusco, S., and Grassi, C. (2019)
Brain insulin resistance and hippocampal plasticity: mechanisms and
biomarkers of cognitive decline, Front. Neurosci., 13,
788, doi: 10.3389/fnins.2019.00788.
120.Irving, A., and Harvey, J. (2021) Regulation of
hippocampal synaptic function by the metabolic hormone leptin:
implications for health and disease, Prog. Lipid Res.,
82, 101098, doi: 10.1016/j.plipres.2021.101098.
121.McGregor, G., Malekizadeh, Y., and Harvey, J.
(2015) Minireview: food for thought:regulation of synaptic function by
metabolic hormones, Mol. Endocrinol., 29, 3-13, doi:
10.1210/me.2014-1328.
122.Lazarov, O., Minshall, R. D., and Bonini, M. G.
(2020) Harnessing neurogenesis in the adult brain-A role in type 2
diabetes mellitus and Alzheimer’s disease, Int. Rev.
Neurobiol., 155, 235-269, doi:
10.1016/bs.irn.2020.03.020.
123.Detka, J., Kurek, A., Basta-Kaim, A., Kubera,
M., Lasoń, W., and Budziszewska, B. (2013) Neuroendocrine link
between stress, depression and diabetes, Pharmacol. Rep.,
65, 1591-1600, doi: 10.1016/s1734-1140(13)71520-2.
124.Doyle, T., Halaris, A., and Rao, M. (2014)
Shared neurobiological pathways between type 2 diabetes and depressive
symptoms: a review of morphological and neurocognitive findings,
Curr. Diab. Rep., 14, 560, doi:
10.1007/s11892-014-0560-7.
125.Lyra E Silva, N. M., Gonçalves, R. A.,
Boehnke, S. E., Forny-Germano, L., Munoz, D. P., and De Felice, F. G.
(2019) Understanding the link between insulin resistance and
Alzheimer’s disease: Insights from animal models, Exp.
Neurol., 316, 1-11, doi:
10.1016/j.expneurol.2019.03.016.
126.Yagi, S., and Galea, L. A. M. (2019) Sex
differences in hippocampal cognition and neurogenesis,
Neuropsychopharmacology, 44, 200-213, doi:
10.1038/s41386-018-0208-4.
127.Koss, W. A., and Frick, K. M. (2017) Sex
differences in hippocampal function, J. Neurosci. Res.,
95, 539-562, doi: 10.1002/jnr.23864.
128.Scharfman, H. E., and MacLusky, N. J. (2017)
Sex differences in hippocampal area CA3 pyramidal cells, J.
Neurosci. Res., 95, 563-575, doi: 10.1002/jnr.23927.
129.Tozzi, A., Bellingacci, L., and Pettorossi, V.
E. (2020) Rapid estrogenic and androgenic neurosteroids effects in the
induction of long-term synaptic changes:implication for early memory
formation, Front. Neurosci., 14, 572511, doi:
10.3389/fnins.2020.572511.
130.Sheppard, P. A. S., Choleris, E., and Galea, L.
A. M. (2019) Structural plasticity of the hippocampus in response to
estrogens in female rodents, Mol. Brain, 12, 22, doi:
10.1186/s13041-019-0442-7.
131.Nicholson, K., MacLusky, N. J., and Leranth, C.
(2020) Synaptic effects of estrogen, Vitam. Horm., 114,
167-210, doi: 10.1016/bs.vh.2020.06.002.
132.Murakami, G., Hojo, Y., Kato, A., Komatsuzaki,
Y., Horie, S., Soma, M., Kim, J., and Kawato, S. (2018) Rapid
nongenomic modulation by neurosteroids of dendritic spines in the
hippocampus: androgen, oestrogen and corticosteroid, J.
Neuroendocrinol., 30, e12561, doi: 10.1111/jne.12561.
133.Gall, C. M., Le, A. A., and Lynch, G. (2021)
Sex differences in synaptic plasticity underlying learning, J.
Neurosci. Res., 101, 764-782, doi: 10.1002/jnr.24844.
134.Kramár, E. A., Babayan, A. H., Gall, C.
M., and Lynch, G. (2013) Estrogen promotes learning-related plasticity
by modifying the synaptic cytoskeleton, Neuroscience,
239, 3-16, doi: 10.1016/j.neuroscience.2012.10.038.
135.Harte-Hargrove, L. C., Maclusky, N. J., and
Scharfman, H. E. (2013) Brain-derived neurotrophic factor-estrogen
interactions in the hippocampal mossy fiber pathway: implications for
normal brain function and disease, Neuroscience, 239,
46-66, doi: 10.1016/j.neuroscience.2012.12.029.
136.Deakk, T., Quinnk, M., Cidlowskik, J. A.,
Victoriak, N. C., Murphyk, A. Z., and Sheridank, J. F. (2015)
Neuroimmune mechanisms of stress: sex differences, developmental
plasticity, and implications for pharmacotherapy of stress-related
disease, Stress, 18, 367-380, doi:
10.3109/10253890.2015.1053451.
137.Kandasamyk, M., Radhakrishnank, R. K.,
Poornimaik Abirami, G. P., Roshank, S. A., Yesudhask, A., Balamuthuk,
K., Prahalathank, C., Shanmugaapriyak, S., Moorthyk, A., Essakk, M. M.,
and Anusuyadevik, M. (2019) Possible existence of the
hypothalamic-pituitary-hippocampal (HPH) axis: a reciprocal
relationship between hippocampal specific neuroestradiol synthesis and
neuroblastosis in ageing brains with special reference to menopause and
neurocognitive disorders, Neurochem. Res., 44, 1781-1795,
doi: 10.1007/s11064-019-02833-1.
138.Resmini, E., Santos, A., and Webb, S. M. (2016)
Cortisol excess and the brain, Front. Horm. Res.,
46, 74-86, doi: 10.1159/000443868.
139.Hill, A. R., and Spencer-Segal, J. L. (2021)
Glucocorticoids and the brain after critical illness,
Endocrinology, 162, bqaa242, doi:
10.1210/endocr/bqaa242.
140.Druzhkova, T. A., Yakovlev, A. A., Rider, F.
K., Zinchuk, M. S., Guekht, A. B., and Gulyaeva, N. V. (2022) Elevated
serum cortisol levels in patients with focal epilepsy, depression, and
comorbid epilepsy and depression, Int. J. Mol. Sci., 23,
10414, doi: 10.3390/ijms231810414.
141.Zhanina, M. Y., Druzhkova, T. A., Yakovlev, A.
A., Vladimirova, E. E., Freiman, S. V., Eremina, N. N., Guekht, A. B.,
and Gulyaeva, N. V. (2022) Development of post-stroke cognitive and
depressive disturbances: associations with neurohumoral indices,
Curr. Issues Mol. Biol., 44, 6290-6305, doi:
10.3390/cimb44120429.
142.Gulyaeva, N. V., Onufriev, M. V., and Moiseeva,
Y. V. (2021) Ischemic stroke, glucocorticoids, and remote hippocampal
damage: a translational outlook and implications for modeling,
Front. Neurosci., 15, 781964, doi:
10.3389/fnins.2021.781964.
143.Komoltsev, I. G., and Gulyaeva, N. V. (2022)
Brain trauma, glucocorticoids and neuroinflammation: dangerous liaisons
for the hippocampus, Biomedicines, 10, 1139, doi:
10.3390/biomedicines10051139.
144.Komoltsev, I. G., Frankevich, S. O.,
Shirobokova, N. I., Volkova, A. A., Onufriev, M. V., Moiseeva, J. V.,
Novikova, M. R., and Gulyaeva, N. V. (2021) Neuroinflammation and
neuronal loss in the hippocampus are associated with immediate
posttraumatic seizures and corticosterone elevation in rats, Int. J.
Mol. Sci., 22, 5883, doi: 10.3390/ijms22115883.
145.Herbert, J., and Lucassen, P. J. (2016)
Depression as a risk factor for Alzheimer’s disease: genes,
steroids, cytokines and neurogenesis – what do we need to know?
Front. Neuroendocrinol., 41, 153-171, doi:
10.1016/j.yfrne.2015.12.001.
146.Linnemann, C., and Lang, U. E. (2020) Pathways
connecting late-life depression and dementia, Front. Pharmacol.,
11, 279, doi: 10.3389/fphar.2020.00279.
147.Gulyaeva, N. V. (2019) Biochemical mechanisms
and translational relevance of hippocampal vulnerability to distant
focal brain injury: the price of stress response, Biochemistry
(Moscow), 84, 1306-1328, doi: 10.1134/S0006297919110087.
148.McEwen, B. S., and Akil, H. (2020) Revisiting
the stress concept: implications for affective disorders, J.
Neurosci., 40, 12-21, doi:
10.1523/JNEUROSCI.0733-19.2019.
149.Xu, W., Yao, X., Zhao, F., Zhao, H., Cheng, Z.,
Yang, W., Cui, R., Xu, S., and Li, B. (2020) Changes in hippocampal
plasticity in depression and therapeutic approaches influencing these
changes, Neural Plast., 2020, 8861903, doi:
10.1155/2020/8861903.
150.Sotiropoulos, I., Silva, J. M., Gomes, P.,
Sousa, N., and Almeida, O. F. X. (2019) Stress and the etiopathogenesis
of Alzheimer’s disease and depression, Adv. Exp.
Med.Biol., 1184, 241-257, doi:
10.1007/978-981-32-9358-8_20.
151.Meyer, M., Lara, A., Hunt, H., Belanoff, J., de
Kloet, E. R., Gonzalez Deniselle, M. C., and De Nicola, A. F. (2018)
The selective glucocorticoid receptor modulator cort 113176 reduces
neurodegeneration and neuroinflammation in wobbler mice spinal cord,
Neuroscience, 384, 384-396, doi:
10.1016/j.neuroscience.2018.05.042.
152.Dalm, S., Karssen, A. M., Meijer, O. C.,
Belanoff, J. K., and de Kloet, E. R. (2019) Resetting the stress system
with a mifepristone challenge, Cell. Mol. Neurobiol., 39,
503-522, doi: 10.1007/s10571-018-0614-5.
153.De Kloet, E. R., de Kloet, S. F., de Kloet, C.
S., and de Kloet, A. D. (2019) Top-down and bottom-up control of
stress-coping, J. Neuroendocrinol., 31, e12675, doi:
10.1111/jne.12675.
154.De Nicola, A. F., Meyer, M., Guennoun, R.,
Schumacher, M., Hunt, H., Belanoff, J., de Kloet, E. R., and Gonzalez
Deniselle, M. C. (2020) Insights into the therapeutic potential of
glucocorticoid receptor modulators for neurodegenerative diseases,
Int. J. Mol. Sci., 21, 2137, doi:
10.3390/ijms21062137.
155.Meyer, M., Kruse, M. S., Garay, L., Lima, A.,
Roig, P., Hunt, H., Belanoff, J., de Kloet, E. R., Deniselle, M. C. G.,
and De Nicola, A. F. (2020) Long-term effects of the glucocorticoid
receptor modulator CORT113176 in murine motoneuron degeneration,
Brain Res., 1727, 146551, doi:
10.1016/j.brainres.2019.146551.31726042.
156.De Kloet, E. R., and Joëls, M. (2023) The
cortisol switch between vulnerability and resilience, Mol.
Psychiatry, doi: 10.1038/s41380-022-01934-8.
157.Daskalakis, N. P., Meijer, O. C., and de Kloet,
E. R. (2022) Mineralocorticoid receptor and glucocorticoid receptor
work alone and together in cell-type-specific manner: implications for
resilience prediction and targeted therapy, Neurobiol. Stress,
18, 100455, doi: 10.1016/j.ynstr.2022.100455.