The Mitochondrial Genome of the Moss Brachythecium rivulare (Hypnales, Brachytheciaceae)
D. V. Goryunov1, M. D. Logacheva1,2, M. S. Ignatov3, I. A. Milyutina1, A. V. Fedorova1, and A. V. Troitsky1*
1Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; E-mail: bobr@belozersky.msu.ru2Extreme Biology Laboratory, Institute of Fundamental Medicine and Biology, Kazan Federal University, 420012 Kazan, Russia
3Main Botanical Garden, Russian Academy of Sciences, 127276 Moscow, Russia
* To whom correspondence should be addressed.
Received September 11, 2017
The mitochondrial genome of the pleurocarpous moss Brachythecium rivulare has been sequenced and annotated. The genome consists of 104,460 base pairs and has approximately the same gene set and organization as other bryophyte mitogenomes. Whole mitochondrial genome comparison between B. rivulare and Physcomitrella patens, Tetraphis pellucida, Anomodon rugelii, and Anomodon attenuatus was performed. The primary cause of bryophyte mitochondrial gene length variation was found to be numerous indels in the introns. Bryophyte mitochondrial gene conservation level was estimated, and it was in a good congruence with the overall phylogeny of bryophytes with the percentage of mitogenome similarity being proportional to the age estimated by phylochronologic analysis. Annotation discrepancies in the analyzed mitogenome sequences were identified. The simple sequence repeat (SSR) content was evaluated, and candidate sites of RNA editing were predicted in the B. rivulare mitochondrial genome.
KEY WORDS: mitochondrial genome, Brachythecium rivulare, Bryophyta, mossesDOI: 10.1134/S0006297917110153
Abbreviations: CDSs, coding sequences; mya, million years ago; NGS, next generation sequencing; ORF, open reading frame; SSR, simple sequence repeat.
Bryophytes sensu lato are the earliest terrestrial group of
plants that still exist on Earth. Bryophyta (sensu stricto), or
mosses, branched off from the stem of the Embryophyta phylogenetic tree
after Marchantiophyta and before the separation of Anthocerotophyta [1-4], although an alternative
topology with Anthocerotophyta in the basal position repeatedly appears
in analyses [5]. Estimates of the date of moss
origin vary greatly from 440 to 710 mya [6].
These nonvascular pioneers of land plants first acquired morphological,
biochemical, and physiological adaptations that enabled the transition
from aquatic to terrestrial habitats. The primary terrestrial biotopes
formed by the bryophytes were important spots for the subsequent
colonization of land by other plant evolutionary lineages. However,
there is no comprehensive scenario of this crucial step in plant
evolution. One approach to clarifying some obscurities of evolution is
comparative genomics. Comparative genomics can be used to investigate
the diversity of genome structure across different groups of living
beings, to identify genome organization conformities, and to understand
the mechanisms and factors of its evolution.
Recently, the development of next generation sequencing (NGS) technologies created new opportunities for genome studies and dramatically changed the methodology of investigation. As a result, many new genome sequences of different organisms have been loaded into the NCBI database. However, bryophyte genomics remains in the early stages of progress in comparison with other groups of plants. Until recently, the nuclear genome sequence was only available for a single moss species, namely Physcomitrella patens [7], and for eight species plastid genomes are known. Mitochondrial genomes from 39 moss species from 11 orders have been deposited in the NCBI GenBank (www.ncbi.nlm.nih.gov) to date. This is significantly fewer than for vascular plants, for which sequences of 164 mitochondrial genomes are present in GenBank.
Although moss chondrioms are rather conservative in structure [3, 8-10], the available data are scarce; six orders represented by only single species. The largest Hypnales order, consisting of 42 families, is represented by only five species from four families.
In the current study, we performed sequencing, assembly, and annotation of the mitochondrial genome of the moss Brachythecium rivulare (Hypnales, Brachytheciaceae) and compared it to the mitogenomes of four other mosses: two hypnaceous Anomodon rugelii NC_016121 [8] and Anomodon attenuatus NC_021931 [3] from Anomodontaceae, and two evolutionarily more distant Physcomitrella patens NC_007945 (Funariales, Funariaceae) [11] and Tetraphis pellucida NC_024290 (Tetraphidaceae) [9] from another class, Tetraphidopsida.
MATERIALS AND METHODS
The Brachythecium rivulare Schimp. plant sample was collected in the Moscow Region in a public park in a Moscow city suburb where permission to collect plants is not required. Approximately 1 µg of total DNA was isolated using a Nucleospin Plant Extraction Kit (Macherey-Nagel, Germany). Two pair-end libraries with insert sizes ca. 164 and 259 bp were obtained. The sequencing procedure was accomplished on an Illumina HiSeq 2000 NGS platform (Illumina, USA). Both library preparation and sequencing were performed following standard Illumina protocols.
The raw sequencing data consisted of approximately 187 and 175 million of 101-bp paired reads. After trimming low-quality read positions and the removal of sequencing adapters, the read quantity was 136.4 and 117.9 million read pairs, respectively. Ten million read pairs from each library were extracted and assembled using Velvet [12] with a k-mer length equal to 91. The assembly consisted of 617 contigs with a total length of 573,364 bp, and the longest contig (104,474 bp in length) was the complete mitochondrial genome with partially overlapping ends and 61× coverage.
After clipping the overlapping contig ends to verify the correctness of the nucleotide sequence ends, we closed it into circular form and then mapped the initial read subset to the genome end junction sequences with Bowtie 2 [13] and used Tablet [14] for SAM file visualization. For multiple nucleotide alignment and analyses of whole moss mitochondrial genomes, the Geneious software package [15] was utilized. For annotation of the assembled B. rivulare mitogenome, the annotated mitochondrial genome sequences of A. attenuatus, A. rugelii, T. pellucida, and P. patens were downloaded from the NCBI website (http://www.ncbi.nlm.nih.gov) and used as references.
For predicting RNA-editing sites in B. rivulare mitochondria, the BLASTx method in PREPACT 2.0 [16] was used. As references, species of 11 angiosperms, two lycopods, and the moss P. patens were used. For simple sequence repeat (SSR) loci identification, IMEx [17] and GMATo [18] tools were applied. Artemis [19], BLAST [20], BioEdit [21], genoPlotR [22], and the CGView Server [23] (http://stothard.afns.ualberta.ca/cgview_server) were also used in data analyses.
RESULTS AND DISCUSSION
Overall structure of the B. rivulare mitochondrial genome. The B. rivulare mitogenome was assembled as a single circular molecule (Fig. 1) and was deposited in the NCBI GenBank under accession number KR732319. The genome consists of 104,460 bp, and it was in the range of values from other studied mosses, except for Sphagnum palustre KC_784957, which has the largest mitogenome (141,276 bp) [3]. The comparison of selected features of the five bryophyte mitochondrial genomes is presented in Table S1 (see Supplement to this paper on the site of the journal (http://protein.bio.msu.ru/biokhimiya) and Springer site (https://link.springer.com/journal/10541)).
Fig. 1. Map of the B. rivulare mitochondrial genome. The circular scheme consists of several rings as follows: 1) (outer) forward strand – coding sequences (CDSs) and introns; 2) forward strand ORFs; 3, 4) the same locuses but located on the reverse strand; 5) GC content distribution across the sequence. The two latter internal rings show a GC skew for the forward and reverse strands as well as the genome coordinates.
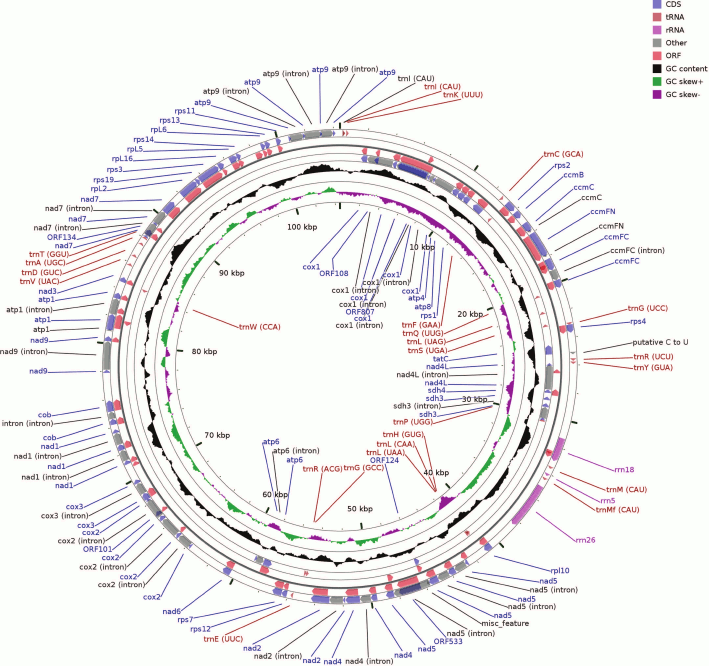
The B. rivulare mitochondrial genome has the same gene set and order as other known bryophyte mitogenomes. The genome contains three genes for rRNAs, 24 genes for tRNAs, 10 genes for the small subunit ribosomal proteins, five genes for the large subunit ribosomal proteins, nine, two, one, three, and five genes for mitochondrial respiratory chain complexes I, II, III, IV, and V, respectively, four genes involved in cytochrome c biogenesis, one gene for the sec-independent protein, three pseudogenes (reverse transcriptase-like protein, rps8 and rps10), and six different ORF genes.
Refinements of annotations of some known bryophyte mitogenomes. Despite the high similarity in the gene content of the compared mosses, some discrepancies in their annotations were identified. For example, the ORF134 gene is not annotated in the P. patens, T. pellucida, and A. attenuatus genomes. Interestingly, there are two insertions of 8 and 1 bp in this locus of P. patens that are spaced by a 100-bp nucleotide sequence. The last insertion returns the translated protein product to the correct reading frame, thus suggesting a functional role for this genome region. In contrast, the substitution of the tryptophan codon (TGG) in B. rivulare for the stop codon (UAG) in A. rugelii, A. attenuatus, and P. patens and the presence of multiple internal stop codons and indels in T. pellucida may indicate pseudogenization of ORF134.
The ORF101 and ORF124 genes are also not annotated in A. attenuatus, P. patens, and T. pellucida. In A. attenuatus, the ORF124 locus has a 17-bp deletion at its 3′ end, resulting in a reading frame shift. Thus, the functional activity and correct annotation of the locus remains unresolved.
ORF807 is not annotated in A. attenuatus. Instead, P. patens and T. pellucida contain ORF622, which is almost completely located in the ORF807 locus coordinates. Importantly, the sequence of this ORF is quite conserved as its identity in B. rivulare and T. pellucida is close to 90%.
ORF108 is 327-bp long in B. rivulare, A. attenuatus, and A. rugelii, but it is not annotated in A. attenuatus. In P. patens, this ORF is almost completely lost, with only 63 bp at the 3′ end. In T. pellucida, there are multiple internal stop codons and indels in its sequence.
ORF533 is not annotated in A. attenuatus.
The rpl10 gene is named ORF187 but not annotated in P. patens, and this gene is annotated as functional in other mosses. However, the question of the existence of functional rpl10 in the P. patens mitochondrial genome is not completely solved, which was described in detail by Kubo and Arimura [24]. There is no direct evidence of activity for this gene in mosses. However, transcripts from rpl10 have been detected in the liverwort Marchantia and the hornwort Megaceros [24]. In mosses, we found differences both in length and sequence near the 3′ end of this gene, resulting in shifting of the reading frame.
In the P. patens, T. pellucida, and A. attenuatus mitogenomes, the rps8, rps10, and rtl pseudogenes are not annotated. The rps10 pseudogene in T. pellucida is almost completely lost.
The analysis of the five moss mitogenome alignments showed additional potential inaccuracies in the annotations. In P. patens and T. pellucida, the cox2 gene intron 3 and exon 4 regions are missing in the annotation. The atp9 gene in T. pellucida may not have the correct 3′ boundary, because it is 78 bp shorter from its 3′ end with a C to U RNA-editing site creating the correct stop codon (UGA). The same situation is evident for the T. pellucida atp1 gene because its size is shorter by 9 bp, with an additional C to U RNA-editing site. In addition, the rpl15 gene name of T. pellucida should be changed to rpl5. This locus is treated under the name rpl5 in the other available moss mitogenomes, and introducing a new one is not necessary.
Characteristics of bryophyte mitochondrial genomes. Gene lengths. The distributions of the lengths of the B. rivulare mitochondrial genes, exons, and introns are shown in Fig. 2. The B. rivulare exon length varies from 8 bp (exon 3 of atp9) to 1314 bp (exon 2 of nad2). The B. rivulare intron length ranges from 390 to 2639 bp (introns 2 and 3 of cox1, respectively).
Fig. 2. Distribution of B. rivulare mitogenome gene length (a), exon length (b), and intron length (c). L, length in base pairs; n, number of corresponding sequences. The length of most of the genes is in the first interval from 0 to 1000 bp (minimal length is 71 bp). Most of the exons lie in the interval from 0 to 200 bp (with a minimal length of 8 bp). The most common intron length ranges from 500 to 1000 bp.
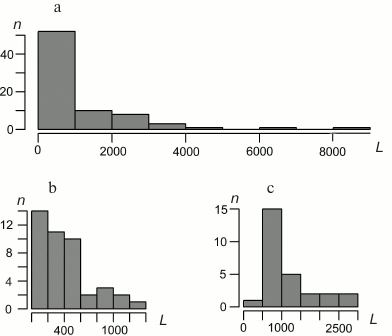
The B. rivulare gene length varies from 71 bp (trnG and trnC) to 8290 bp (cox1) (Fig. 2). Thus, the longest B. rivulare genes are cox1, nad5 (6341 bp), and cox2 (4550 bp). The same genes are the longest in A. attenuatus and A. rugelii. In P. patens, the longest genes are cox1 (7602 bp), nad5 (6095 bp), and nad7 (3210 bp). In T. pellucida, the longest genes are cox1 (9252 bp), atp9 (4372 bp), and atp1 (3281 bp).
The genes that are the most variable in length of the four moss mitochondrial genomes are atp9 (from 3253 bp in A. attenuatus to 4372 bp in T. pellucida), cox1 (from 7602 bp in P. patens to 9252 bp in T. pellucida), cob (from 1753 bp in B. rivulare to 2175 bp in P. patens), cox2 (from 3119 bp in P. patens to 4601 bp in A. attenuatus and A. rugelii), atp1 (from 2684 bp in A. attenuatus to 3281 bp in T. pellucida), and nad9 (from 1711 bp in T. pellucida to 2629 bp in A. attenuatus and A. rugelii).
The main reason for the differences in gene size is the presence of numerous indels in the introns. For example, P. patens has two large deletions in intron 4 of cox1 (106 and 500 bp), single deletions in introns 2 and 3 of nad5 (136 and 73 bp, respectively), three large deletions in intron 1 of nad4L (271, 45, and 73 bp), and many smaller deletions. Tetraphis pellucida has long insertions in intron 1 of cox1 as well as in introns 1 and 3 of atp9.
Gene size variation in ribosomal RNA genes was also identified in rrn18 (from 1587 bp in P. patens to 1738 bp in A. attenuatus) and rrn26 (from 2946 bp in P. patens to 3387 bp in B. rivulare). Anomodon attenuatus has an insertion in the 18S rDNA (16 bp). Moreover, there are two deletions in the rrn18 gene of P. patens (131 and 4 bp). In the 26S rDNA of P. patens, a number of deletions (the largest deletions were 73 and 365 bp) and one insertion (3 bp) were found.
RNA editing. The editing of transcripts in mitochondria occurs in all land plants except marchantiid liverworts. RNA-editing frequencies vary widely across land plants, see [25-27] and references therein. C→U editing dominates among seed plants, mosses, and nonmarchantiid liverworts, whereas frequent U→C transition by amination is frequent in mosses, hornworts, lycophytes, and ferns.
For predicting RNA-editing sites in B. rivulare mitochondria by PREPACT 2.0, species of 11 angiosperms, two lycopods, and the moss P. patens were used as references. As a result, 47 and 189 potential sites were predicted by 100 and 75% of references, respectively. The maximum number of hits was revealed for the Cucurbitaceae species Citrullus lanatus (336) and Cucurbita pepo (335). Using P. patens as a reference, 27 C→U and 75 U→C events were identified (Table S2, see Supplement).
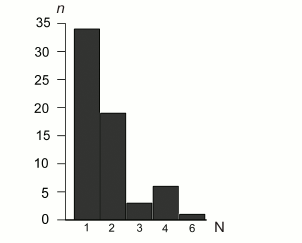
SSR content. Following the more stringent criteria of perfect SSR loci identification (minimal number of repeating units ≥10 for mononucleotides, ≥5 for dinucleotides, ≥4 for trinucleotides, and ≥3 for tetra-, penta-, and hexanucleotides), 63 SSR loci were identified in the B. rivulare mitochondrial genome. Three SSR loci in coding regions, and 60 SSR loci are in noncoding regions. The total perfect SSR length is 734 bp, which is approximately 0.7% of the total genome size. The average SSR loci size is 11.65 bp. The distribution of SSR sequences between different classes is shown in Fig. 3 and Table S3 (see Supplement).
Fig. 3. Distribution of SSR repeat unit length in the B. rivulare mitogenome. The numbers of base pairs in different microsatellite classes identified in the analyzed genome are on the horizontal axis. The number of loci in each SSR category (1, 2, 3, 4, and 6) is shown on the y-axis. The mononucleotide repeats are the majority of identified SSR sequences (34 loci). Hexanucleotides are the least represented (only one locus was found).
Gene conservation level in bryophyte mitogenomes. Liu et al. [3] demonstrated a great conservation of both structure and sequence of mitochondrial genomes of mosses, and our data agree with this finding. To estimate the variability of the different loci in the B. rivulare mitogenome, we compared the different loci in terms of the Geneious similarity measure to orthologous loci (excluding pseudogenes, RNA, and ORF genes) from P. patens, A. attenuatus, A. rugelii, and T. pellucida (table). The most conservative gene is rpl16 when performing pairwise mitogenome comparisons of B. rivulare with P. patens, A. attenuatus, and A. rugelii, and the most conservative gene is rpl6 when comparing B. rivulare and T. pellucida. The most variable gene when comparing B. rivulare with A. rugelii and A. attenuatus is cob (as mentioned above because of the presence of a deletion in intron 1 in B. rivulare) encoding the ubiquinol-cytochrome c oxidoreductase subunit. Compared to the P. patens gene set, the most variable gene is nad4L (encoding the NADH dehydrogenase 4L protein) because of the existence of multiple deletions in the P. patens nad4L intron 1. Compared to T. pellucida, the most variable gene is the atp9 gene.
Similarity of nucleotide sequences of B. rivulare genes with
corresponding orthologs in four other mosses (%)
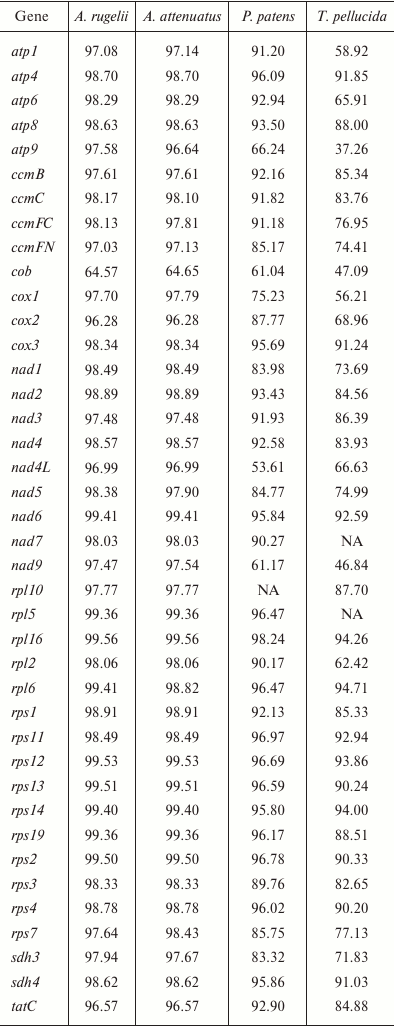
Note: NA, absence of gene annotation in the compared genomes.
The gene similarity between B. rivulare and A. rugelii varies from 64.57% (cob) to 99.56% (rpl16), and the mean similarity is 97.62%. The gene similarity between B. rivulare and A. attenuatus ranges from 64.65% (cob) to 99.56% (rpl16), and the mean similarity is 97.54%. When comparing B. rivulare and P. patens, the gene similarity varies from 53.61% (nad4L) to 98.24 (rpl16), and the mean similarity is 88.56%. Finally, the gene similarity between B. rivulare and T. pellucida varies from 37.26% (atp9) to 94.71% (rpl6), and the mean similarity is 78.87%.
Thus, similarity of the Hypnales species mitogenomes is 97.6%, whereas for species from different orders it is 78.9-88.6%. This result is in good agreement with the overall phylogeny of bryophytes, and the percentage of mitogenome similarity is correlated to the age estimated from the phylochronologic analysis [28].
Acknowledgments
We are grateful to D. A. Alexeyevsky, K. V. Mikhailov, V. V. Aleshin, and A. S. Kasianov for advice and recommendations in NGS data analysis and valuable discussion. Also, we would like to thank Paul Stothard for assistance in preparation of the figures.
This study was funded by the Russian Foundation for Basic Research (project No. 15-04-06027) and the Russian Science Foundation (project No. 14-50-00029, construction and sequencing the DNA libraries).
REFERENCES
1.Lewis, L. A., Mishler, B. D., and Vilgalys, R.
(1997) Phylogenetic relationships of the liverworts (Hepaticae), a
basal embryophyte lineage, inferred from nucleotide sequence data of
the chloroplast gene rbcL, Mol. Phylogenet. Evol.,
7, 377-393.
2.Samigullin, T. Kh., Yacentyuk, S. P., Degtyaryeva,
G. V., Valieho-Roman, K. M., Bobrova, V. K., Capesius, I., Martin, W.
F., Troitsky, A. V., Filin, V. R., and Antonov, A. S. (2002) Paraphyly
of bryophytes and close relationship of hornworts and vascular plants
inferred from chloroplast rDNA spacers sequence analysis,
Arctoa, 11, 31-43.
3.Liu, Y., Medina, R., and Goffinet, B. (2014) 350 My
of mitochondrial genome stasis in mosses, an early land plant lineage,
Mol. Biol. Evol., 31, 2586-2591.
4.Qiu, Y. L., Li, L., Wang, B., Chen, Z., Knoop, V.,
Groth-Malonek, M., Dombrovska, O., Lee, J., Kent, L., Rest, J.,
Estabrook, G. F., Hendry, T. A., Taylor, D. W., Testa, C. M., Ambros,
M., Crandall-Stotler, B., Duff, R. J., Stech, M., Frey, F., Quandt, D.,
and Davis, C. C. (2006) The deepest divergences in land plants inferred
from phylogenomic evidence, Proc. Natl. Acad. Sci. USA,
103, 15511-15516.
5.Wickett, N. J., Mirarab, S., Nguyen, N., Warnow,
T., Carpenter, E., Matasci, N., Ayyampalayam, S., Barker, M. S.,
Burleigh, J. G., Gitzendanner, M. A., Ruhfel, B. R., Wafula, E., Der,
J. P., Graham, S. W., Mathews, S., Melkonian, M., Soltis, D. E.,
Soltis, P. S., Miles, N. W., Rothfels, C. J., Pokorny, L., Shaw, A. J.,
DeGironimo, L., Dennis, W., Stevenson, D. W., Surek, B., Villarreal, J.
C., Roure, B., Philippe, H., dePamphilis, C. W., Chen, T., Deyholos, M.
K., Baucom, R. S., Toni, M., Kutchan, T. M., Augustin, M. M., Wang, J.,
Zhang, Y., Tian, Z., Yan, Z., Wu, X., Sun, X., Wong, G. K.-S., and
Leebens-Mack, J. (2014) Phylotranscriptomic analysis of the origin and
early diversification of land plants, Proc. Natl. Acad. Sci.
USA, 111, E4859-E4868.
6.Magallon, S., and Hilu, K. W. (2009) Land plants
(Embryophyta), in The Timetree of Life (Hedges, S. B., and
Kumar, S., eds.) Oxford University Press, pp. 133-137.
7.Rensing, S. A., Lang, D., Zimmer, A. D., Terry, A.,
Salamov, A., Shapiro, H., Nishiyama, T., Perroud, P.-F., Lindquist, E.
A., Kamisugi, Y., Tanahashi, T., Sakakibara, K., Fujita, T., Oishi, K.,
Shin-I., T., Kuroki, Y., Toyoda, A., Suzuki, Y., Hashimoto, S.-i.,
Yamaguchi, K., Sugano, S., Kohara, Y., Fujiyama, A., Anterola, A.,
Aoki, S., Ashton, N., Barbazuk, W. B., Barker, E., Bennetzen, J. L.,
Blankenship, R., Sung Hyun Cho, Dutcher, S. K., Estelle, M., Fawcett,
J. A., Gundlach, H., Hanada, K., Heyl, A., Hicks, K. A., Hughes, J.,
Lohr, M., Mayer, K., Melkozernov, A., Murata, T., Nelson, D. R., Pils,
B., Prigge, M., Reiss, B., Renner, T., Rombauts, S., Rushton, P. J.,
Sanderfoot, A., Schween, G., Shin-Han Shiu, Stueber, K., Theodoulo, F.
L., Tu, H., van de Peer, Y., Verrier, P. J., Waters, E., Wood, A.,
Lixing Yang, Cove, D., Cuming, A. C., Hasebe, M., Lucas, S., Mishler,
B. D., Reski, R., Grigoriev, I. V., Quatrano, R. S., and Boore, J. L.
(2008) The Physcomitrella genome reveals evolutionary insights
into the conquest of land by plants, Science, 319,
64-69.
8.Liu, Y., Xue, J.-Y., Wang, B., Li, L., and Qiu,
Y.-L. (2011) The mitochondrial genomes of the early land plants
Treubia lacunosa and Anomodon rugelii: dynamic and
conservative evolution, PLoS One, 6, e25836.
9.Bell, N. E., Boore, J. L., Mishler, B. D., and
Hyvonen, J. (2014) Organellar genomes of the four-toothed moss,
Tetraphis pellucida, BMC Genom., 15, 383.
10.Liu, Y., Cox, C. J., Wang, W., and Goffinet, B.
(2014) Mitochondrial phylogenomics of early land plants: mitigating the
effects of saturation, compositional heterogeneity, and codon-usage
bias, Syst. Biol., 63, 862-878.
11.Terasawa, K., Odahara, M., Kabeya, Y., Kikugawa,
T., Sekine, Y., Fujiwara, M., and Sato, N. (2007) The mitochondrial
genome of the moss Physcomitrella patens sheds new light on
mitochondrial evolution in land plants, Mol. Biol. Evol.,
24, 699-709.
12.Zerbino, D. R., and Birney, E. (2008) Velvet:
algorithms for de novo short read assembly using de Bruijn
graphs, Genome Res., 18, 821-829.
13.Lingmead, B., Trapnell, C., Pop, M., and
Salzberg, S. L. (2009) Ultrafast and memory-efficient alignment of
short DNA sequences to the human genome, Gen. Biol., 10,
R25.
14.Milne, I., Stephen, G., Bayer, M., Cock, P. J.
A., Pritchard, L., Cardle, L., Shaw, P. D., and Marshall, D. (2013)
Using Tablet for visual exploration of second-generation sequencing
data, Brief. Bioinformatics, 14, 193-202.
15.Kearse, M., Moir, R., Wilson, A., Stones-Havas,
S., Cheung, M., Sturrock, S., Buxton, S., Cooper, A., Markowitz, S.,
Duran, C., Thierer, T., Ashton, B., Meintjes, P., and Drummond, A.
(2012) Geneious Basic: an integrated and extendable desktop software
platform for the organization and analysis of sequence data,
Bioinformatics, 28, 1647-1649.
16.Lenz, H., and Knoop, V. (2013) PREPACT 2.0:
predicting C-to-U and U-to-C RNA editing in organelle genome sequences
with multiple references and curated RNA editing annotation,
Bioinform. Biol. Insights, 7, 1-19.
17.Mudunuri, S. B., and Nagarajaram, H. A. (2007)
IMEx: Imperfect Microsatellite Extractor, Bioinformatics,
23, 1181-1187.
18.Wang, X., Lu, P., and Luo, Z. (2013) GMATo: a
novel tool for the identification and analysis of microsatellites in
large genomes, Bioinformation, 9, 541-544.
19.Rutherford, K., Parkhill, J., Crook, J.,
Horsnell, T., Rice, P., Rajandream, M.-A., and Barrell, B. (2000)
Artemis: sequence visualization and annotation, Bioinformatics,
16, 944-945.
20.Altschul, S. F., Gish, W., Miller, W., Myers, E.
W., and Lipman, D. J. (1990) Basic local alignment search tool, J.
Mol. Biol., 215, 403-410.
21.Hall, T. A. (1999) BioEdit: a user-friendly
biological sequence alignment editor analysis program for Windows
95/98/NT, Nucleic Acids Symp. Ser., 41, 95-98.
22.Guy, L., Kultima, J. R., and Andersson, S. G.
(2010) GenoPlotR: comparative gene and genome visualization in R,
Bioinformatics, 26, 2334-2335.
23.Grant, J. R., and Stothard, P. (2008) The CGView
server: a comparative genomics tool for circular genomes, Nucleic
Acids Res., 36, W181-W184.
24.Kubo, N., and Arimura, S.-I. (2010) Discovery of
a functional rpl10 gene in diverse plant mitochondrial genomes
and its functional replacement by a nuclear gene for chloroplast
RPL10 in two lineages of angiosperms, DNA Res.,
17, 1-9.
25.Groth-Malonek, M., Wahrmund, U., Polsakiewicz,
M., and Knoop, V. (2007) Evolution of a pseudogene: exclusive survival
of a functional mitochondrial nad7 gene supports
Haplomitrium as the earliest liverwort lineage and proposes a
secondary loss of RNA editing in Marchantiidae, Mol. Biol.
Evol., 24, 1068-1074.
26.Knoop, V., Volkmar, U., Hecht, J., and Grewe, F.
(2011) Mitochondrial genome evolution in the plant lineage, in Plant
Mitochondria, Advances in Plant Biology (Kempken, F., ed.)
Springer, N.Y., pp. 3-29.
27.Rudinger, M., Volkmar, U., Lenz, H.,
Groth-Malonek, M., and Knoop, V. (2012) Nuclear DYW-type PPR gene
families diversify with increasing RNA editing frequencies in liverwort
and moss mitochondria, J. Mol. Evol., 74, 37-51.
28.Newton, A. E., Wikstrom, N., Bell, N., Forrest,
L. L., and Ignatov, M. S. (2007) Dating the diversification of the
pleurocarpous mosses, in Pleurocarpous Mosses: Systematics and
Evolution (Newton, A. E., and Tangney, R. S., eds.) Systematics
Association Special, CRC Press, Boca Raton, FL, pp. 337-366.
Supplementary Tables (PDF)