REVIEW: Quantitative Aspects of RNA Silencing in Metazoans
A. M. Sergeeva1,2, N. Pinzón Restrepo1, and H. Seitz1*
1IGH du CNRS (UPR 1142), 141 rue de la Cardonille, 34396 Montpellier, France; E-mail: herve.seitz@igh.cnrs.fr2Current address: Institute of Molecular Genetics, Russian Academy of Sciences, pl. Kurchatova 2, 123182 Moscow, Russia; E-mail: asergeeva@img.ras.ru
* To whom correspondence should be addressed.
Received January 21, 2013
Small regulatory RNAs (microRNAs, siRNAs, and piRNAs) exhibit several unique features that clearly distinguish them from other known gene regulators. Their genomic organization, mode of action, and proposed biological functions raise specific questions. In this review, we focus on the quantitative aspect of small regulatory RNA biology. The original nature of these small RNAs accelerated the development of novel detection techniques and improved statistical methods and promoted new concepts that may unexpectedly generalize to other gene regulators. Quantification of natural phenomena is at the core of scientific practice, and the unique challenges raised by small regulatory RNAs have prompted many creative innovations by the scientific community.
KEY WORDS: small RNAs, RNA silencing, RNA interferenceDOI: 10.1134/S0006297913060072
Three classes of small regulatory RNAs have been described in metazoans: microRNAs (miRNAs), small interfering RNAs (siRNAs) and Piwi-interacting RNAs (piRNAs). All these small RNAs interact with effector proteins (members of the Ago protein subfamily for miRNAs and siRNAs, members of the Piwi protein subfamily for piRNAs; these two protein subfamilies have a very similar domain architecture, but they can be easily discriminated based on their sequence). Guiding these effector proteins to specific targets, they generally act as repressors of gene expression (see [1] for an extensive review): miRNAs in metazoans typically trigger degradation and translation inhibition of target mRNAs; siRNAs guide the endonucleolytic cleavage of transcripts from repeated and non-repeated sequences (a process called “RNA interference”, RNAi); and piRNAs are necessary for gametogenesis (in particular, transposons are often derepressed in the germ line of mutants of the piRNA pathway).
These classes not only differ by their function, but also by their biogenesis: while most miRNAs are matured by two endonucleases of the RNase III family (Drosha and Dicer) from stem-loop-folded precursors, siRNAs are generated by Dicer-catalyzed cleavage of long double-stranded RNAs (dsRNA), and piRNAs seem to derive from single-stranded precursors matured in an RNase III-independent manner [1].
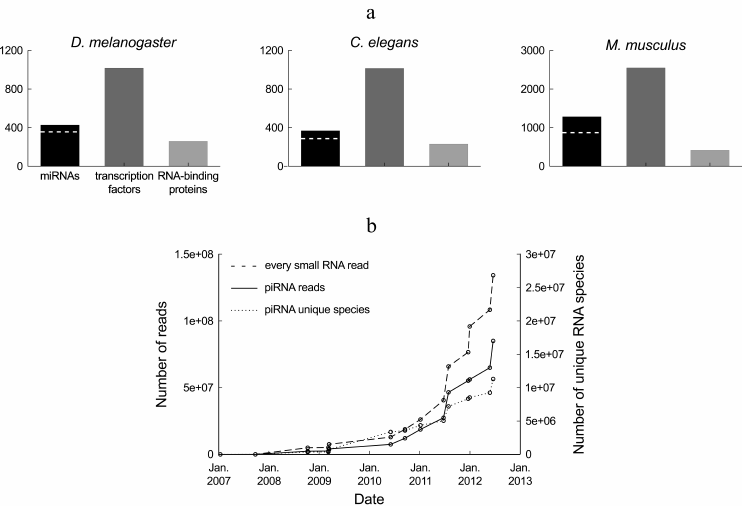
In terms of diversity, small regulatory RNAs constitute a major class of gene regulators. miRNAs are only half as diverse as transcription factors and more diverse than RNA-binding proteins (Fig. 1a), with a few hundreds of miRNA genes in a typical metazoan genome. A unified database has been created to store miRNA sequences, as well as various information regarding their genomic organization, expression level, and predicted targets: that database, called miRBase (http://microrna.sanger.ac.uk/sequences/), is updated approximately twice a year with novel miRNAs [2]. piRNAs and endogenous siRNAs are incomparably more diverse, with millions of reported piRNA species and tens of thousands of reported siRNA species in the analyzed model organisms.
Not only small regulatory RNAs are very diverse in sequence, but they are also poorly specific: the main determinant of miRNA and siRNA targets is the “seed” (nt 2-7 of the small RNA, counted from the 5′ end), which has to be perfectly complementary to a target RNA for the target to be repressed, and affected genes sometimes even exhibit seed match imperfections [3-7]. Such a short binding site is very frequent in transcriptomes – even when only conserved miRNA seed matches are considered, more than 60% of human coding genes are predicted to be targeted by miRNAs [8].
miRNA and siRNA targets are repressed by different mechanisms in animals, depending on the extent of the complementarity between the small RNA and its target, as well as on the identity of the effector Ago protein bound to the small RNA: when the Ago protein possesses an endonucleolytic site and when the two RNAs are perfectly paired in the center of the duplex, the target is usually cleaved [9-12]. Additional conditions may apply, as has been shown in plants (reviewed in [13]).
Fig. 1. Diversity of small regulatory RNAs in metazoans. a) Number of known miRNAs and miRNA families (according to miRBase, release 19 [2]), transcription factor genes (according to the DBD database, release 2.0 [14]) and RNA-binding proteins (according to the RBPDP database, release 1.3 [15]) in three model organisms. The number of miRNA families is indicated by a dashed white line. Members of a miRNA family share the same seed, and hence they can target the same genes (actual redundancy between family members depends on the similarity of their expression pattern and on the contribution of non-seed sequence to target silencing). The redundancy rate between transcription factors or between RNA-binding proteins is unknown. b) Number of Drosophila ovarian piRNA reads and number of unique Drosophila ovarian piRNA sequences deposited at the NCBI GEO database from 2007 to the Autumn 2012. This chart summarizes sequence counts from wild-type or heterozygous Drosophila ovaries, or from a cultured cell line derived from the ovary soma (untreated or treated with control, extragenomic siRNAs): GEO data series GSE9138 [16], GSE6734 [17], GSE13081 [18], GSE15186 [19], GSE15137 [20], GSE24108 [21], GSE22067 [22], GSE26407 [23], GSE30088 [24], GSE30955, GSE34728, GSE34506 [25], GSE38728 [26] and GSE38089 [27]. The dashed line indicates the total number of deposited small RNA reads from Drosophila ovarian samples. For the datasets where piRNAs were not purified by immunoprecipitation of Piwi proteins, the pool of piRNAs was approximated by the population of 23-29-mers in the small RNA library.
In this review, we will focus on a few questions related to the quantitative analysis of the properties of small RNAs. Measuring natural phenomena is at the core of scientific practice, and several aspects of small RNA biology (their genomic organization, their mode of action, and their proposed biological roles) raise original and interesting questions on quantification methods, on the significance of their numerical outputs, and on the implementation of traditional statistical methods.
DEFINITION OF SMALL RNA REPERTOIRES DEPENDS ON DETECTION
SENSITIVITY
miRNA vs. miRNA*: an artifact of low coverage? Most miRNAs are generated by the cleavage of a pre-miRNA hairpin by the Dicer endonuclease. That reaction liberates a short RNA duplex, where the miRNA is paired to another small RNA, named the “miRNA*”. The duplex is then loaded on a protein of the Ago subfamily and is subsequently unwound [28-32]. One strand remains stably associated to the Ago protein (that strand is called the “guide strand”), while the other one (“passenger strand”) is discarded and rapidly degraded. Loading of a miRNA/miRNA* duplex on an Ago protein is usually asymmetric, with the strand whose 5′ end is less stably paired being preferentially selected as the guide strand [33, 34]: that small RNA is then strongly stabilized relatively to the other strand.
That feature of miRNA biogenesis explains why one strand (the miRNA) is usually more abundant than the other strand (the miRNA*) at steady state. When the variety of miRNAs was first described, the miRNA* usually escaped detection by cloning or Northern blot [35-37], and its existence was only proven on a poorly asymmetric miRNA/miRNA* duplex (worm miR-56 [36]). With the advent of deep sequencing, which is sensitive enough to capture both miRNA and miRNA* sequences in samples where they are reasonably expressed [38], the distinction between miRNAs and miRNA*s turned from qualitative (one strand was observed and not the other) to quantitative (the miRNA is the more abundant strand, the miRNA* is the less abundant strand).
Investigation of a large variety of biological samples then revealed that miRNA/miRNA* ratios could vary greatly across tissues and species, sometimes to the point that the miRNA* can be more abundant than its miRNA in some samples [39-42]. Together with the observation that miRNA* sequence is often under selective pressure [43] and that miRNA* can be selectively loaded on a specific Ago effector [44-46], this notion led to a fundamental redefinition of miRNAs: there is probably no qualitative difference between miRNAs and miRNA*s, and both strands should probably be considered functional – the latest releases of miRBase no longer distinguish miRNAs from miRNA*s.
Definition of the piRNA and siRNA pool is sequencing depth-dependent. piRNAs and siRNAs are much more diverse in sequence than miRNAs. But each individual piRNA and siRNA tends to be poorly expressed: in a deep-sequencing library containing millions of piRNA reads (or thousands of siRNA reads), most sequences are usually read only once; replicating the library will, most probably, not detect the same set of low-abundance RNA species. After several years of investigation, and tens of millions of sequenced small RNAs, the complete pool of Drosophila ovary piRNAs is certainly not yet known: the number of detected piRNA species does not seem to be reaching a plateau (Fig. 1b).
While piRNAs are mostly expressed in gonads, siRNAs are detected in various organs [47-52]. They originate from specific loci, whose transcripts can form monomolecular or bimolecular double-stranded RNA (dsRNA), and it is believed that they are generated by the cleavage of these double-stranded RNAs by Dicer. The resulting pools of siRNAs are highly diverse, covering the sequence of the double-stranded precursor.
Consequently, recognition of the existence of most piRNAs and siRNAs depends heavily on sequencing depth. Attempts to inventory piRNAs [53] are probably of little use currently, as the list of existing piRNAs is not expected to stabilize as fast as the list of existing miRNAs.
The overwhelming diversity of piRNAs and siRNAs exceeds the number of known genes (including transposable elements) by several orders of magnitude. This situation is probably unique in biology, with regulatory molecules being much more diverse than their potential targets. In the case of siRNAs, it is clear that genes can be repressed by endogenous siRNAs and every individual siRNA probably contributes to the repression of complementary RNAs [43, 52]. The actual mode of action of piRNAs is much less obvious. One possibility is that piRNAs really only target transposons and other genes, and numerous distinct piRNAs interact with each target. Another possibility is that piRNAs do not target genes, but genomic sequences per se; from this point of view, each individual piRNA would target its own genomic locus (as well as homologous sequences if it can work in trans). Such a function could explain why the location of piRNA clusters tends to be conserved while their sequence is poorly conserved [54, 55].
GENOMIC ANNOTATION OF SMALL RNA GENES REVEALS GREAT HETEROGENEITY
IN THEIR ABUNDANCE AND DIVERSITY
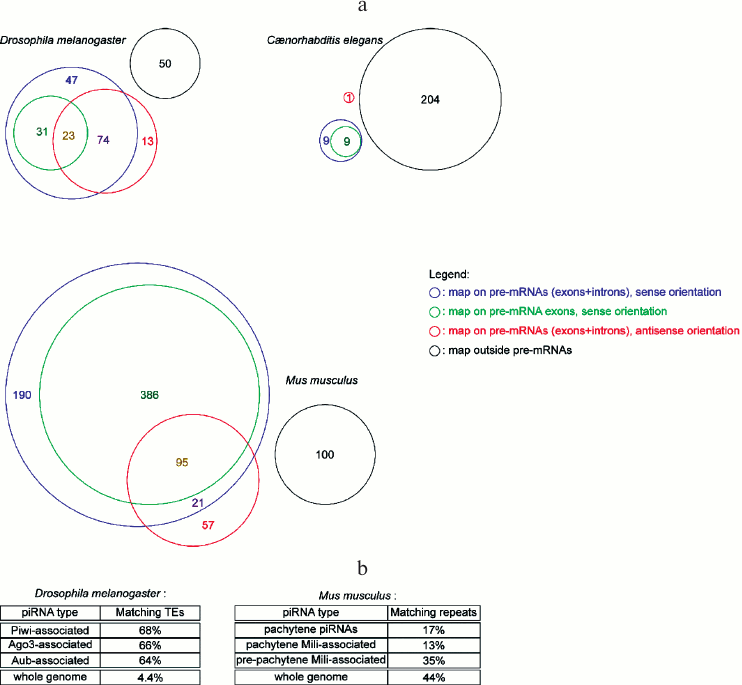
miRNA abundance cannot be predicted from their genomic organization. Many miRNA genes in metazoans are hosted in coding genes (either in their introns or in their exons), and most frequently in the sense orientation (some miRNA genes are located in the overlap of a sense/anti-sense gene pair, hence they map on pre-mRNAs both in the sense and the anti-sense orientation) (Fig. 2a; see color insert). Pre-miRNA hairpins falling outside annotated pre-mRNAs are usually named “intergenic”, even though, strictly speaking, the pre-miRNA is located in a gene (the miRNA gene), hence it is not intergenic – “intergenic” tends to be used as a synonym of “inter-coding genes”. While it was generally assumed that intronic miRNAs are co-transcribed with their host gene, some exceptions exist, where the intronic miRNA has its own promoter, located in the intron of the host gene [56].
Even when a miRNA is co-transcribed with a host gene, their expression levels do not necessarily correlate: in the past years, several mechanisms of post-transcriptional regulation of miRNA expression have been described. The Lin28 protein appears to regulate both Drosha and Dicer-catalyzed processing of specific miRNA precursors [57-60]. The exact mechanism was initially disputed; though it is formally possible that Lin28 also has alternative activities, at least it is clear that mammalian Lin28 recruits the poly(U) polymerase Tut4 (and its close homolog, Tut7) on the pre-let-7 pre-miRNA, and poly-uridylation of the pre-miRNA inhibits its cleavage by Dicer [61-63]. Polymerase Pup-2 appears to exert a similar Lin28-guided repressive action on let-7 biogenesis in C. elegans [64]. Reciprocally, Tut4 (as well as other poly(U) polymerases) can also mono-uridylate pre-miRNAs when Lin28 is absent: addition of a single uridine on the 3′ end of a pre-miRNA can facilitate its processing by Dicer in specific circumstances, hence promoting miRNA maturation [65].
Another post-transcriptional phenomenon can distort the correlation between miRNA and host gene mRNA abundances: regulated stabilization or destabilization of mature miRNAs. Addition of adenosines at the 3′ end of miR-122 by the mammalian poly(A) polymerase Gld-2 stabilizes that miRNA, while other miRNAs appear to be unaffected [66]. Other miRNAs could also be adenylated by that enzyme, though their abundance does not seem to be sensitive to their adenylation status – rather, 3′ adenylation may affect their specific activity [67]. Reciprocally in Drosophila, the addition of uridines and adenosines at the 3′ end of miRNAs triggers their degradation by a 3′ to 5′ exonuclease. These miRNA “tailing” and “trimming” reactions are regulated by a combination of factors: they only affect the small RNAs loaded on the Ago2 effector protein, and only when they encounter a perfectly (or almost perfectly) matched RNA target [68]. Another regulated miRNA-degrading activity has been reported in worms, where exonuclease Xrn-2-catalyzed miRNA turnover is inhibited by the presence of target mRNAs for the miRNA [69]. Finally, human polynucleotide phosphorylase (an exonuclease) is also able to degrade specifically some miRNAs in cultured cells, suggesting a function in the regulation of miRNA abundance [70].
Genome mapping statistics of piRNAs and siRNAs. piRNAs are often presented as transposon-matching small RNAs; while there is indeed enrichment for transposable element sequences in piRNA populations, a large subset of piRNAs map on non-repeated sequences (Fig. 2b; see color insert). In worms it is even more the case, where a single transposon (Tc3) appears to be regulated by piRNAs [71, 72].
Transposons are de-repressed in mutants for the piRNA pathway in fly [73, 74] and in mouse [75-77]. Yet no transposon de-repression was observed in zebrafish Danio rerio; it has been proposed that the loss of germ cells in the analyzed mutant may have complicated the analysis [78]. The fact that piRNAs frequently match transposon sequences, together with their apparent role in transposon repression in fly and in mouse, may suggest that their main function is the silencing of these mutagenic, mobile elements. Several observations also suggested that piRNAs could repress non-transposable genes [79, 80], though it remains to be established why these effects are specific to just a few target genes. The diversity of detected piRNAs, as well as the apparent tolerance of these phenomena to mismatches between piRNAs and their targets, would rather predict that most genes would be affected.
Endogenous siRNAs also frequently match transposons, as well as other repeated sequences (such as satellites and other tandem repeats) and non-repeated genes [47-50, 52, 81]. While piRNAs are mostly restricted to the gonads, siRNAs are detected in non-gonadal tissues [47, 48, 51, 52]. If transposon silencing were the only biological role of piRNAs, piRNAs could appear dispensable (piRNA-repressed transposons could be silenced by siRNAs, and the whole piRNA pathway would be useless). Several recent findings shed a new light on the possible reasons for the coexistence of the two pathways: in worms, both systems cooperate, with piRNAs triggering the generation of siRNAs specific for the piRNA target [82], then promoting a piRNA-independent, nuclear siRNA-dependent repression [83]. In flies, the situation appears to be symmetrical: after introduction in the genome, the Penelope transposon is first silenced by siRNAs. RNAi-based repression appears to be incomplete, and a transposon copy ends up landing in a piRNA cluster, thus promoting a piRNA-based, more efficient repression [84].
Fig. 2. Genomic distribution of small RNA genes. a) Diversity of the genomic distribution of miRNAs in metazoans. Numbers in the colored sectors indicate the number of pre-miRNAs falling in each category (pre-miRNA genomic coordinates are from miRBase release 19, dated August 2012; genomic coordinates for pre-mRNAs and their exons are from the UCSC Genome Browser, using genome assemblies BDGP R5 for D. melanogaster, WS220 for C. elegans, and GRCm38 for M. musculus). b) Percentage of transposable element (TE)-matching reads in deep-sequencing piRNA libraries. Drosophila melanogaster data was extracted from the NCBI GEO datasets GSM154620, GSM154621, and GSM154622 [17]. Mus musculus statistics are from [85] (for pachytene piRNAs), [86] (for pachytene Mili-associated piRNAs), and [75] (for pre-pachytene Mili-associated piRNAs). “Whole genome” indicates the percentage of genomic sequence covered by transposable elements or repeats.
ASSESSING SMALL RNA QUANTIFICATION METHODS
Several methods are currently used for the quantification of small RNAs. While amplification-free methods (such as Northern blotting and microarrays) tend to be less sensitive than PCR-based assays (such as qRT-PCR and deep-sequencing), they are also less prone to PCR biases that can affect the reliability of measured small RNA abundances.
Northern blotting: an amplification-free procedure. Northern blot analysis is a poorly sensitive and low-throughput technique, but with a very precise outcome. This technique involves size-separation of RNA molecules followed by transfer and cross-linking to a nylon membrane. RNAs of interest are then detected by hybridization with labeled complementary nucleic acid probes (when the probe is in large excess, the intensity of the radioactive signal is proportional to the amount of RNA on the membrane). When calibrated with synthetic RNA oligonucleotides with the same sequence as the detected small RNA, Northern blotting allows a straightforward measurement of RNA abundance.
Northern blot also displays the size of small RNAs at a single nucleotide resolution, allowing the discrimination of precursor and mature species. The standard Northern blot procedure has also been adapted in order to enhance short RNA detection [87, 88]. This adapted procedure takes advantage of the chemical properties of known small regulatory RNAs (which all bear a 5′ monophosphate): cross-linking small RNAs to the membrane through their 5′ phosphate, instead of the usual UV-induced cross-linking all along the small RNA sequence, improves RNA accessibility to the probe, resulting in a better sensitivity.
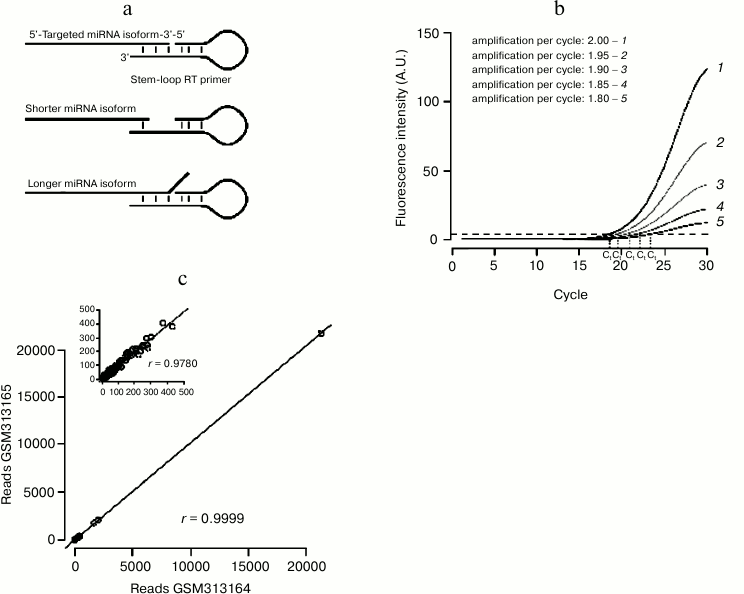
qRT-PCR: a sensitive technique with specific drawbacks. RT-PCR-based assays benefit from the great sensitivity of PCR that can even detect miRNAs from single cell lysate [89]. As miRNAs are too short to accommodate a primer pair, specific protocols had to be designed, where adapter sequences are ligated or polymerized at each end of the small RNA (reviewed in [90]). An alternative, popular method involves a reverse transcription primer whose short interaction with the miRNA is stabilized by a stem-loop structure (Fig. 3a). The double-stranded structure of the stem-loop stabilizes the pairing of the primer to the miRNA, enhancing the stacking interaction between base pairs. This technique is thus very sensitive to heterogeneities on the 3′ end of the miRNA (which are very common in vivo [91]): each primer detects only one 3′ isoform, thus underestimating the total amount of the quantified miRNA.
Another issue can bias qRT-PCR-based measurements: the method is frequently used to calculate the relative amount of the RNA of interest, normalized to a control RNA [92]. That method simply assumes that both RNAs are perfectly amplified by their primer pairs during the analyzed PCR cycles. In fact different primer pairs will have different amplification efficiency; a small difference on the value of amplification efficiency can greatly affect the result of the measurement (Fig. 3b). More accurate methods have been developed, which estimate amplification efficiency from the recorded PCR run (reviewed in [93]).
The extreme sensitivity of RT-PCR may actually also become its main weakness: unintended RNA species may exhibit partial complementarity to the primers and be amplified, resulting in an artifactual qRT-PCR signal. Such unspecific amplification could explain the apparent detection of mammalian-specific miR-134 in Xenopus [94].
Small RNA microarrays. The short size of small RNAs also causes specific challenges for their quantification by microarray that required the development of specific protocols (see [95] for an extensive review). Cross-hybridization is a major issue, as many miRNAs are highly similar to each other; introduction of chemically modified probes (with locked nucleic acid modifications) allowed high stringency hybridization conditions that result in highly specific probe signals.
Microarrays can only detect small RNAs whose probes are present on the array, thus limiting the analysis to known, targeted RNA sequences. That limitation can be overturned by the use of custom-made microarrays, whose probes can tile entire genomic regions (hence detecting every small RNA originating from these loci, provided that post-transcriptional maturation does not affect their sequence): that technique was successfully used to quantify the expression of piRNAs in Drosophila [96].
Deep sequencing: a high-throughput method for small RNA identification and quantification. Deep sequencing cDNA libraries prepared from purified small RNAs (“small RNA-Seq”) allows the detection of novel, unknown RNA sequences. In principle, small RNA-Seq cannot only be used for the identification of small RNA species, but also for their quantification: the number of sequenced cDNA reads is so large (typically ≥ 10 millions on a routine basis nowadays) that many small RNAs can be read hundreds of times each. These numbers are large enough to be representative of the abundance of the small RNA in the analyzed sample.
Technical reproducibility of the small RNA-Seq procedure can be evaluated by the comparison of technical replicates: when assessed, replicate-to-replicate correlation was very high ([97], also see Fig. 3c). These experiments showed that the sequencing reaction itself is highly reproducible.
Fig. 3. Small RNA quantification methods. a) Stem-loop-primed RT is specific for just one 3′ isoform of the assessed miRNA. Heterogeneity on the position of the miRNA 3′ end decreases the affinity of the primer for the miRNA, either because of the shortening of the annealed segment (middle panel) or because of steric hindrance between the miRNA 3′ end and the primer (bottom panel). b) A small error on the estimation of PCR amplification efficiency yields large errors on the estimation of threshold cycle “Ct”. c) NCBI GEO datasets GSM313164 and GSM313165 are two technical replicates of the same small RNA library (a single cDNA library was prepared from ago2 mutant fly heads and sequenced twice on the same instrument [46]). Synthetic spiked-in RNAs were excluded from the analysis. Correlation across replicates was assessed by Pearson’s product moment correlation coefficient (denoted as r). Correlation is very high, even when only the least abundant RNAs are considered (less than 500 reads in each library; cf inset).
PITFALLS IN STATISTICAL TREATMENTS
Identification of affected genes is sensitive to the choice of cutoffs. Classifying affected genes after perturbation of RNA silencing pathways (e.g. after transfecting or inhibiting a miRNA in cultured cells) is not as straightforward as it may seem at first glance. Affected genes are usually identified by the amplitude of their change in expression and the associated p-value: genes displaying a fold-change in gene expression above a chosen cutoff, as well as a p-value below a chosen cutoff, are flagged as “deregulated”. Indeed each criterion alone is not sufficient to describe the behavior of the gene: the fold-change may be large, yet the gene is not significantly affected (with a high p-value; this is the case in particular when replicate values are strongly dispersed); reciprocally, the p-value may be small, yet the fold-change could be modest (hence the gene is hardly affected, even though that change appears to be significant, i.e. not due to technical noise). In sum, the fold-change describes how much the gene is affected, and the p-value measures the confidence one can have in the measured fold-change.
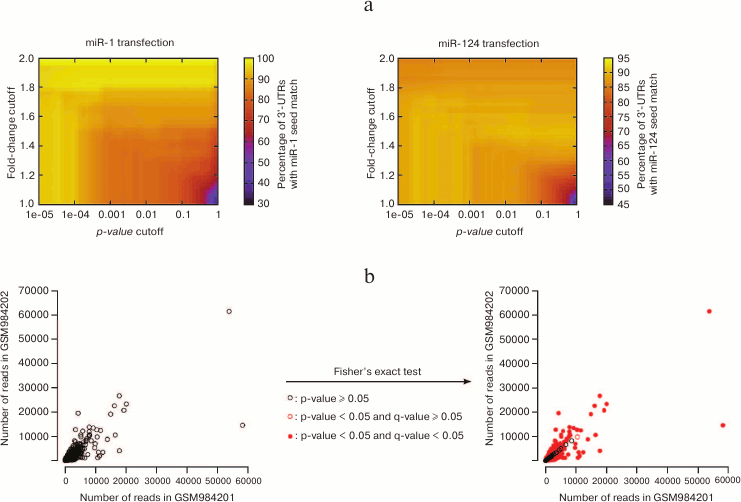
Consequently, the identification of affected genes relies on the choice of these two cutoffs; modifying these cutoffs will re-define the list of “affected genes”, which can affect downstream analyses. This concept is illustrated in Fig. 4a (see color insert), on the data that first established the importance of seed matches in the identification of miRNA targets at a genome-wide scale: using a particular cutoff on the p-value (and no cutoff on the fold-change) Lim et al. [4] noticed that the 3′ UTRs of miRNA-repressed genes are enriched in seed matches for the transfected miRNA. Enrichment is sensitive to the values of the cutoffs, with the most stringent cutoffs (in terms of fold-change as well as in terms of p-value) yielding larger enrichments: this observation confirms that seed match is indeed a good predictor of the affected genes; it also shows that the most strongly affected genes are direct miRNA targets (affected genes that do not bear seed matches are probably under the control of direct miRNA targets, and the perturbation in gene expression is attenuated when it is transmitted from direct to indirect targets, at the analyzed time points).
Comparison of the outcome of a variety of cutoff values is thus required to ascertain that the chosen values are not arbitrary, and that the result of downstream analyses is robust to changes in the cutoff.
Fig. 4. Issues with statistical treatment of high-throughput datasets. a) Lim et al. [4] performed the first high-throughput assessment of the importance of seed matches in miRNA target identification (quantifying mRNA expression in HeLa cells following miR-1 or miR-124 transfection). With a cutoff of 0.001 on the p-value and no cutoff on the fold-change (hence, a cutoff of 1), thousands of genes were affected, and ≈80 to 90% of their 3′ UTRs contained seed matches for the transfected miRNA. These values range from less than 50 to 100% with alternative cutoff values. b) NCBI GEO datasets GSM984201 and GSM984202 are two biological replicates of Drosophila melanogaster first instar larva small RNAs from the modENCODE project. Each point represents an individual small RNA. Their spreading around the diagonal indicates differences in abundance across the two replicates. For most small RNAs, these differences appear significant according to Fisher’s exact test (red points on the right panel), even after correction for multiple hypothesis testing with the Bioconductor qvalue package [98] (filled red points).
Criteria for differential expression in an RNA-Seq experiment. When RNA abundance is quantified by RNA-Seq (be it a Small RNA-Seq experiment, where each read is the full-length sequence of a small RNA; or an mRNA RNA-Seq experiment, where mRNAs are fragmented before sequencing, then re-assembled computationally), the experimental output is a number of reads per gene (hence a discrete, “digital” score, in contrast with the continuous distribution of microarray signals for instance).
The significance of deviations in the distribution of a discrete variable across samples is classically assessed by Fisher’s exact test. That test is very sensitive when the numbers of counts are large, which is the case in an RNA-Seq experiment – consequently, most differences will be flagged “significant” for the most abundant RNAs, even when the compared samples are biological replicates of the same genotype and same experimental conditions, and even when p-values are adjusted for multiple hypothesis testing (Fig. 4b; see color insert).
Hence RNA-Seq is sensitive enough, and Fisher’s exact test is powerful enough, to detect biological variability between individuals (or pools of individuals) of the same genotype, treated the same way. While this sensitivity is certainly a good thing in many cases, it also has its downside: when comparing two different samples (say, a wild-type and a mutant), a biologist usually wants to identify the genes that are differentially expressed because of the intended difference between the samples (here, the genotype) and biological variability within each genotype is then perceived as a source of experimental noise, to be ignored.
Sorting out biological variability from the effect of the intended difference between samples implies two things:
– several biological replicates of each experimental condition have to be analyzed (in order to estimate the variability within each group of replicates, and to compare it to the variability between the groups);
– novel statistical methods have to be used to assess significance, instead of Fisher’s exact test. Several methods now exist to address that problem: improving the distribution model for read counts [99]; correcting the distribution model and adjusting normalization on a gene-specific basis [100]; correcting the distribution model and deriving additional descriptive parameters from the actual datasets [101, 102].
KINETICS OF RNA SILENCING
In vitro reconstitution of the RNAi reaction was achieved as early as 1999, and it allowed a precise investigation of its biochemical process [103]. In vitro, the catalytic rate for the target cleavage reaction falls between 10–3 and 10–2 s–1, with perfectly matched targets being cleaved faster than mismatched targets [104]. Such detailed analyses cannot be achieved in vivo, where siRNA and target concentration is difficult to control. Reports from various experimental systems indicate that, in the tested conditions, the RNAi reaction occurs within minutes, or tens of minutes, after introduction of a long double-stranded RNA trigger. For example in Drosophila embryos, RNAi-mediated repression of an injected mRNA is almost fully efficient when the target and the double-stranded trigger are co-injected, and injecting the trigger 10 to 30 min prior to target injection hardly improves RNAi efficiency; and ≥90% of the target RNA is degraded within the first 10 min [105]. Translation repression and small RNA-guided exonucleolytic degradation (which are the most common modes of action of metazoan miRNAs) could only be recapitulated in vitro more recently [106-108]. Analysis of these phenomena has been complicated by the fact that translational repression and decay are apparently achieved by several mechanisms, whose preponderance depends on the identity of the Ago protein [109], the sequence and structure of the miRNA/mRNA duplex [110], and perhaps additional factors (discussed in [111]). In vitro recapitulation of target repression (possibly through several of these mechanisms) showed that, with the tested concentration of miRNA and mRNA, target translation was unaffected by the miRNA during the first 15 to 20 min, then repression progressively mounted during the first hour [106, 107].
The chronology of miRNA-guided repression could also be explored in vivo: early zebrafish development is particularly suited for these studies, as a single miRNA (miR-430) constitutes most of the early embryo miRNA pool (97% of all miRNAs 4 h post fertilization (hpf) and 88% 12 hpf [112]), and its expression begins at a very precise time point (between 3 and 4 hpf [113]). Analysis of that model miRNA indicated that target mRNAs are translationally silenced before they start to be degraded: target translation is repressed as early as 4 hpf without any measurable mRNA loss, then mRNA decay is evident at 6 hpf [114].
Such a precise experimental dissection has not been possible in other systems, but the kinetics of target repression after introduction of a synthetic small RNA could also be assessed in cultured cells. It is expected that the kinetics of the decrease of a target mRNA depends on its intrinsic half-life; the kinetics of the decrease of a target protein should also depend on the half-life of the protein itself – consequently, some heterogeneity is expected among miRNA targets. For instance, target repression kinetics can be measured during differentiation of the C2C12 myoblast cell line into myotubes. These cells start to express muscle-specific miR-1 and miR-206 between the first and second days of differentiation. For most tested targets, repression of mRNA abundance is already maximal on day 2 and repression of protein abundance takes 2 to 3 more days [115]. Similar kinetics was reported for targets of miRNAs that are upregulated during mouse ES cell differentiation [116].
The dynamics of target repression can also be followed after artificial introduction of synthetic small RNAs. For example, siRNAs promote the repression of “off-targets” (mRNAs that exhibit fortuitous seed matches to the transfected siRNA; they are thought to be repressed by the same mechanism as miRNA targets), whose abundance decreases in a few hours. Jackson et al. [3] provided the first experimental measurement of off-target repression, showing that they reach maximal silencing ca. 20 h after siRNA transfection. Similarly, when a synthetic small RNA mimicking a natural miRNA is paired to a passenger strand, the transfected duplex can repress genes that exhibit binding sites for the small RNA. That experiment is not very well suited to identify the targets of the natural miRNA: only the mRNAs expressed in that cell type can be assessed, and the transfected concentrations can be non-physiological. But the kinetics of mRNA repression following such an experiment may be indicative of mRNA repression kinetics, even though the analyzed mRNAs are not the actual targets.
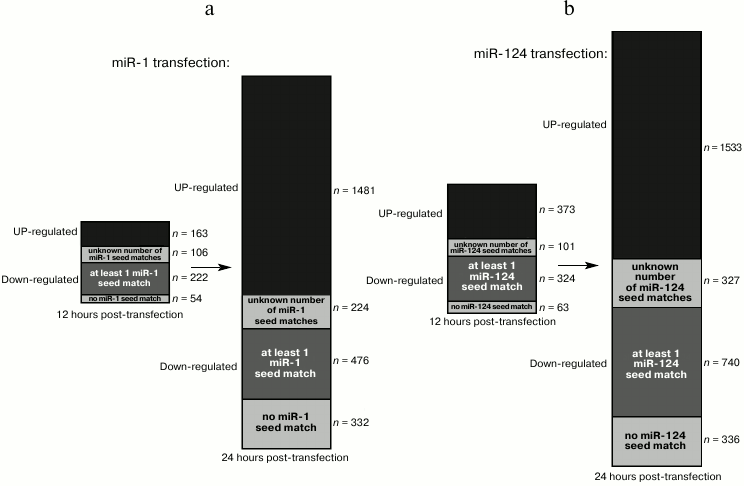
Transfection of HeLa cells with a synthetic miR-1 or miR-124 duplex represses dozens of genes within 12 h [4]. Most of these genes are downregulated, and a large fraction of those exhibit seed matches for the transfected small RNA. Twelve hours later, an even larger set of genes is affected, including many upregulated genes (and among downregulated genes, the proportion of seed-matched RNAs decreases), suggesting that many indirect targets became affected between 12 and 24 h after transfection (Fig. 5, a and b).
Fig. 5. Kinetics of direct and indirect targets in a small RNA transfection experiment. The bars show the number of affected genes 12 h (left bar) or 24 h (right bar) after transfection of miR-1 (a) or miR-124 (b) in HeLa cells [4]. The upregulated genes are shown in black, and the downregulated genes are shown in gray (with several shades of gray). Among downregulated genes, a variable fraction exhibit seed matches for the transfected RNA in their 3′ UTR (for some genes, 3′ UTR information was missing, hence their number of seed matches is unknown).
In the reciprocal experiment (where miRNA expression is constant, but target expression is induced at the first time point), translational repression also precedes mRNA degradation [117], though it should be kept in mind that this experiment does not allow the measurement of the kinetics of target repression – it rather measures the kinetics of target translation after induction, in the presence of a repressing miRNA.
Intriguing in vivo observations suggest that miRNA-mediated regulation could also be much faster: abundance of several miRNAs in mouse retinal neurons is regulated by light, and the abundance of some miRNAs respond very rapidly to changes in retina illumination (maximal reduction in miRNA abundance is achieved in less than 1 h [118]). Non-retinal neurons also exhibit rapid miRNA turnover (with maximal miRNA loss being reached in 2 to 4 h) when their activity is blocked [118]. If this regulation of miRNA abundance has a biological function, it may suggest that some of their targets are able to respond with similarly fast kinetics.
AMPLITUDE OF RNA SILENCING
Efficiency of RNAi. Endonucleolytic cleavage of a target RNA generally reduces target expression more efficiently than translational repression and mRNA exonucleolytic decay [3, 119-121]. Gene silencing by exogenous siRNAs frequently exceeds 80% in cultured cells, but the efficiency of a given RNAi experiment depends on many factors: first, as RNAi targets mRNAs, its efficiency depends on target protein stability (if the target protein is very stable, depleting its mRNA will only slowly translate into a loss of the protein). But RNAi efficiency also depends on the secondary structure of the targeted sequence [122-124], siRNA duplex asymmetry (that determines the relative incorporation of the guide and passenger strands in mature RISC [33, 34, 125]), and target mRNA stability [126].
The activity of endogenous siRNAs can also be measured, comparing target expression in a wild type and in an RNAi-defective strain. For instance, the mus308 gene in Drosophila is repressed 1-3 times by endogenous siRNAs [43, 52]. Of note, 5′ RACE experiments (as well as their high-throughput counterpart, degradome library sequencing [127]), which are frequently used to demonstrate that a target RNA is indeed cleaved in vivo, are not quantitative: detection of a product of the RNAi reaction does not indicate the proportion of target RNAs that were actually cleaved [13].
miRNA-mediated repression is modest. Early description of the effect of miRNAs on target protein expression suggested that repression of the lin-14 gene by the lin-4 miRNA was almost complete [128]. But Western blotting is poorly quantitative, and the effect of lin-4 on its targets has been precisely quantified only at the level of mRNA abundance for two targets, revealing a ≈5-fold reduction [129].
Numerous studies tentatively measured the effect of miRNAs on target expression, but only a minority did so by quantifying the effect of the inactivation of miRNAs – most articles rather over-expressed miRNAs (by transfecting synthetic miRNAs in cultured cells). Transfecting high doses of miRNA can yield large target repression values, just because the intracellular miRNA dose is supra-physiological. Transfecting exogenous small RNAs can also trigger nonspecific effects by titrating the endogenous miRNA machinery [130].
The most convincing analyses quantified the effect of miRNA loss on the proteome. In a pioneer experiment, Nakahara et al. [131] estimated protein abundance in wild type and dcr-1 mutant oocytes in Drosophila. As Dcr-1 is necessary for maturation of fly miRNAs, mutant oocytes are devoid of any miRNA. Bidimensional gel electrophoresis allowed the identification of differentially expressed proteins, which were identified by mass spectrometry. That study pointed at 41 upregulated proteins in dcr-1 mutant oocytes (as well as 51 downregulated proteins, which may be indirect targets of oocyte miRNAs), out of 1003 detected proteins. The presumptive oocyte miRNA targets were variably upregulated, from 1.6- to 69-fold (with an average fold-change of 3.5). A major limitation of that analysis is that many of these deregulated proteins could not be identified by mass spectrometry; as the experiment was carried out almost one generation after dcr-1 gene disruption, it is also possible that some of these deregulated genes are indirect miRNA targets (gene networks have probably been perturbed by the absence of miRNAs), perhaps explaining why some of these targets were not predicted computationally.
Quantitative mass spectrometry provided an efficient method for simultaneous identification and quantification of proteins after perturbation of miRNA abundance. The SILAC technique consists in metabolic labeling of the proteins in each sample by different isotopes, followed by mass spectrometric analysis. Inhibiting the let-7b miRNA in HeLa cells, Selbach et al. [6] used SILAC to show that thousands of genes were repressed (directly or indirectly) by that miRNA in HeLa, and typical let-7b-mediated repression ranges from 1 to 1.5, never more than 2. A similar experiment performed in vivo in mice mutated for the miR-223 gene revealed that this miRNA (which is highly expressed in neutrophils) represses dozens of genes in neutrophils, but once again target repression is limited (≤1.5-fold for most of them [7]).
These high-throughput proteomics experiments also allowed a direct comparison of computational miRNA target predictions with experimental results: it turned out that, among the prediction programs available at the time, PicTar and TargetScan are the most efficient, yet ≈40-66% of their predicted targets were not significantly derepressed. These experiments also showed that mRNA quantification is a reasonable way of identifying miRNA targets, as most affected genes are repressed both in terms of protein and mRNA abundance ([6, 7], see also [132]). This last observation could prove very useful from a practical point of view, as high-throughput mRNA quantification is much more accessible than high-throughput protein quantification, yet it must be kept in mind that protein and mRNA regulation do not always correlate after an experimental perturbation of gene expression [133].
Cell-to-cell variability in miRNA silencing. Recent technological developments allowed single cell analysis for a variety of cellular processes. These experiments revealed cell-to-cell variability among (apparently) homogeneous cell populations. Two main sources of variability can be delimited: intrinsic noise in biochemical processes and deterministic factors [134]. Intrinsic noise arises from the low number of components involved in many cellular processes (e.g. for single-copy genes, there are only two instances of the gene in the nucleus of a cell in the G1 phase): in these conditions, the law of large numbers does not apply and the outcome of biochemical events is submitted to random fluctuations. Deterministic factors are due to uncontrollable heterogeneities in the cell population (e.g. all the cells are not in the same physiological state when transfected or analyzed, resulting in various cellular responses).
Single cell measurements of RNA silencing revealed dramatic differences in miRNA abundance or activity. Estimation of the variability in miRNA abundance in individual murine ES cells could be achieved by sensitive single-cell measurement by qRT-PCR. Among 15 individual cells, miR-16 abundance varied up to ≈2-fold, and similar variations were scored among five individual cells for several other miRNAs [89]. Cell-to-cell variability in miRNA abundance (as well as, potentially, other sources of heterogeneity) leads to heterogeneous silencing efficiency: using an internally normalized system (where two fluorescent proteins are transcribed from the same, bidirectional promoter; one protein is targeted by an endogenously-expressed miRNA while the other protein is not repressed by the miRNA), Mukherji et al. [135] could precisely measure miR-20-mediated repression on a single cell basis. That experiment revealed a very large variability in target repression efficiency: cells expressing low levels of target mRNA silenced them almost completely, while cells with a higher target expression repressed them only moderately. When the reporter bears a single bulged miRNA binding site, cells expressing the lowest levels of target repressed them ≈5-10 times more than cells expressing the highest levels.
The physiological bases for this heterogeneity are still unknown; it has been proposed that the heterogeneous association of mitochondria with P-bodies (where miRNA-target mRNAs accumulate) could contribute to the cellular heterogeneity in silencing efficiency [136].
miRNA DIVERSITY AND SO-CALLED “ORGANISMAL
COMPLEXITY”
Several authors have claimed that the number of miRNA genes (or, more generally, the number of non-coding genes) correlates positively with organismal complexity [137-139]. Indeed, the acquisition of a few additional regulators, which can act in combination, creates an incommensurably large variety of available regulatory networks – for a limited cost in terms of genetic innovation [140].
Yet the premises of this argumentation do not rely on the most objective observations. Before “complexity” can be correlated to anything, it needs to be quantified (then the correlation between the measurement of complexity, and the number of miRNA genes, can be assessed by statistical means). Unfortunately, many authors do not try to quantify complexity, they do not even attempt to define it – rather, it is often postulated that some species are more complex than others (e.g. vertebrates would be more complex than insects) without further definition or justification.
More convincingly, several complexity scores have been proposed that aim at quantifying organismal complexity [141, 142], yet these scoring systems are based on the presence or absence of an arbitrary list of characters, which all turn out to be found in vertebrates (presence of a urogenital system, of a skull, number of neurons in the organism, ...). The fact that vertebrate species are assigned a high score in such a scheme is tautological, and hardly informative.
Another, more objective method relies on the estimation of the diversity of cell types in each species [143, 144]. A major technical challenge in that approach is that a “cell type” is not rigorously defined, and ultimately depends on morphological criteria, assessed by the specialists of each analyzed species – hence, different phyla are scored by different observers [144]. Quantifying cell types also faces a more profound limitation: differentiated cells in a multicellular organism originate from the fertilized egg, through cell division and differentiation. Differentiation is a continuous process, hence the definition of each differentiation intermediate depends on arbitrary cutoffs, and the number of recorded cell types can vary from 1 (when every step of the differentiation process is seen as a different metabolic state of the same cell type) to an infinity (when each small difference is considered to define a new cell type).
While no convincing method has been proposed to measure “complexity” of a species, “evolution” of a species can be much more precisely and objectively defined: every living species apparently derives from the same ancestor (dubbed LUCA, for Last Universal Common Ancestor; see [145] for a review). The “evolution” of a species can be measured by its evolutionary age; as every modern-day species have the same starting and ending points, they all have the same evolutionary age, i.e. they would be equally evolved. From this point of view, the number of miRNA genes did increase with evolution (when the miRNA machinery first appeared), but the most evolved species exhibit a wide diversity of miRNA gene number, ranging from 0 (for current organisms devoid of miRNAs) to at least several hundreds bona fide miRNA genes [41].
CONCLUSION
Small regulatory RNAs exhibit several features that distinguish them from other gene regulators. First, the large diversity of siRNA and piRNA sequences exceeds that of any other known class of regulators, and even the diversity of coding genes, raising intriguing questions on the specificity of their biological roles. Second, all three classes were discovered recently: the first miRNA was discovered in 1993 and the diversity of miRNAs was recognized only in 2001 [35-37, 146]. piRNAs and endogenous siRNAs were discovered during the 2000-2010 decade (reviewed in [1]). Discovery of these regulators coincided with the development of high-throughput techniques for the detection and quantification of small RNAs and their targets, rapidly translating into a high-throughput, quantitative assessment of their molecular functions. It took several decades after the discovery of transcription factors before their mode of action could be probed at such a high resolution – in fact, the poor specificity of most transcription factors (with binding sites defined by short DNA sequence motifs [147]) probably means that each of them also controls hundreds or thousands of genes, as illustrated by recent high-throughput experiments (e.g. [148]). While these notions probably mean that a wide paradigm shift is expected in our perception of the role of transcription factors, they have always been familiar to the small RNA community [149].
Yet several key quantitative questions still deserve more attention. Factors controlling the abundance of miRNAs (regulation of transcription, processing, and stability) have been analyzed at a small scale, following serendipitous discoveries. These questions have hardly been touched for siRNAs and piRNAs. Regarding their biological function, several worrying issues still await a clear answer. In particular, the modest effect of miRNAs on their predicted targets (smaller than well-tolerated fluctuations in gene expression) apparently implies that most of these targets are not repressed enough to be functionally affected – suggesting that the interaction between miRNAs and their targets has been misunderstood [150]. Fortunately, powerful analysis techniques and sophisticated statistical procedures are being developed, allowing a rigorous measurement of the regulatory effect of small RNAs.
A. M. S. was supported by a PhD fellowship from La Ligue Contre le Cancer. Work in the laboratory of H. S. is supported by a Career Development Award from the Human Frontier Science Program and by an ATIP-Avenir grant from the CNRS.
REFERENCES
1.Ghildiyal, M., and Zamore, P. D. (2009) Nat.
Rev. Genet., 10, 94-108.
2.Kozomara, A., and Griffiths-Jones, S. (2011)
Nucleic Acids Res., 39, D152-157.
3.Jackson, A. L., Bartz, S. R., Schelter, J.,
Kobayashi, S. V., Burchard, J., Mao, M., Li, B., Cavet, G., and
Linsley, P. S. (2003) Nat. Biotechnol., 21, 635-637.
4.Lim, L. P., Lau, N. C., Garrett-Engele, P.,
Grimson, A., Schelter, J. M., Castle, J., Bartel, D. P., Linsley, P.
S., and Johnson, J. M. (2005) Nature, 433, 769-773.
5.Lewis, B. P., Burge, C. B., and Bartel, D. P.
(2005) Cell, 120, 15-20.
6.Selbach, M., Schwanhausser, B., Thierfelder, N.,
Fang, Z., Khanin, R., and Rajewsky, N. (2008) Nature,
455, 58-63.
7.Baek, D., Villen, J., Shin, C., Camargo, F. D.,
Gygi, S. P., and Bartel, D. P. (2008) Nature, 455,
64-71.
8.Friedman, R. C., Farh, K. K., Burge, C. B., and
Bartel, D. P. (2009) Genome Res., 19, 92-105.
9.Hutvagner, G., and Zamore, P. D. (2002)
Science, 297, 2056-2060.
10.Doench, J. G., Petersen, C. P., and Sharp, P. A.
(2003) Genes Dev., 17, 438-442.
11.Meister, G., Landthaler, M., Patkaniowska, A.,
Dorsett, Y., Teng, G., and Tuschl, T. (2004) Mol. Cell,
15, 185-197.
12.Liu, J., Carmell, M. A., Rivas, F. V., Marsden,
C. G., Thomson, J. M., Song, J. J., Hammond, S. M., Joshua-Tor, L., and
Hannon, G. J. (2004) Science, 305, 1437-1441.
13.Brodersen, P., and Voinnet, O. (2009) Nat.
Rev. Mol. Cell Biol., 10, 141-148.
14.Wilson, D., Charoensawan, V., Kummerfeld, S. K.,
and Teichmann, S. A. (2008) Nucleic Acids Res., 36,
D88-92.
15.Cook, K. B., Kazan, H., Zuberi, K., Morris, Q.,
and Hughes, T. R. (2011) Nucleic Acids Res., 39,
D301-308.
16.Yin, H., and Lin, H. (2007) Nature,
450, 304-308.
17.Brennecke, J., Aravin, A. A., Stark, A., Dus, M.,
Kellis, M., Sachidanandam, R., and Hannon, G. J. (2007) Cell,
128, 1089-1103.
18.Brennecke, J., Malone, C. D., Aravin, A. A.,
Sachidanandam, R., Stark, A., and Hannon, G. J. (2008) Science,
322, 1387-1392.
19.Malone, C. D., Brennecke, J., Dus, M., Stark, A.,
McCombie, W. R., Sachidanandam, R., and Hannon, G. J. (2009)
Cell, 137, 522-535.
20.Saito, K., Inagaki, S., Mituyama, T., Kawamura,
Y., Ono, Y., Sakota, E., Kotani, H., Asai, K., Siomi, H., and Siomi, M.
C. (2009) Nature, 461, 1296-1299.
21.Haase, A. D., Fenoglio, S., Muerdter, F.,
Guzzardo, P. M., Czech, B., Pappin, D. J., Chen, C., Gordon, A., and
Hannon, G. J. (2010) Genes Dev., 24, 2499-2504.
22.Rozhkov, N. V., Aravin, A. A., Zelentsova, E. S.,
Schostak, N. G., Sachidanandam, R., McCombie, W. R., Hannon, G. J., and
Evgen’ev, M. B. (2010) RNA, 16, 1634-1645.
23.Qi, H., Watanabe, T., Ku, H. Y., Liu, N., Zhong,
M., and Lin, H. (2011) J. Biol. Chem., 286,
3789-3797.
24.Zamparini, A. L., Davis, M. Y., Malone, C. D.,
Vieira, E., Zavadil, J., Sachidanandam, R., Hannon, G. J., and Lehmann,
R. (2011) Development, 138, 4039-4050.
25.Grentzinger, T., Armenise, C., Brun, C., Mugat,
B., Serrano, V., Pelisson, A., and Chambeyron, S. (2012) Genome
Res., 22, 1877-1888.
26.Olivieri, D., Senti, K. A., Subramanian, S.,
Sachidanandam, R., and Brennecke, J. (2012) Mol. Cell,
47, 954-969.
27.Preall, J. B., Czech, B., Guzzardo, P. M.,
Muerdter, F., and Hannon, G. J. (2012) RNA, 18,
1446-1457.
28.Matranga, C., Tomari, Y., Shin, C., Bartel, D.
P., and Zamore, P. D. (2005) Cell, 123, 607-620.
29.Rand, T. A., Petersen, S., Du, F., and Wang, X.
(2005) Cell, 123, 621-629.
30.Miyoshi, K., Tsukumo, H., Nagami, T., Siomi, H.,
and Siomi, M. C. (2005) Genes Dev., 19, 2837-2848.
31.Kawamata, T., Seitz, H., and Tomari, Y. (2009)
Nat. Struct. Mol. Biol., 16, 953-960.
32.Yoda, M., Kawamata, T., Paroo, Z., Ye, X.,
Iwasaki, S., Liu, Q., and Tomari, Y. (2010) Nat. Struct. Mol.
Biol., 17, 17-23.
33.Schwarz, D. S., Hutvagner, G., Du, T., Xu, Z.,
Aronin, N., and Zamore, P. D. (2003) Cell, 115,
199-208.
34.Khvorova, A., Reynolds, A., and Jayasena, S. D.
(2003) Cell, 115, 209-216.
35.Lagos-Quintana, M., Rauhut, R., Lendeckel, W.,
and Tuschl, T. (2001) Science, 294, 853-858.
36.Lau, N. C., Lim, L. P., Weinstein, E. G., and
Bartel, D. P. (2001) Science, 294, 858-862.
37.Lee, R. C., and Ambros, V. (2001) Science,
294, 862-864.
38.Griffiths-Jones, S., Saini, H. K., van Dongen,
S., and Enright, A. J. (2008) Nucleic Acids Res., 36,
D154-158.
39.Ro, S., Park, C., Young, D., Sanders, K. M., and
Yan, W. (2007) Nucleic Acids Res., 35, 5944-5953.
40.DeWit, E., Linsen, S. E., Cuppen, E., and
Berezikov, E. (2009) Genome Res., 19, 2064-2074.
41.Chiang, H. R., Schoenfeld, L. W., Ruby, J. G.,
Auyeung, V. C., Spies, N., Baek, D., Johnston, W. K., Russ, C., Luo,
S., Babiarz, J. E., Blelloch, R., Schroth, G. P., Nusbaum, C., and
Bartel, D. P. (2010) Genes Dev., 24, 992-1009.
42.Griffiths-Jones, S., Hui, J. H., Marco, A., and
Ronshaugen, M. (2011) EMBO Rep., 12, 172-177.
43.Okamura, K., Phillips, M. D., Tyler, D. M., Duan,
H., Chou, Y. T., and Lai, E. C. (2008) Nat. Struct. Mol. Biol.,
15, 354-363.
44.Okamura, K., Liu, N., and Lai, E. C. (2009)
Mol. Cell, 36, 431-444.
45.Czech, B., Zhou, R., Erlich, Y., Brennecke, J.,
Binari, R., Villalta, C., Gordon, A., Perrimon, N., and Hannon, G. J.
(2009) Mol. Cell, 36, 445-456.
46.Ghildiyal, M., Xu, J., Seitz, H., Weng, Z., and
Zamore, P. D. (2010) RNA, 16, 43-56.
47.Ghildiyal, M., Seitz, H., Horwich, M. D., Li, C.,
Du, T., Lee, S., Xu, J., Kittler, E. L., Zapp, M. L., Weng, Z., and
Zamore, P. D. (2008) Science, 320, 1077-1081.
48.Kawamura, Y., Saito, K., Kin, T., Ono, Y., Asai,
K., Sunohara, T., Okada, T. N., Siomi, M. C., and Siomi, H. (2008)
Nature, 453, 793-797.
49.Tam, O. H., Aravin, A. A., Stein, P., Girard, A.,
Murchison, E. P., Cheloufi, S., Hodges, E., Anger, M., Sachidanandam,
R., Schultz, R. M., and Hannon, G. J. (2008) Nature, 453,
534-538.
50.Watanabe, T., Totoki, Y., Toyoda, A., Kaneda, M.,
Kuramochi-Miyagawa, S., Obata, Y., Chiba, H., Kohara, Y., Kono, T.,
Nakano, T., Surani, M. A., Sakaki, Y., and Sasaki, H. (2008)
Nature, 453, 539-543.
51.Okamura, K., Chung, W. J., Ruby, J. G., Guo, H.,
Bartel, D. P., and Lai, E. C. (2008) Nature, 453,
803-806.
52.Czech, B., Malone, C. D., Zhou, R., Stark, A.,
Schlingeheyde, C., Dus, M., Perrimon, N., Kellis, M., Wohlschlegel, J.
A., Sachidanandam, R., Hannon, G. J., and Brennecke, J. (2008)
Nature, 453, 798-802.
53.Sai Lakshmi, S., and Agrawal, S. (2008)
Nucleic Acids Res., 36, D173-177.
54.Ruby, J., Jan, C., Player, C., Axtell, M., Lee,
W., Nusbaum, C., Ge, H., and Bartel, D. (2006) Cell, 127,
1193-1207.
55.Lau, N. C., Seto, A. G., Kim, J.,
Kuramochi-Miyagawa, S., Nakano, T., Bartel, D. P., and Kingston, R. E.
(2006) Science, 313, 363-367.
56.Monteys, A. M., Spengler, R. M., Wan, J.,
Tecedor, L., Lennox, K. A., Xing, Y., and Davidson, B. L. (2010)
RNA, 16, 495-505.
57.Newman, M. A., Thomson, J. M., and Hammond, S. M.
(2008) RNA, 14, 1539-1549.
58.Viswanathan, S. R., Daley, G. Q., and Gregory, R.
I. (2008) Science, 320, 97-100.
59.Heo, I., Joo, C., Cho, J., Ha, M., Han, J., and
Kim, V. N. (2008) Mol. Cell, 32, 276-284.
60.Rybak, A., Fuchs, H., Smirnova, L., Brandt, C.,
Pohl, E. E., Nitsch, R., and Wulczyn, F. G. (2008) Nat. Cell
Biol., 10, 987-993.
61.Heo, I., Joo, C., Kim, Y. K., Ha, M., Yoon, M.
J., Cho, J., Yeom, K. H., Han, J., and Kim, V. N. (2009) Cell,
138, 696-708.
62.Hagan, J. P., Piskounova, E., and Gregory, R. I.
(2009) Nat. Struct. Mol. Biol., 16, 1021-1025.
63.Thornton, J. E., Chang, H. M., Piskounova, E.,
and Gregory, R. I. (2012) RNA, 18, 1875-1885.
64.Lehrbach, N. J., Armisen, J., Lightfoot, H. L.,
Murfitt, K. J., Bugaut, A., Balasubramanian, S., and Miska, E. A.
(2009) Nat. Struct. Mol. Biol., 16, 1016-1020.
65.Heo, I., Ha, M., Lim, J., Yoon, M. J., Park, J.
E., Kwon, S. C., Chang, H., and Kim, V. N. (2012) Cell,
151, 521-532.
66.Katoh, T., Sakaguchi, Y., Miyauchi, K., Suzuki,
T., Kashiwabara, S., Baba, T., and Suzuki, T. (2009) Genes Dev.,
23, 433-438.
67.Burroughs, A. M., Ando, Y., de Hoon, M. J.,
Tomaru, Y., Nishibu, T., Ukekawa, R., Funakoshi, T., Kurokawa, T.,
Suzuki, H., Hayashizaki, Y., and Daub, C. O. (2010) Genome Res.,
20, 1398-1410.
68.Ameres, S. L., Horwich, M. D., Hung, J. H., Xu,
J., Ghildiyal, M., Weng, Z., and Zamore, P. D. (2010) Science,
328, 1534-1539.
69.Chatterjee, S., and Großhans, H. (2009)
Nature, 461, 546-549.
70.Das, S. K., Sokhi, U. K., Bhutia, S. K., Azab,
B., Su, Z. Z., Sarkar, D., and Fisher, P. B. (2010) Proc. Natl.
Acad. Sci. USA, 107, 11948-11953.
71.Batista, P. J., Ruby, J. G., Claycomb, J. M.,
Chiang, R., Fahlgren, N., Kasschau, K. D., Chaves, D. A., Gu, W.,
Vasale, J. J., Duan, S., Conte, D. J., Luo, S., Schroth, G. P.,
Carrington, J. C., Bartel, D. P., and Mello, C. C. (2008) Mol.
Cell, 31, 67-78.
72.Das, P. P., Bagijn, M. P., Goldstein, L. D.,
Woolford, J. R., Lehrbach, N. J., Sapetschnig, A., Buhecha, H. R.,
Gilchrist, M. J., Howe, K. L., Stark, R., Matthews, N., Berezikov, E.,
Ketting, R. F., Tavare, S., and Miska, E. A. (2008) Mol. Cell,
31, 79-90.
73.Sarot, E., Payen-Groschene, G., Bucheton, A., and
Pelisson, A. (2004) Genetics, 166, 1313-1321.
74.Kalmykova, A. I., Klenov, M. S., and Gvozdev, V.
A. (2005) Nucleic Acids Res., 33, 2052-2059.
75.Aravin, A. A., Sachidanandam, R., Girard, A.,
Fejes-Toth, K., and Hannon, G. J. (2007) Science, 316,
744-747.
76.Carmell, M. A., Girard, A., van de Kant, H. J.,
Bourc’his, D., Bestor, T. H., de Rooij, D. G., and Hannon, G. J.
(2007) Dev. Cell, 12, 503-514.
77.Kuramochi-Miyagawa, S., Watanabe, T., Gotoh, K.,
Totoki, Y., Toyoda, A., Ikawa, M., Asada, N., Kojima, K., Yamaguchi,
Y., Ijiri, T. W., Hata, K., Li, E., Matsuda, Y., Kimura, T., Okabe, M.,
Sakaki, Y., Sasaki, H., and Nakano, T. (2008) Genes Dev.,
22, 908-917.
78.Houwing, S., Kamminga, L. M., Berezikov, E.,
Cronembold, D., Girard, A., van den Elst, H., Filippov, D. V.,
Blaser, H., Raz, E., Moens, C. B., Plasterk, R. H., Hannon, G. J.,
Draper, B. W., and Ketting, R. F. (2007) Cell, 129,
69-82.
79.Rouget, C., Papin, C., Boureux, A., Meunier, A.
C., Franco, B., Robine, N., Lai, E. C., Pelisson, A., and Simonelig, M.
(2010) Nature, 467, 1128-1132.
80.Rajasethupathy, P., Antonov, I., Sheridan, R.,
Frey, S., Sander, C., Tuschl, T., and Kandel, E. R. (2012) Cell,
149, 693-707.
81.Chung, W. J., Okamura, K., Martin, R., and Lai,
E. C. (2008) Curr. Biol., 18, 795-802.
82.Bagijn, M. P., Goldstein, L. D., Sapetschnig, A.,
Weick, E. M., Bouasker, S., Lehrbach, N. J., Simard, M. J., and Miska,
E. A. (2012) Science, 337, 574-578.
83.Ashe, A., Sapetschnig, A., Weick, E. M.,
Mitchell, J., Bagijn, M. P., Cording, A. C., Doebley, A. L., Goldstein,
L. D., Lehrbach, N. J., Le Pen, J., Pintacuda, G., Sakaguchi, A.,
Sarkies, P., Ahmed, S., and Miska, E. A. (2012) Cell,
150, 88-99.
84.Rozhkov, N. V., Schostak, N. G., Zelentsova, E.
S., Yushenova, I. A., Zatsepina, O. G., and Evgen’ev, M. B.
(2013) Mol. Biol. Evol., 30, 397-408.
85.Girard, A., Sachidanandam, R., Hannon, G., and
Carmell, M. A. (2006) Nature, 442, 199-202.
86.Aravin, A., Gaidatzis, D., Pfeffer, S.,
Lagos-Quintana, M., Landgraf, P., Iovino, N., Morris, P., Brownstein,
M. J., Kuramochi-Miyagawa, S., Nakano, T., Chien, M., Russo, J. J., Ju,
J., Sheridan, R., Sander, C., Zavolan, M., and Tuschl, T. (2006)
Nature, 442, 203-207.
87.Pall, G. S., Codony-Servat, C., Byrne, J.,
Ritchie, L., and Hamilton, A. (2007) Nucleic Acids Res.,
35, e60.
88.Pall, G. S., and Hamilton, A. J. (2008) Nat.
Protoc., 3, 1077-1084.
89.Tang, F., Hajkova, P., Barton, S. C., Lao, K.,
and Surani, M. A. (2006) Nucleic Acids Res., 34, e9.
90.Benes, V., and Castoldi, M. (2010)
Methods, 50, 244-249.
91.Seitz, H., Ghildiyal, M., and Zamore, P. D.
(2008) Curr. Biol., 18, 147-151.
92.Livak, K. J., and Schmittgen, T. D. (2001)
Methods, 25, 402-408.
93.Derveaux, S., Vandesompele, J., and Hellemans, J.
(2010) Methods, 50, 227-230.
94.Han, L., Wen, Z., Lynn, R. C., Baudet, M. L.,
Holt, C. E., Sasaki, Y., Bassell, G. J., and Zheng, J. Q. (2011)
Mol. Brain, 4, 40.
95.Yin, J. Q., Zhao, R. C., and Morris, K. V. (2008)
Trends Biotechnol., 26, 70-76.
96.Vagin, V. V., Sigova, A., Li, C., Seitz, H.,
Gvozdev, V., and Zamore, P. D. (2006) Science, 313,
320-324.
97.Fahlgren, N., Sullivan, C. M., Kasschau, K. D.,
Chapman, E. J., Cumbie, J. S., Montgomery, T. A., Gilbert, S. D.,
Dasenko, M., Backman, T. W., Givan, S. A., and Carrington, J. C. (2009)
RNA, 15, 992-1002.
98.Dabney, A., Storey, J. D., and with
assistance from Gregory R. Warnes (2011) Q-Value: Q-Value
Estimation for False Discovery Rate Control, R package version
1.26.0.
99.Srivastava, S., and Chen, L. (2010) Nucleic
Acids Res., 38, e170.
100.Langmead, B., Hansen, K. D., and Leek, J. T.
(2010) Genome Biol., 11, R83.
101.Anders, S., and Huber, W. (2010) Genome
Biol., 11, R106.
102.Robinson, M. D., McCarthy, D. J., and Smyth, G.
K. (2010) Bioinformatics, 26, 139-140.
103.Tuschl, T., Zamore, P. D., Lehmann, R., Bartel,
D. P., and Sharp, P. A. (1999) Genes Dev., 13,
3191-3197.
104.Haley, B., and Zamore, P. D. (2004) Nat.
Struct. Mol. Biol., 11, 599-606.
105.Yang, D., Lu, H., and Erickson, J. W. (2000)
Curr. Biol., 10, 1191-1200.
106.Mathonnet, G., Fabian, M. R., Svitkin, Y. V.,
Parsyan, A., Huck, L., Murata, T., Biffo, S., Merrick, W. C.,
Darzynkiewicz, E., Pillai, R. S., Filipowicz, W., Duchaine, T. F., and
Sonenberg, N. (2007) Science, 317, 1764-1767.
107.Wakiyama, M., Takimoto, K., Ohara, O., and
Yokoyama, S. (2007) Genes Dev., 21, 1857-1862.
108.Thermann, R., and Hentze, M. W. (2007)
Nature, 447, 875-878.
109.Iwasaki, S., Kawamata, T., and Tomari, Y.
(2009) Mol. Cell, 34, 58-67.
110.Aleman, L. M., Doench, J., and Sharp, P. A.
(2007) RNA, 13, 385-395.
111.Fabian, M. R., Sonenberg, N., and Filipowicz,
W. (2010) Annu. Rev. Biochem., 79, 351-379.
112.Chen, P. Y., Manninga, H., Slanchev, K., Chien,
M., Russo, J. J., Ju, J., Sheridan, R., John, B., Marks, D. S.,
Gaidatzis, D., Sander, C., Zavolan, M., and Tuschl, T. (2005) Genes
Dev., 19, 1288-1293.
113.Giraldez, A. J., Mishima, Y., Rihel, J.,
Grocock, R. J., Van Dongen, S., Inoue, K., Enright, A. J., and
Schier, A. F. (2006) Science, 312, 75-79.
114.Bazzini, A. A., Lee, M. T., and Giraldez, A. J.
(2012) Science, 336, 233-237.
115.Goljanek-Whysall, K., Pais, H., Rathjen, T.,
Sweetman, D., Dalmay, T., and Munsterberg, A. (2012) J. Cell
Sci., 125, 3590-3600.
116.Ciaudo, C., Servant, N., Cognat, V., Sarazin,
A., Kieffer, E., Viville, S., Colot, V., Barillot, E., Heard, E., and
Voinnet, O. (2009) PLoS Genet., 5, e1000620.
117.Bethune, J., Artus-Revel, C. G., and
Filipowicz, W. (2012) EMBO Rep., 13, 716-723.
118.Krol, J., Busskamp, V., Markiewicz, I.,
Stadler, M. B., Ribi, S., Richter, J., Duebel, J., Bicker, S., Fehling,
H. J., Schubeler, D., Oertner, T. G., Schratt, G., Bibel, M., Roska,
B., and Filipowicz, W. (2010) Cell, 141, 618-631.
119.Zeng, Y., Yi, R., and Cullen, B. R. (2003)
Proc. Natl. Acad. Sci. USA, 100, 9779-9784.
120.Yekta, S., Shih, I. H., and Bartel, D. P.
(2004) Science, 304, 594-596.
121.Mullokandov, G., Baccarini, A., Ruzo, A.,
Jayaprakash, A. D., Tung, N., Israelow, B., Evans, M. J.,
Sachidanandam, R., and Brown, B. D. (2012) Nat. Methods,
9, 840-846.
122.Patzel, V., Rutz, S., Dietrich, I., Koberle,
C., Scheffold, A., and Kaufmann, S. H. (2005) Nat. Biotechnol.,
23, 1440-1444.
123.Westerhout, E. M., and Berkhout, B. (2007)
Nucleic Acids Res., 35, 4322-4330.
124.Shao, Y., Chan, C. Y., Maliyekkel, A.,
Lawrence, C. E., Roninson, I. B., and Ding, Y. (2007) RNA,
13, 1631-1640.
125.Tafer, H., Ameres, S. L., Obernosterer, G.,
Gebeshuber, C. A., Schroeder, R., Martinez, J., and Hofacker, I. L.
(2008) Nat. Biotechnol., 26, 578-583.
126.Larsson, E., Sander, C., and Marks, D. (2010)
Mol. Syst. Biol., 6, 433.
127.Addo-Quaye, C., Eshoo, T. W., Bartel, D. P.,
and Axtell, M. J. (2008) Curr. Biol., 18, 758-762.
128.Wightman, B., Ha, I., and Ruvkun, G. (1993)
Cell, 75, 855-862.
129.Bagga, S., Bracht, J., Hunter, S., Massirer,
K., Holtz, J., Eachus, R., and Pasquinelli, A. E. (2005) Cell,
122, 553-563.
130.Khan, A. A., Betel, D., Miller, M. L., Sander,
C., Leslie, C. S., and Marks, D. S. (2009) Nat. Biotechnol.,
27, 549-555.
131.Nakahara, K., Kim, K., Sciulli, C., Dowd, S.
R., Minden, J. S., and Carthew, R. W. (2005) Proc. Natl. Acad. Sci.
USA, 102, 12023-12028.
132.Guo, H., Ingolia, N. T., Weissman, J. S., and
Bartel, D. P. (2010) Nature, 466, 835-840.
133.Bonaldi, T., Straub, T., Cox, J., Kumar, C.,
Becker, P. B., and Mann, M. (2008) Mol. Cell, 31,
762-772.
134.Snijder, B., and Pelkmans, L. (2011) Nat.
Rev. Mol. Cell Biol., 12, 119-125.
135.Mukherji, S., Ebert, M. S., Zheng, G. X.,
Tsang, J. S., Sharp, P. A., and van Oudenaarden, A. (2011) Nat.
Genet., 43, 854-859.
136.Huang, L., Mollet, S., Souquere, S.,
Le Roy, F., Ernoult-Lange, M., Pierron, G., Dautry, F., and Weil,
D. (2011) J. Biol. Chem., 286, 24219-24230.
137.Taft, R. J., Pheasant, M., and Mattick, J. S.
(2007) Bioessays, 29, 288-299.
138.Heimberg, A. M., Sempere, L. F., Moy, V. N.,
Donoghue, P. C., and Peterson, K. J. (2008) Proc. Natl. Acad. Sci.
USA, 105, 2946-2950.
139.Cacchiarelli, D., Santoni, D., and Bozzoni, I.
(2008) RNA Biol., 5, 120-122.
140.Berezikov, E. (2011) Nat. Rev. Genet.,
12, 846-860.
141.Aburomia, R., Khaner, O., and Sidow, A. (2003)
J. Struct. Funct. Genom., 3, 45-52.
142.Technau, U. (2008) Nature, 455,
1184-1185.
143.Valentine, J. W., Collins, A. G., and Meyer, C.
P. (1994) Paleobiology, 20, 131-142.
144.Bell, G., and Mooers, A. O. (1997) Biol. J.
Linn Soc., 60, 345-363.
145.Glansdorff, N., Xu, Y., and Labedan, B. (2009)
Res. Microbiol., 160, 522-528.
146.Lee, R. C., Feinbaum, R. L., and Ambros, V.
(1993) Cell, 75, 843-854.
147.Sandelin, A., Alkema, W., Engstrom, P.,
Wasserman, W. W., and Lenhard, B. (2004) Nucleic Acids Res.,
32, D91-94.
148.Schmidt, D., Wilson, M. D., Ballester, B.,
Schwalie, P. C., Brown, G. D., Marshall, A., Kutter, C., Watt, S.,
Martinez-Jimenez, C. P., Mackay, S., Talianidis, I., Flicek, P., and
Odom, D. T. (2010) Science, 328, 1036-1040.
149.Bartel, D. P., and Chen, C. Z. (2004) Nat.
Rev. Genet., 5, 396-400.
150.Seitz, H. (2009) Curr. Biol., 19,
870-873.