Role of Protein L25 and Its Contact with Protein L16 in Maintaining the Active State of Escherichia coli Ribosomes in vivo
A. Y. Anikaev, A. B. Isaev, A. V. Korobeinikova, M. B. Garber, and G. M. Gongadze*
Institute of Protein Research, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia; fax: +7 (495) 514-0218; E-mail: gongadze@vega.protres.ru* To whom correspondence should be addressed.
Received July 6, 2015; Revision received July 22, 2015
A ribosomal protein of the L25 family specifically binding to 5S rRNA is an evolutionary feature of bacteria. Structural studies showed that within the ribosome this protein contacts not only 5S rRNA, but also the C-terminal region of protein L16. Earlier we demonstrated that ribosomes from the ΔL25 strain of Escherichia coli have reduced functional activity. In the present work, it is established that the reason for this is a fraction of functionally inactive 50S ribosomal subunits. These subunits have a deficit of protein L16 and associate very weakly with 30S subunits. To study the role of the contact of these two proteins in the formation of the active ribosome, we created a number of E. coli strains containing protein L16 with changes in its C-terminal region. We found that some mutations (K133L or K127L/K133L) in this protein lead to a noticeable slowing of cell growth and decrease in the activity of their translational apparatus. As in the case of the ribosomes from the ΔL25 strain, the fraction of 50S subunits, which are deficient in protein L16, is present in the ribosomes of the mutant strains. All these data indicate that the contact with protein L25 is important for the retention of protein L16 within the E. coli ribosome in vivo. In the light of these findings, the role of the protein of the L25 family in maintaining the active state of the bacterial ribosome is discussed.
KEY WORDS: 5S rRNA-binding protein L25, ribosomal protein L16, RNA–protein interactions, ribosome, translation, Escherichia coliDOI: 10.1134/S0006297916010028
A great body of data regarding ribosome formation and function has been accumulated over 50 years of study. Crystallographic studies of ribosomes and their functional complexes have made a significant contribution to understanding of these processes [1-9]. However, the roles of most ribosomal proteins and their intermolecular contacts in the function of the translation apparatus are still poorly explored. Evolution of the ribosome structure of different organisms additionally complicates understanding of their roles. Thus, ribosomes of contemporary organisms contain 60-80 proteins: among them, only 34 are conserved in all domains of life, while the others are evolutionary acquisitions of Archaea, Bacteria, or Eukarya [10, 11].
5S rRNA-binding ribosomal protein L25 from Escherichia coli and its homologs, the L25 protein family [12], are features of the bacterial translation apparatus. At the same time, a gene for this protein is found in the majority but not in all bacteria [11, 13]. One of the representatives of the family, protein CTC from Bacillus subtilis, is only temporarily associated with the ribosome, as it is synthesized in the cell only under stressful conditions [14, 15]. Moreover, it was shown that even protein L25 of E. coli is not essential for survival of the cells [16, 17]. It is known from structural data that protein L25 of E. coli (or the 5S rRNA-binding domain of its homologs) occupies an isolated position in the central protuberance of the 50S subunit and does not make direct contact with translation ligands (tRNA, mRNA, and protein factors) [1-9, 18]. This might give the impression that a protein of the L25 family is not required for the functioning of the bacterial ribosome. However, we showed that, although E. coli cells lacking protein L25 (ΔL25 strain) survive, they grow significantly more slowly than the control cells, and their ribosomes are less efficient in protein synthesis in vivo and in vitro [17]. These results indicate that this protein is still required for efficient ribosome functioning, but they do not reveal its precise role.
Until recently, it was believed that protein L25 interacts only with 5S rRNA within the E. coli ribosome. Crystallographic studies of bacterial ribosomes revealed a contact of the protein of the L25 family with protein L16, in which the C-terminal tail of the latter participates [1, 2, 19]. The first reports indicating that protein L16 could be a key component of the ribosome appeared 30 years ago. Thus, it followed from results on the reconstruction of 50S ribosomal subunit in vitro that protein L16 is very important for the formation of a number of functional sites of the subunit and its conformational changes upon assembly [20-22]. Later, it was shown that L16 is one of the proteins that are strictly required for survival of E. coli [16]. Structural studies of the bacterial ribosomes mostly confirmed the conclusions made earlier, revealing extensive contacts of protein L16 with important regions of 23S rRNA and a direct contact with A-site tRNA [1, 2, 6]. At the same time, we recently showed that the presence in the ribosome of protein L25 mutant form, which is unable to bind 5S rRNA, affects positively the activity of the translation apparatus in vivo [23]. Therefore, it was assumed that protein L25 has an indirect effect on the functional activity of the E. coli ribosome via the contact with protein L16. In the present work, we found the reason for the reduced efficiency in protein biosynthesis of ribosomes from the ΔL25 strain and tested the effect of some mutations in the C-terminal region of protein L16 on the formation of functionally active bacterial ribosomes in vivo.
MATERIALS AND METHODS
Strains and microbiological methods. Standard microbiological techniques described in [24] were used in this work. Growth characteristics as well as activity of the translation apparatus of the cells of each strain were estimated based on results of 3-4 independent experiments. Each measurement was done three times, and the relative error of the measurements did not exceed 5-7%. For β-galactosidase analysis, cells were grown to A600 = 0.2, and production of the enzyme was induced by addition of isopropyl-β-D-thiogalactopyranoside to 1 mM. Accumulation of the enzyme in the culture was detected by the color reaction of the conversion of 2-nitrophenyl-β-D-galactopyranoside. The β-galactosidase activity was normalized to the cell mass [24]. Strains used or created in this work are presented in Table S1 in the Supplement (see Supplement to this paper on the site of the journal (http://protein.bio.msu.ru/biokhimiya) and of Springer Publisher (Link.springer.com)). Oligonucleotides used in the work are listed in Table S2 in the Supplement. The E. coli cells were grown at appropriate temperature in LB medium supplemented with 10 µg/ml chloramphenicol when needed.
Introduction of point mutations into the chromosomal gene for protein L16. Directed changes into the E. coli chromosome were introduced by the “recombineering” technique [25]. First, a DNA cassette was obtained by two-step PCR using KOD Hot Start DNA Polymerase (Novagen, USA). The DNA cassette was composed of the open reading frame (ORF) of the rplP gene or its mutant form, a 12-nt spacer (carrying the regulatory Shine–Dalgarno sequence), and the ORF of the chloramphenicol acetyltransferase gene (cat), as well as short (~40 nt) sequences on the 5′- and 3′-ends that are homologous to the regions flanking the rplP gene in the chromosome and are required for the subsequent recombination. In the first step, the ORF of the cat gene was amplified. The DNA of strain KNB800 (Table S1 in the Supplement) was used as the template in PCR. The forward primer was AI 01-F for creation of the control strain (wild-type L16) and L16-K133L-F for the strain with the K133L mutation in L16 (Table S2 in the Supplement). In both cases, the reverse primer was AI 02-R (Table S2). In the second step, the ORF of the rplP gene or its mutant form was amplified. The DNA of strain MG1655 (Table S1) was used as the template in PCR. The forward primer was AI 03-F (Table S2). The reverse primer was the PCR-product obtained in the first step. To prepare strains containing protein L16 with mutations K127A or K127L/K133L, we used a modified scheme for creation of the DNA-cassette. In the first step, the ORFs of the cat and rplP genes were amplified separately in two different PCRs. To amplify the ORF of the rplP gene, we used primers AI 03-F and L16-X-R (where X is the corresponding mutation) (Table S2) and the DNA of strain MG1655 as template (Table S1). To amplify the ORF of the cat gene, we used primers AI 02-R and L16-X-F (where X is the corresponding mutation) (Table S2) and the DNA of strain KNB800 as template (Table S1). The primers L16-X-F and L16-X-R are complementary to each other; thus, the 3′-end of the PCR-product with the ORF of the rplP gene was complementary to the 5′-end of the PCR-product with the ORF of the cat gene. In the second step, the full-length DNA-cassette was amplified, wherein the PCR products obtained in the first step annealing to each other were used as template. The forward primer was AI 03-F; the reverse primer was AI 02-R (Table S2). The resulting DNA cassettes were recombined into the chromosome of cells of strain DY330 [25]. Selection was carried out on LB-agar in the presence of chloramphenicol. Checking and sequencing of the rplP locus were performed by PCR with primers rplP-check-F and rplP-check-R. After that, mutant alleles were transferred into the chromosome of strain MG1655 (Table S1) by bacteriophage P1 transduction as described [26]. Strain AMrB01 containing allele rplP-1::cat (wild-type rplP ORF) (Table S1) was used as a control in all experiments.
Preparation of ribosomes and ribosomal subunits. Ribosomes from cells of control and mutant strains were obtained according to published procedures [27, 28] with modifications described in [17, 23]. Samples of ribosomes were obtained from the crude cell extract S30 by centrifugation at 290,000g at 4°C for 3-4 h (10 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 0.2 mM EDTA, and 3 mM 2-mercaptoethanol (buffer A) with 100 mM NH4Cl). In some experiments, the concentration of MgCl2 in buffer A was increased to 20 mM. For additional purification, ribosomes were centrifuged at 290,000g at 4°C for 5-6 h through 30% (w/w) sucrose prepared in buffer A with 500 mM NH4Cl. To separate 70S ribosomes and dissociated ribosomal subunits, samples of the ΔL25 ribosomes and the ribosomes containing mutant protein L16 were centrifuged at 87,000g at 4°C for 13-14 h in 10-30% (w/w) sucrose density gradients prepared in buffer A with 100 mM NH4Cl. Active subunits from the fraction of the 70S ribosomes were obtained by technique described in [28]. All samples of ribosomes and ribosomal subunits were kept in buffer A with 100 mM NH4Cl and 10% glycerol at –70°C.
Analysis of ribosomes and ribosomal proteins. Analysis of distribution of ribosomal particles in samples of ribosomes and cell extracts S30 was performed by centrifugation at 270,000g at 4°C for 80 min in a 5-20% (w/w) sucrose gradient prepared in buffer A with 100 mM NH4Cl. Experiments on the association of ribosomal subunits were performed as follows. Equal amounts of 30S and 50S subunits were mixed in buffer A containing 100 mM NH4Cl and varying concentrations of MgCl2 (3-25 mM) and incubated at 37°C for 30 min. Then, the mixtures were analyzed by centrifugation in 5-20% (w/w) sucrose gradient as described above. Ribosomes were treated with puromycin according to published procedures [29, 30]. To release peptides from ribosomes, crude cell extract S30 was incubated for 15 min at 37°C in buffer B (20 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 150 mM NH4Cl, 3 mM 2-mercaptoethanol, 0.2 mM EDTA) with 1 mM puromycin. As a control, crude extract was incubated in the same manner but without puromycin. Then the samples were analyzed by centrifugation in the sucrose density gradient as described above. Translation of luciferase mRNA in vitro was performed according to a published technique [31] with modifications described in [17]. To analyze the protein composition of ribosomes, 2D gel electrophoresis (system IV) was used as described in [32].
RESULTS AND DISCUSSION
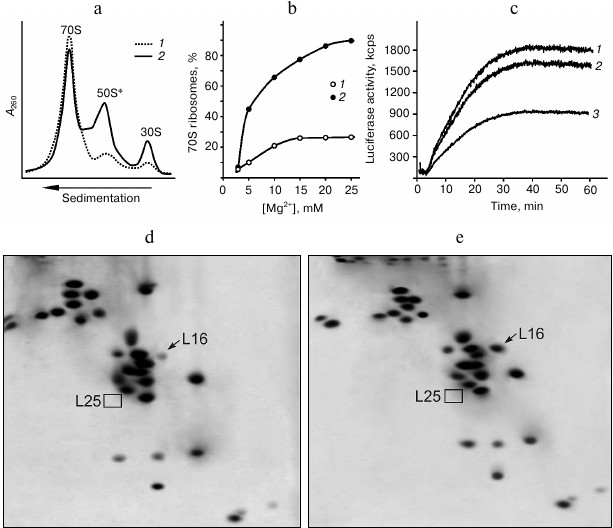
Changing properties of E. coli ribosomes due to lack of protein L25. Earlier, we showed that ribosomes from the cells of ΔL25 strain contain all of the proteins except for L25, and their subunits are present predominantly in the associated form [17]. However, as shown in the same work, the ΔL25 ribosomes are considerably less efficient in protein synthesis than control ribosomes, both in vivo and in vitro. In the present work, detailed analysis of the properties of the ribosomes from the ΔL25 strain revealed the basis of their reduced functional activity. To do this, ribosomes from the mutant and control strains were purified from cell components using standard methods (see details in “Materials and Methods”). We found that the purified ribosomes from the ΔL25 strain contain a significant amount (from 40 to 50%) of dissociated subunits, in contrast to the control ribosomes (Fig. 1a). In addition, the 50S subunits from this fraction have a defect that hinders their association with 30S subunits into ribosome even at high concentrations (over 20 mM) of Mg2+ (Fig. 1b). Since ΔL25 ribosomes have reduced functional activity [17], it was logical to assume that the indicated fraction of 50S subunits causes this. Therefore, we compared the functional activity of the 70S fractions from both control and ΔL25 ribosomes in vitro. As seen in Fig. 1c, in contrast to the total sample of ΔL25 ribosomes, the 70S fraction of these ribosomes is practically of the same efficiency in polypeptide synthesis as the control sample. To determine the reason for functional inactivity of the part of the ΔL25 ribosomes, the protein composition of the defective 50S subunits was analyzed. It turned out that these 50S subunits have a deficit of protein L16 (Fig. 1d), unlike the active 50S subunits isolated from the 70S fraction (Fig. 1e) containing this protein in about equimolar amount. As previously shown in a number of experiments on reconstruction of 50S subunits in vitro, protein L16 is critical for the formation of the functionally active bacterial ribosome [20-22]. Therefore, we conclude that the inactive fraction of the 50S subunits lacking protein L16 is indeed the reason for reduced functional activity of the ribosomes isolated from the ΔL25 strain. However, this result gives rise to a number of new questions. First, if these defective 50S ribosomal subunits are present in cells, why was it not detected in the cytoplasmic fraction in our previous work [17]? Second, are these defective subunits assembled in the cell or do they become inactive during functioning? Third, is protein L16 present in the defective 50S subunits before purification?
Fig. 1. Properties of ribosomes from the ΔL25 strain of E. coli. a) Sedimentation profiles of purified ribosomes of control (1) and ΔL25 (2) strains. b) Effect of Mg2+ concentration on the association of ribosomal subunits. For the analysis, subunits from the fraction of dissociated (50S* and 30S) ΔL25 ribosomes (1) and subunits of wild-type ribosomes (2) were used. с) Synthesis of luciferase in the E. coli cell-free translation system containing 70S fraction of the control (1) and ΔL25 (2) ribosomes or total preparation of the ΔL25 ribosomes (3). d, e) Analysis of protein composition of defective (50S*) and active (from 70S fraction) 50S ribosomal subunits from the ΔL25 strain, respectively. The positions of proteins L16 and L25 are indicated on the electrophoregrams.
It was previously found that after gentle disruption of bacterial cells, a major part of the ribosomes in the crude cell extract remains associated with mRNA and peptidyl-tRNA (so-called “charged” ribosomes) [29, 30, 33]. These ligands increase the stability of the associated ribosomal subunits. We assumed that in the crude cell extract this defect of the ribosomes from ΔL25 strain could be masked by translation ligands. Puromycin treatment of the charged ribosomes is known to lead to release of peptides that, in turn, weakens the contact of deacylated tRNA and mRNA with the ribosome [29, 30, 34]. Wild-type bacterial ribosomal subunits remain in the associated state after such treatment [30, 33]. We tested the effect of puromycin on the stability of the association of the ribosomes in the crude cell extract of the ΔL25 strain (Fig. 2a). It is seen that ΔL25 ribosomes contain a minor amount of free subunits before puromycin treatment. After such treatment, in contrast to the control ribosomes, almost half of the ΔL25 ribosomes dissociate into subunits. This sedimentation profile is very similar to the profile of ΔL25 ribosomes after standard purification (Figs. 1a and 2a). We analyzed the protein composition of the 50S subunits dissociated after treatment of ΔL25 ribosomes with puromycin (Fig. 2b). These subunits, as well as the defective subunits after purification (Fig. 1d), are deficient in protein L16. It is most likely that the fraction of the defective 50S subunits loses protein L16 before their isolation and purification. Furthermore, in the cytoplasmic fraction most of these subunits are found in 70S ribosomes containing peptidyl-tRNA. Thus, the defect of 50S subunits is masked in the cytoplasmic fraction. Considering these results, it is unlikely that the 50S subunits having this functional defect could be formed during assembly and then involved in translation. Therefore, our findings suggest that the loss of functional activity of the 50S subunit of ΔL25 ribosomes can occur in the process of translation, generating a fraction of arrested ribosomes.
Fig. 2. Effect of puromycin on properties of ribosomes in crude cell extract (S30) of E. coli ΔL25 strain. a) Sedimentation profiles of ribosomes from S30 of the ΔL25 strain before (1) and after (2) puromycin treatment. The profile of ribosomes from wild-type strain after puromycin treatment is presented as a control (3). b) Analysis of protein composition of 50S subunits (50S*) appearing after incubation of S30 of the ΔL25 strain with puromycin. The positions of proteins L16 and L25 are indicated on the electrophoregram. c) Model of a part of the structure of the central protuberance of the ribosome. Ribosomal proteins, 5S rRNA, and helices of 23S rRNA discussed in the text are indicated. The structure of the E. coli ribosome (PDB code 2AW4) was used to build the model.
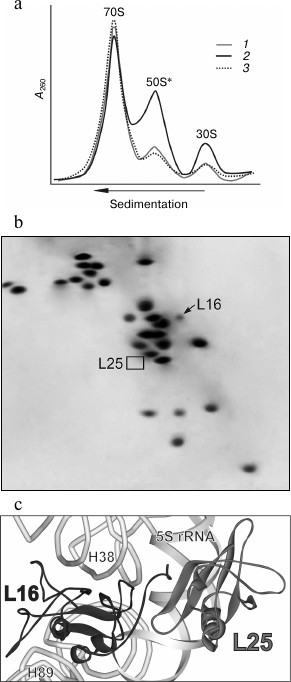
It follows from structural studies of the bacterial ribosomes that several amino acid residues of the C-terminal tail of protein L16 may be involved in the interaction with protein L25 (Fig. 2c) [1, 2, 19]. At the same time, protein L16 has many contacts (via more than half of its surface) with high molecular weight rRNA in the ribosome of all domains of life [1, 2, 6, 19, 35, 36]. Most of these contacts are within the functionally important helices 89 (the helix which is adjacent to the peptidyl transferase center) and 38 (A-site finger) of the 23S rRNA (Fig. 2c). According to crystallographic and biochemical data, the function of the ribosome is accompanied by conformational changes in the structure of its components [2-9, 37-42]. In particular, such changes were found in helices 38 and 89 of 23S rRNA upon interaction of the bacterial ribosome with translation factors. Therefore, taking into account our data, we suggest that rRNA conformational rearrangements in the translating ribosome of the ΔL25 strain weaken the intermolecular contacts of protein L16 that, in turn, leads to release of this protein from the ribosome.
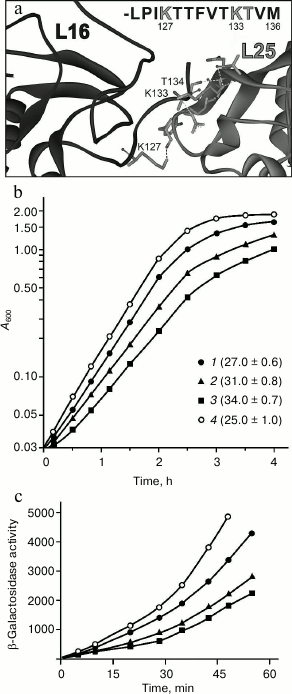
Effect of mutations in the C-terminal region of protein L16 on its retention in the ribosome. The data give rise to a reasonable question: does a rather small area contact between proteins L25 and L16 contribute significantly to the retention of the latter in the ribosome? In this section, we attempt to answer this question. It was decided to check how changes in the contact region of these proteins would affect the formation and function of the ribosome in vivo. Within the E. coli ribosome, polar atoms of the main chain of one of the β-sheets of protein L25 and atoms of the side groups of K127, K133, and T134 of the C-terminal tail of protein L16 can be involved in the formation of H-bonds between the indicated proteins [1] (Fig. 3a). In this situation, changes in the indicated region of protein L25 could affect the formation of its tertiary structure. At the same time, mutations in the flexible tail of L16 should not affect its spatial structure. Therefore, changes were introduced into protein L16. To exclude at once several intermolecular H-bonds and most efficiently hinder the approach of the L16 C-terminal region to protein L25, we decided to replace K127 and/or K133 in protein L16 by a massive hydrophobic residue of leucine (Fig. 3a). The indicated lysine residues were also replaced by alanine as an additional control. Using the “recombineering” approach, the wild-type gene of protein L16 (rplP) was replaced by its mutant form. It turned out that the single mutation K127A in protein L16 has practically no effect on the growth characteristic of the cells (Fig. 3b). At the same time, the single mutation K133L or double mutation K127L/K133L in protein L16 results in a noticeable (25 or 35%, respectively) slowing of cell growth (Fig. 3b). The changes in the cell growth agree well with the data on functional activity of their ribosomes in vivo (Fig. 3c). These data suggest that the changes introduced into protein L16 are reflected directly on the activity of the translational machinery of the bacterial cell.
Fig. 3. Effect of mutations in protein L16 on E. coli cell growth and properties of their ribosomes. a) Contact area of proteins L16 and L25 in the E. coli ribosome (PDB code 2AW4). At the top of the figure, the amino acid sequence of the C-terminal region of protein L16 is presented, in which residues that form hydrogen bonds with protein L25 are marked in gray. b) Growth of cells containing protein L16 with mutations: K127A (1), K133L (2), or K127L/K133L (3). The strain bearing wild-type protein L16 (4) was used as a control. Doubling time (in minutes) of a given strain is indicated on the right from the symbol in parentheses. c) Functional activity of ribosomes of the strains in vivo (designations as in panel (b)). Activity of β-galactosidase is presented in Miller units.
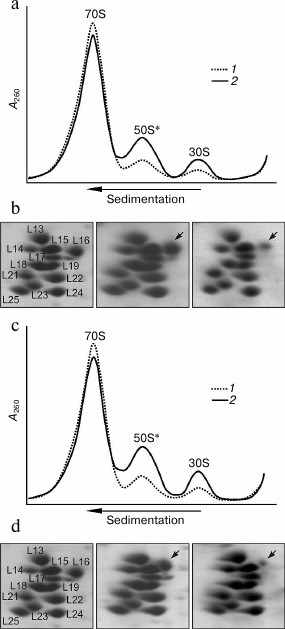
Taking into account these results, the properties of the ribosomes of two mutant strains containing ribosomal protein L16 with replacement K133L or K127L/K133L were analyzed (Fig. 4). It is seen that in contrast to the preparation of the ribosomes from the control strain in which the amount of dissociated subunits is usually less than 10%, in the analogous preparations of both mutant strains the fraction of dissociated subunits is noticeably more (~20-25%) (Fig. 4, a and c). At the same time, the amount of dissociated subunits that can be already observed in the cytoplasmic fraction of the mutant strains (Fig. 4, a and c) changes slightly after the standard purification of the ribosomes or upon using high concentration of Mg2+ (20 mM) in the solution during their isolation (see details in “Materials and Methods”). These results indicate that, in contrast to the ribosomes from the ΔL25 strain, these mutant ribosomes contain a fraction of weakly associating subunits even before their purification. Besides that, the double mutation in protein L16 leads to a higher effect than the single mutation (Fig. 4, a and c). We analyzed the protein composition of the 50S subunits from the fractions of the 70S ribosomes and the dissociated subunits. As seen in Fig. 4, b and d (middle panel), in the 70S fraction the large ribosomal subunits contain proteins L16 and L25 in about equimolar amount. At the same time, when comparing the electrophoregrams of the control and the mutant samples (left and middle panels) it is noticeable that the changes introduced into protein L16 lead to a corresponding change in its mobility. These findings suggest that both studied mutant forms of protein L16 could be effectively incorporated into the ribosome in vivo. Moreover, these mutant forms of protein L16 are retained tightly in most of the purified and dissociated into subunits ribosomes. At the same time, as seen in the right panels of Fig. 4, b and d, the fraction of defective 50S subunits from both mutant strains has a significant lack of protein L16. Furthermore, the effect of the double mutation in protein L16 is considerably more than that of the single mutation.
Fig. 4. Effect of mutations in C-terminal region of protein L16 on properties of E. coli ribosomes. a) Sedimentation profiles of the ribosomes from crude cell extract (S30) of wild-type strain (1) and the strain containing protein L16 with mutation K133L (2). b) Analysis of protein composition of 50S subunits from ribosomes of wild-type strain (left panel), active subunits from 70S fraction (middle panel) and defective (50S*) fraction (right panel) of ribosomes from mutant (K133L) strain (corresponding fragments of 2D-electrophoregrams are presented). The arrow marks the position of the mutant protein L16. c) Sedimentation profiles of ribosomes from S30 of the wild-type strain (1) and the strain containing protein L16 with mutation K127L/K133L (2). d) Analysis of protein composition of 50S subunits from ribosomes of the wild-type strain (left panel), active subunits from 70S fraction (middle panel), and defective (50S*) fraction (right panel) of ribosomes from the mutant (K127L/K133L) strain. The arrow marks the position of the mutant protein L16.
Thus, the mutations in the C-terminal region of protein L16 as well as the absence of protein L25 in the bacterial cell lead to appearance in the ribosomes of a fraction of defective 50S subunits lacking protein L16. These results indicate that the contact between proteins L25 and L16 is important for the retention of the latter in the ribosome in vivo. At the same time, as seen from the presented results, the amount of the defective 50S subunits detected in the ΔL25 ribosomes is significantly more than in the ribosomes containing mutant protein L16. This difference might be explained by the by the size of the changes introduced into the ribosome: in one case, point changes in one of the proteins, and, in another case, deletion of the whole protein. In the absence of protein L25, the corresponding region of the ribosome could become more accessible to solvent molecules that, in turn, could lead to destabilization of the intermolecular contacts of protein L16.
It was assumed earlier that the protein of the L25 family appeared in the bacterial ribosome for additional stabilization of a functionally important region including the 5S rRNA, helix 38 of the 23S rRNA, and protein L16 (Fig. 2c) [13]. It is known from structural data that helix 38 of high molecular weight rRNA and E-loop of the 5S rRNA form an extensive evolutionarily conservative contact in the ribosome of all domains of life [1, 2, 19, 35, 36]. In archaeal and eukaryotic ribosomes, this RNA–RNA contact is stabilized by three proteins – L10e (L16 homolog), L21e (a homolog in bacteria has not been found), and L30, which interact simultaneously with structural elements of both rRNAs [35, 36]. Bacterial homologs of two of these proteins are smaller and form tight contacts only with one of the ribosomal RNAs [1, 2, 11, 13, 19]. However, the bacterial ribosome acquired the protein of the L25 family, which interacts with the E-loop of the 5S rRNA and the C-terminal region of protein L16 [13]. Moreover, the contact area of protein L16 is significantly greater with multi-domain members of the L25 family (e.g. Thermus thermophilus and Deinococcus radiodurans) than with the single-domain protein L25 of E. coli [1, 2, 19]. In addition, the multi-domain protein of the L25 family forms a direct contact with helix 38 of the 23S rRNA. Thus, based on the results of the structural studies, the protein of the L25 family should make a significant contribution to formation and stability of the functionally important structural region of the bacterial ribosome mentioned above. The results of this work confirm this assumption.
The authors are grateful to A. P. Korepanov for providing the ΔL25 strain of E. coli, and to S. V. Nikonov and O. S. Nikonov for fruitful discussion of the results.
This work was financially supported by the Russian Foundation for Basic Research (project No. 13-04-00587) and the Program “Molecular and Cell Biology” of the Russian Academy of Sciences Presidium.
REFERENCES
1.Schuwirth, B. S., Borovinskaya, M. A., Hau, C. W.,
Zhang, W., Vila-Sanjurjo, A., Holton, J. M., and Cate, J. H. D. (2005)
Structures of the bacterial ribosome at 3.5 Å resolution,
Science, 310, 827-834.
2.Selmer, M., Dunham, C. M., Murphy, F. V., IVth,
Weixlbaumer, A., Petry, S., Kelley, A. C., Weir, J. R., and
Ramakrishnan, V. (2006) Structure of the 70S ribosome complexed with
mRNA and tRNA, Science, 313, 1935-1942.
3.Korostelev, A., Trakhanov, S., Laurberg, M., and
Noller, H. F. (2006) Crystal structure of a 70S ribosome–tRNA
complex reveals functional interactions and rearrangements,
Cell, 126, 1065-1077.
4.Yusupova, G., Jenner, L., Rees, B., Moras, D., and
Yusupov, M. (2006) Structural basis for messenger RNA movement on the
ribosome, Nature, 444, 391-394.
5.Laurberg, M., Asahara, H., Korostelev, A., Zhu, J.,
Trakhanov, S., and Noller, H. F. (2008) Structural basis for
translation on the 70S ribosome, Nature, 454,
852-857.
6.Voorhees, R. M., Weixlbaumer, A., Loakes, D.,
Kelley, A. C., and Ramakrishnan, V. (2009) Insights into substrate
stabilization from snapshots of the peptidyl transferase center of the
intact 70S ribosome, Nat. Struct. Mol. Biol., 16,
528-533.
7.Schmeing, T. M., Voorhees, R. M., Kelley, A. C.,
Gao, Y. G., Murphy, F. V., IVth, Weir, J. R., and Ramakrishnan, V.
(2009) The crystal structure of the ribosome bound to EF-Tu and
aminoacyl-tRNA, Science, 326, 688-694.
8.Gao, Y. G., Selmer, M., Dunham, C. M., Weixlbaumer,
A., Kelley, A. C., and Ramakrishnan, V. (2009) The structure of the
ribosome with elongation factor G trapped in the posttranslocation
state, Science, 326, 694-699.
9.Dunkle, J. A., Wang, L., Feldman, M. B., Pulk, A.,
Chen, V. B., Kapral, G. J., Noeske, J., Richardson, J. S., Blanchard,
S. C., and Cate, J. H. D. (2011) Structures of the bacterial ribosome
in classical and hybrid states of tRNA binding, Science,
332, 981-984.
10.Lecompte, O., Ripp, R., Thierry, J. C., Moras,
D., and Poch, O. (2002) Comparative analysis of ribosomal proteins in
complete genomes: an example of reductive evolution at the domain
scale, Nucleic Acids Res., 30, 5382-5390.
11.Benson, D. A., Karsch-Mizrachi, I., Lipman, D.
J., Ostell, J., and Wheeler, D. L. (2008) GenBank, Nucleic Acids
Res., 36, 25-30.
12.Ban, N., Beckmann, R., Cate, J. H., Dinman, J.
D., Dragon, F., Ellis, S. R., Lafontaine, D. L., Lindahl, L., Liljas,
A., Lipton, J. M., McAlear, M. A., Moore, P. B., Noller, H. F., Ortega,
J., Panse, V. G., Ramakrishnan, V., Spahn, C. M., Steitz, T. A.,
Tchorzewski, M., Tollervey, D., Warren, A. J., Williamson, J. R.,
Wilson, D., Yonath, A., and Yusupov, M. (2014) A new system for naming
ribosomal proteins, Curr. Opin. Stuct. Biol., 24,
1-5.
13.Gongadze, G. M., Korepanov, A. P., Korobeinikova,
A. V., and Garber, M. B. (2008) Bacterial 5S rRNA-binding proteins of
the CTC family, Biochemistry (Moscow), 73,
1405-1417.
14.Hecker, M., and Volker, U. (1990) General stress
proteins in Bacillus subtilis, FEMS Microbiol. Ecol.,
74, 197-214.
15.Schmalisch, M., Langbein, I., and Stulke, J.
(2002) The general stress protein CTC of Bacillus subtilis is a
ribosomal protein, J. Mol. Microbiol. Biotechnol., 4,
495-501.
16.Baba, T., Ara, T., Hasegawa, M., Takai, Y.,
Okumura, Y., Baba, M., Datsenko, K. A., Tomita, M., Wanner, B. L., and
Mori, H. (2006) Construction of Escherichia coli K-12 in-frame,
single-gene knockout mutants: the Keio collection, Mol. Syst.
Biol., 2, 1-11.
17.Korepanov, A. P., Gongadze, G. M., Garber, M. B.,
Court, D. L., and Bubunenko, M. G. (2007) Importance of the 5S
rRNA-binding ribosomal proteins for cell viability and translation in
Escherichia coli, J. Mol. Biol., 366,
1199-1208.
18.Lotti, M., Noah, M., Stoffler-Meilicke, M., and
Stoffler, G. (1989) Localization of L4, L5, L20 and L25 on the
ribosomal surface by immune-electron microscopy, Mol. Gen.
Genet., 216, 245-253.
19.Harms, J., Schluenzen, F., Zarivach, R., Bashan,
A., Gat, S., Agmon, I., Bartels, H., Franceschi, F., and Yonath, A.
(2001) High resolution structure of the large ribosomal subunit from a
mesophilic eubacterium, Cell, 107, 679-688.
20.Kazemie, M. (1976) Binding of aminoacyl-tRNA to
reconstituted subparticles of Escherichia coli large ribosomal
subunits, Eur. J. Biochem., 67, 373-378.
21.Teraoka, H., and Nierhaus, K. H. (1978) Protein
L16 induces a conformational change when incorporated into a
L16-deficient core derived from Escherichia coli ribosomes,
FEBS Lett., 88, 223-227.
22.Tate, W. P., Schulze, H., and Nierhaus, K. H.
(1983) The importance of the Escherichia coli ribosomal protein
L16 for the reconstitution of the peptidyl-tRNA hydrolysis activity of
peptide chain termination, J. Biol. Chem., 258,
12810-12815.
23.Anikaev, A. Y., Korepanov, A. P., Korobeinikova,
A. V., Kljashtorny, V. G., Piendl, W., Nikonov, S. V., Garber, M. B.,
and Gongadze, G. M. (2014) Mutant forms of Escherichia coli
protein L25 unable to bind to 5S rRNA are incorporated efficiently into
the ribosome in vivo, Biochemistry (Moscow), 79,
826-835.
24.Miller, J. H. (1972) Experiments in Molecular
Genetics, Cold Spring Harbor Laboratory Press, N. Y.
25.Yu, D., Ellis, H. M., Lee, E. C., Jenkins, N. A.,
Copeland, N. G., and Court, D. L. (2000) An efficient recombination
system for chromosome engineering in Escherichia coli, Proc.
Natl. Acad. Sci. USA, 97, 5978-5983.
26.Thomason, L. C., Bubunenko, M., Costantino, N.,
Wilson, H., Oppenheim, A., Datta, S., and Court, D. L. (2007)
Recombineering: genetic engineering in bacteria using homologous
recombination, Curr. Protoc. Mol. Biol., doi:
10.1002/0471142727.mb0116s78.
27.Erbe, R. W., Nau, M. M., and Leder, P. (1969)
Translation and translocation of defined RNA messengers, J. Mol.
Biol., 38, 441-460.
28.Staehelin, T., Maglott, D. M., and Monro, R. E.
(1969) On the catalytic center of peptidyl transfer: a part of the 50S
ribosome structure, Cold Spring Harb. Symp. Quant. Biol.,
34, 39-48.
29.Schlessinger, D., Mangiarotti, G., and Apirion,
D. (1967) The formation and stabilization of 30S and 50S ribosome
couples in Escherichia coli, Proc. Natl. Acad. Sci. USA,
58, 1782-1789.
30.Kohler, R. A., Ron, E. Z., and Davis, B. D.
(1968) Significance of the free 70S ribosomes in Escherichia
coli extracts, J. Mol. Biol., 36, 71-82.
31.Kolb, V. A., Makeyev, E. V., and Spirin, A. S.
(2000) Co-translational folding of an eukaryotic multidomain protein in
a prokaryotic translation system, J. Biol. Chem., 275,
16597-16601.
32.Madjar, J., Michel, S., Cozzone, A., and Reboud,
J. (1979) A method to identify individual proteins in four different
two-dimensional electrophoresis systems: application to E. coli
ribosomal proteins, Anal. Biochem., 92, 174-182.
33.Algranati, I. D., Gonzalez, N. S., and Bade, E.
G. (1969) Physiological role of 70S ribosome in bacteria, Proc.
Natl. Acad. Sci. USA, 62, 574-580.
34.Cannon, M. (1967) The ribosomal binding site for
peptidyl-transfer-ribonucleic acid, Biochem. J., 104,
934-946.
35.Klein, D. J., Moore, P. B., and Steitz, T. A.
(2004) The roles of ribosomal proteins in the structure assembly, and
evolution of the large ribosomal subunit, J. Mol. Biol.,
340, 141-177.
36.Ben-Shem, A., Garreau de Loubresse, N., Melnikov,
S., Jenner, L., Yusupova, G., and Yusupov, M. (2011) The structure of
the eukaryotic ribosome at 3.0 Å resolution, Science,
334, 1524-1529.
37.Wilson, K. S., Ito, K., Noller, H. F., and
Nakamura, Y. (2000) Functional sites of interaction between release
factor RF1 and the ribosome, Nat. Struct. Biol., 7,
866-870.
38.La Teana, A., Gualerzi, C. O., and Dahlberg, A.
E. (2001) Initiation factor IF2 binds to the α-sarcin loop and
helix 89 of Escherichia coli 23S ribosomal RNA, RNA,
7, 1173-1179.
39.Scarlett, D. J., McCaughan, K. K., Wilson, D. N.,
and Tate, W. P. (2003) Mapping functionally important motifs SPF and
GGQ of the decoding release factor RF2 to the Escherichia coli
ribosome by hydroxyl radical footprinting, J. Biol. Chem.,
278, 15095-15104.
40.Wilson, K. S., and Nechifor, R. (2004)
Interactions of translation factor EF-G with the bacterial ribosome
before and after mRNA translocation, J. Mol. Biol., 337,
15-30.
41.Sergiev, P. V., Bogdanov, A. A., and Dontsova, O.
A. (2005) How can elongation factors EF-G and EF-Tu discriminate the
functional state of the ribosome using the same binding site? FEBS
Lett., 579, 5439-5442.
42.Kiparisov, S. V., Sergiev, P. V., Bogdanov, A.
A., and Dontsova, O. A. (2006) The structural changes in the ribosome
during the elongation cycle, Mol. Biol. (Moscow), 40,
755-768.
Supplementary Tables S1 and S2 (PDF)