Biochemical Characterization of Human Enteropeptidase Light Chain
M. E. Gasparian*, V. G. Ostapchenko, D. A. Dolgikh, and M. P. Kirpichnikov
Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, ul. Miklukho-Maklaya 16/10, 117997 Moscow, Russia; fax: (7-495) 336-8011; E-mail: marine_gasparian@yahoo.com* To whom correspondence should be addressed.
Received July 7, 2005; Revision received October 17, 2005
The synthetic gene encoding human enteropeptidase light chain (L-HEP) was cloned into plasmid pET-32a downstream from the gene of fusion partner thioredoxin immediately after the DNA sequence encoding the enteropeptidase recognition site. The fusion protein thioredoxin (Trx)/L-HEP was expressed in Escherichia coli BL21(DE3). Autocatalytic cleavage of the fusion protein and activation of recombinant L-HEP were achieved by solubilization of inclusion bodies and refolding of Trx/L-HEP fusion protein. The kinetic parameters of human and bovine enteropeptidases in the presence of different concentrations of Ca2+ and Na+ for cleavage of the specific substrate GD4K-na and nonspecific substrates such as small ester Z-Lys-SBzl and chromogenic substrates Z-Ala-X-Arg-pNA have been comparatively analyzed. It is demonstrated that positively charged ions increased the Michaelis constant (Km) for cleavage of specific substrate GD4K-na, while the catalytic constant (kcat) remained practically unchanged. L-HEP demonstrated secondary specificity to the chromogenic substrate Z-Ala-Phe-Arg-pNA with kcat/Km 260 mM-1·sec-1. Enzymatic activity of L-HEP was suppressed by inhibitors of trypsin-like and cysteine (E-64), but not metallo-, amino-, or chymotrypsin-like proteinases. L-HEP was active over a broad range of pH (6-9) with optimum activity at pH 7.5, and it demonstrated high stability to different denaturing agents.
KEY WORDS: human enteropeptidase, recombinant fusion proteins, specificity, kinetic parameters, protease inhibitorsDOI: 10.1134/S0006297906020015
Abbreviations: L-HEP) recombinant human enteropeptidase light chain; L-BEP) recombinant bovine enteropeptidase light chain purified from E. coli; Trx/L-HEP) thioredoxin/human enteropeptidase light chain fusion protein; Trx/hEGF) thioredoxin/human epidermal growth factor fusion protein; Z-Lys-SBzl) thiobenzyl benzyloxy-carbonyl-L-lysinate; GD4K-na) Gly-Asp-Asp-Asp-Asp-Lys-beta-naphthylamide; Z-Ala-X-Arg-pNA) benzyloxycarbonyl-L-alanine-X-L-arginine-p-nitroanilide; STI) soybean trypsin inhibitor; DTNB) 5,5´-dithiobis-(2-nitrobenzoic acid); TPCK) N-tosyl-L-phenylalanine chloromethyl ketone; TLCK) N-tosyl-L-lysine chloromethyl ketone.
Enteropeptidase (synonym: enterokinase) (EC 3.4.21.9) is a serine
protease of the intestinal brush border that activates trypsinogen by
cleavage of the N-terminal peptide following the sequence
(Asp)4-Lys. As a result, active trypsin is generated which
activates many pancreatic zymogens [1]. Native
dimer of enteropeptidase has been purified from porcine [2], bovine [3, 4], and human [5] intestine. In
all cases, enteropeptidase appears to be a disulfide-linked heterodimer
that is derived from a single-chain precursor; it is composed of the
heavy (82-140 kD) and the light (35-62 kD) chains. The light
chain is a catalytic subunit of enteropeptidase that consists of a
chymotrypsin-like serine protease domain. Human enteropeptidase mRNA
expression is limited to the proximal small intestine, and the protein
is found in enterocytes of the duodenum and proximal jejunum [6, 7]. Congenital deficiency of
enteropeptidase causes severe intestinal malabsorption with diarrhea,
vomiting, and growth failure. These abnormalities can be treated
successfully by supplementation with pancreatic extract [8, 9]. Mutations in the
proenteropeptidase gene are the molecular cause of congenital
enteropeptidase deficiency [10]. The high degree
of specificity exhibited by enteropeptidase makes it an ideal reagent
for cleaving bacterially produced fusion proteins. The expression and
purification of the catalytic subunit of bovine enteropeptidase from
E. coli [11], the methylotrophic yeast
Pichia pastoris [12], the filamentous
fungus Aspergillus niger [13], and
Saccharomyces cerevisiae [14] have been
described. In all cases, the recombinant proteins demonstrated high and
remarkably specific protease activity. Kinetic parameters of wild type
and various mutant forms have been determined, and this revealed a
critical substrate-binding segment outside the active site [15]. Finally, the crystal structure of bovine
enteropeptidase light chain complexed with an analog of the
trypsinogen-activation peptide has been solved to 2.3 Å
resolution [15].
In this paper, we report cloning and expression in E. coli of the synthetic gene encoding the human enteropeptidase catalytic subunit with subsequent refolding and purification. The influence of ionic strength, pH, different denaturing agents, and protease inhibitors has been investigated for the cleavage of synthetic substrates as well as of fusion proteins by L-HEP (recombinant human enteropeptidase light chain) and L-BEP (recombinant bovine enteropeptidase light chain purified from E. coli).
MATERIALS AND METHODS
Materials. Host strain E. coli BL21(DE3) and plasmid pET-32a were from Novagen (USA). GD4K-na (Gly-Asp-Asp-Asp-Asp-Lys-beta-naphthylamide), Z-Lys-SBzl (thiobenzyl benzyloxy-carbonyl-L-lysinate), STI (soybean trypsin inhibitor), aprotinin, leupeptin, antipain, E-64, benzamidine hydrochloride, chymostatin, pepstatin A, bestatin, TPCK (N-tosyl-L-phenylalanine chloromethyl ketone), TLCK (N-tosyl-L-lysine chloromethyl ketone), STI-agarose, and DTNB (5,5´-dithiobis-(2-nitrobenzoic acid)) were from Sigma (USA). Recombinant bovine enteropeptidase light chain, restriction enzymes, and DNA polymerase were from New England Biolabs (USA).
Synthesis of the L-HEP gene and its cloning into pET-32a vector. The DNA sequence encoding the light chain of human enteropeptidase (705 bp) was synthesized from 26 oligonucleotides (35-43 nucleotides each) by polymerase chain reaction (PCR) as described before [16]. The amplified DNA sequence was cleaved by BglII and HindIII and cloned into plasmid pET-32a at BglII/HindIII restriction sites downstream to the gene of the fusion partner thioredoxin following the DNA sequence encoding (Asp)4-Lys. The L-HEP gene sequence was confirmed by automatic sequencing on an ABI Prism-310 Genetic Analyzer (Applied Biosystems, USA).
Expression, renaturation, and purification of L-HEP. Escherichia coli BL21(DE3) cells were transformed by plasmid pET-32a/L-HEP. Expression, renaturation, and purification of L-HEP were done as described in our previous paper [16]. Total autocatalytic cleavage of the Trx/L-HEP (thioredoxin/human enteropeptidase light chain fusion protein) was observed after 2 h dialysis against 0.1 M Tris-HCl (pH 8.0) containing 1 mM CaCl2 at room temperature. NaCl to the final concentration of 500 mM was added to the dialyzed protein, and the active enzyme was purified by affinity chromatography on STI-agarose. The purified protein was stored in buffer containing 0.1 M Tris-HCl (pH 8.0), 50% glycerol, and 500 mM NaCl at -20şC. The concentration of L-HEP was determined spectrophotometrically by absorbance at 280 nm using a molar extinction coefficient of 54,450 M-1·cm-1 calculated from the known amino acid sequence using the ProtParam tool on the ExPASy server (www.expasy.org).
The rate of hydrolysis of the fluorogenic substrate GD4K-na was determined as described previously [17]. Assays were performed at 37şC in 1.5 ml buffer containing 0.01-1.0 mM GD4K-na, 25 mM Tris-HCl (pH 8.4), and 10% dimethylsulfoxide with different concentrations of CaCl2. The reaction was initiated by adding enteropeptidase (final concentration 0.7 nM). The rate of beta-naphthylamine formation was determined from the increment of fluorescence (lambdaex = 337 nm and lambdaem = 420 nm) monitored continuously for 5 min using MPF-4 fluorimeter (Hitachi, Japan).
Rates of hydrolysis of synthetic chromogenic tripeptides Z-Ala-X-Arg-pNA (benzyloxycarbonyl-L-alanine-X-L-arginine-p-nitroanilide) were measured in 1 ml buffer containing 0.1 M Tris-HCl (pH 8.0), 5% dimethylsulfoxide, with concentration range of the substrates 0.01-1 mM. Reactions were started by addition of L-HEP to the final concentration of 4 nM, and absorbance of the released p-nitroaniline was recorded at 405 nm continuously for 5 min. The kcat and Km values for all substrates were determined from Lineweaver-Burk plots.
Effects of protease inhibitors and denaturing reagents on L-HEP activity. The reaction of hydrolysis of thiobenzyl benzyloxycarbonyl-L-lysinate (Z-Lys-SBzl) was used for measurements of L-HEP activity in the presence of protease inhibitors and denaturing reagents as described previously [18]. Assays were performed at room temperature in 1.5 ml buffer containing 0.1 M Tris-HCl (pH 8.0), 260 µM DTNB, and 66 µM Z-Lys-SBzl. After addition of the enzyme (final concentration 0.5 nM) the rate of 3-carboxy-4-nitrothiophenoxide production was measured continuously for 5-30 min from the absorbance changes at 412 nm, using the extinction coefficient 13,600 M-1·cm-1.
Cleavage of fusion proteins by L-HEP and L-BEP. Trx/hEGF (thioredoxin/human epidermal growth factor fusion protein) was partially purified from inclusion bodies by affinity chromatography on Ni-NTA agarose. Digestions by L-HEP and L-BEP were held in buffer containing 50 mM Tris-HCl (pH 8.0) in the presence of different denaturing agents during 20 h at room temperature. After digestion, samples were analyzed on Tris-Tricine SDS-PAGE [19]. Protein electrophoretic profiles in gels were stained with Coomassie Blue G-250, and the bands were scanned using an ImageMaster VDS densitometer (Amersham Pharmacia Biotech, USA). The Trx/hEGF concentration was determined by the Bradford method [20].
RESULTS AND DISCUSSION
L-HEP gene expression in E. coli and L-HEP purification. The DNA sequence encoding the light chain of human enteropeptidase (GenBank Accession Number U09860) was synthesized from 26 oligonucleotides by PCR and cloned into plasmid pET-32a after the DNA sequence encoding enteropeptidase recognition site [16]. High level expression of Trx/L-HEP (100 mg from 100 ml culture) was achieved in E. coli strain BL21(DE3). Most (90%) of the fusion protein was insoluble and formed inclusion bodies. After solubilization of inclusion bodies in 6 M guanidine-HCl, Trx/L-HEP fusion protein was refolded using oxidized and reduced glutathione as described in our previous work [16]. Refolded protein cleaved autocatalytically. Active enzyme was purified by affinity chromatography on STI-agarose. This approach yielded more than 1 mg of highly active enzyme from 100 ml of E. coli culture (Table 1).
Table 1. Yield of purified active L-HEP
after refolding of Trx/L-HEP fusion protein

Note: Values are from five independent experiments.
Estimation of L-HEP and L-BEP kinetic parameters for cleavage of the specific substrate GD4K-na at different concentrations of Ca2+ and Na+. Conventionally, kinetic parameters for the specific substrate GD4K-na cleavage by bovine enteropeptidase light chain have been determined in the presence of 10 mM CaCl2 [15, 21]. Our measurements have shown that the maximum cleavage rate is observed at 0.02 mM CaCl2 both for L-HEP and L-BEP with the specificity constants (kcat/Km) of 3464 and 347 mM-1*sec-1, respectively. For hydrolysis of the substrate at 10 mM CaCl2, 4.5-fold lower values of specificity constants were observed (Table 2, Fig. 1a). It has been shown that Ca2+ mainly increases Km, while kcat remains practically unchanged. The rate of GD4K-na hydrolysis was inhibited with 100 and 250 mM NaCl by 70 and 90%, respectively (data not shown). Similar results were obtained for cleavage of Trx/hEGF fusion at different concentrations of CaCl2 (Fig. 1b). Even 1 mM CaCl2 slightly diminished the rate of Trx/hEGF hydrolysis, while 5 and 10 mM CaCl2 reduced it by 50 and 80%, respectively. The absence of Ca2+ stimulation of enteropeptidase activity is consistent with Leu and Thr at positions 70 and 80 in the polypeptide chain (chymotrypsin numbering), while Glu at these positions are involved in formation of a Ca2+ binding site in thrombin and trypsin [22].
Table 2. Kinetic parameters for the cleavage
of the specific substrate GD4K-na by L-HEP and L-BEP
Note: Values for Km and kcat are
expressed as mean ± S.D. of three independent determinations.
*Data from [15].
However, addition of 10 mM CaCl2 had no effect on the rate of hydrolysis of nonspecific substrates Z-Lys-SBzl and Z-Ala-X-Phe-Arg-pNA (data not shown), and NaCl at very high concentration (up to 1-5 M) increased (2-2.5-fold) the rate of their hydrolysis by the enteropeptidase (Table 3). This suggests that Ca2+ or Na+ successfully shields substrate (Asp)4 negatively charged side chains thereby impairing its binding to the positively charged substrate-binding RRRK site of the enzyme. One should note that despite high amino acid sequence homology of human and bovine enteropeptidase light chains (84%), L-HEP cleaved the specific substrate GD4K-na almost 10 times faster than L-BEP at any concentration of CaCl2.Fig. 1. Effect of CaCl2 on the hydrolysis rate of the GD4K-na peptide and Trx/hEGF fusion protein cleavage by L-HEP and L-BEP. a) The rate of GD4K-na hydrolysis at different concentrations of CaCl2 or 1 mM EDTA (point 0 mM in the diagram). b, c) Separate samples containing 15 µg of Trx/hEGF fusion were incubated with the same relative activity units of enzymes (L-HEP and L-BEP) for 16 h at 25°C in 0.1 M Tris-HCl, pH 8.0, at different concentrations of CaCl2 (b) or at 0.02 mM CaCl2 for various time intervals (c). Samples were analyzed by 12% Tris-Tricine SDS-PAGE and the gel was stained with Coomassie brilliant blue.
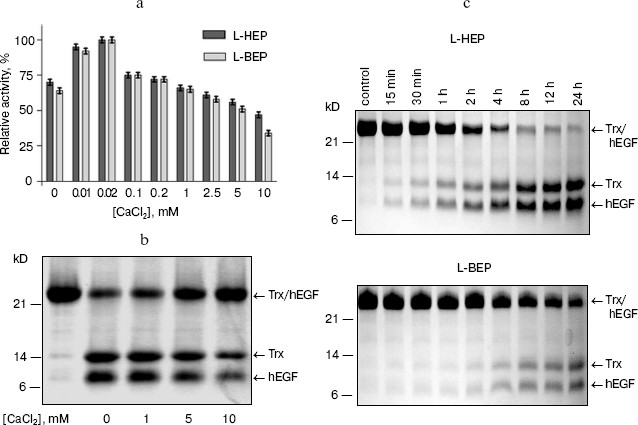
Table 3. Influence of different denaturing
agents on the rate of Z-Lys-SBzl hydrolysis by L-HEP
Note: Data are presented as means ± S.D. from at least three
independent experiments.
In our previous work, we have shown that the kinetic parameters for cleavage of the small ester Z-Lys-SBzl by human and bovine enzymes are practically identical [16]. This observation let us carry out a comparative analysis of cleavage rates of different fusion proteins, in particular, Trx/hEGF by L-HEP and L-BEP (Fig. 1c). After quantification of Trx/hEGF bands on SDS-PAGE gels by densitometry, the rates of hydrolysis of Trx/hEGF fusion by L-HEP and L-BEP (2.1 and 0.22 mmol/min per mg, respectively) were calculated. Additional studies with site-directed mutagenesis are required to explain higher (about an order of magnitude) activity of L-HEP in comparison with L-BEP.
Secondary substrate specificity of L-HEP. Kinetic parameters of L-HEP were determined for hydrolysis of the thioester Z-Lys-SBzl and some synthetic tripeptides (Table 4). Kinetic parameters for thioester Z-Lys-SBzl cleavage by L-HEP and L-BEP were practically identical. Hydrolysis of the tripeptide substrates For-Ala-Phe-Arg-pNA and For-Ala-Phe-Lys-pNA indicated a 2.5-fold Arg to Lys preference in P1 position. L-HEP demonstrated relatively high affinity to chromogenic tripeptides with hydrophobic residues in the P2 position (Phe > Tyr > Leu > Trp). For example, Z-Ala-Phe-Arg-pNA was hydrolyzed with quite high specificity constants kcat/Km 260 and 140 mM-1·sec-1 by L-HEP and L-BEP, respectively. We believe that a substrate with even greater specificity constant can be obtained by optimizing amino acids at positions P3 and P4 of the substrate. Similar value of kcat/Km = 212 mM-1·sec-1 was obtained for native porcine enteropeptidase toward Z-Phe-Arg-MCA [23].
Table 4. Kinetic parameters for the cleavage
of nonspecific substrates by L-HEP
Note: Data are presented as mean ± S.D. from at least three
independent experiments.
Calcium ions (up to 20 mM) had no effect on hydrolysis rate of Z-Lys-SBzl and tripeptides, and NaCl (1-3 M) only stimulated 2.0-2.5 times the rate of hydrolysis (Table 3). All hereinabove data indicated that the binding site for Z-Ala-X-Arg-pNA tripeptides differs from that for the specific substrate GD4K-na. It remains possible that other targets playing an important physiological role may exist for the human enteropeptidase proteolytic domain under certain conditions.
Interestingly, in the phylogenetic tree of 84 serine proteases of the chymotrypsin family enteropeptidase was found in one group with hepsin, plasma kallikrein, prostasin, and factor XI, which prefer Arg and Phe at the P1 and P2 positions of substrates, respectively [22]. This suggests that P1Arg-P2Phe secondary substrate specificity was the original one for enteropeptidase during evolution. Detailed analysis of the evolution of trypsinogen activation peptide demonstrated that the (Asp)4-Lys sequence in mammalian trypsinogen has evolved to inhibit autoactivation and enhance cleavage by enteropeptidase [24].
Influence of denaturing agents on the enzymatic activity of L-HEP. The stability of L-HEP to denaturing agents was examined using Z-Lys-SBzl (Table 3) and Trx/hEGF fusion protein (Fig. 2) as substrates. The enzyme appeared highly resistant to detergents like Triton X-100, Tween 20, and SDS. L-HEP hydrolyzed Z-Lys-SBzl in 20% ethanol preserving about 60% of activity. High concentrations of imidazole (250 mM) and dithiothreitol (20 mM), usually used in purification of fusion proteins, were acceptable for cleavage of both Z-Lys-SBzl and Trx/hEGF fusion proteins. High concentrations of NaCl (up to 5 M) or KCl (up to 3 M) only stimulated cleavage of the nonspecific substrates Z-Lys-SBzl and Z-Ala-Phe-Arg-pNA, while cleavage of the specific substrate GD4K-na was strongly inhibited even by low concentrations of NaCl (100 mM and more, data not shown).
Effect of protease inhibitors on L-HEP. The effect of various protease inhibitors on L-HEP was studied using the thioester Z-Lys-SBzl as a substrate (Table 5). The activity of enteropeptidase was strongly inhibited by trypsin-like serine protease inhibitors. STI and aprotinin inhibited the enzymes in a time-dependent manner (with equilibrium established in 30-60 min), while leupeptin, antipain, and benzamidine did it instantaneously. L-HEP activity was inhibited very poorly by TLCK and was practically not influenced by chymotrypsin-like protease (chymostatin), aminoprotease (bestatin), metalloprotease (EDTA), or aspartic protease (pepstatin) inhibitors. Surprisingly, cysteine protease inhibitor E-64 inhibited L-HEP efficiently. Earlier the same inhibition by E-64 was demonstrated for porcine enteropeptidase [23] and beta-trypsin [25]. It has been shown by computer modeling that inhibition of beta-trypsin was particularly determined by the interaction of the guanidine-terminal group of E-64 with S1-Asp189 side chain. In contrast, in cysteine proteinases the epoxide ring from the opposite terminus of E-64 reacts with the catalytic site thiolate anion [26]. Inhibition of enteropeptidase can be additionally related to electrostatic interaction of the carboxy-terminal group of E-64 with the 96-99 (RRRK) loop of L-HEP.Fig. 2. Effect of different denaturing agents on the cleavage rate of Trx/hEGF fusion protein by L-HEP. Samples containing 15 µg of Trx/hEGF fusion were incubated for 20 h at 25°C in buffer containing 0.1 M Tris-HCl (pH 8.0) and different concentrations of imidazole, NaCl, or Triton X-100.
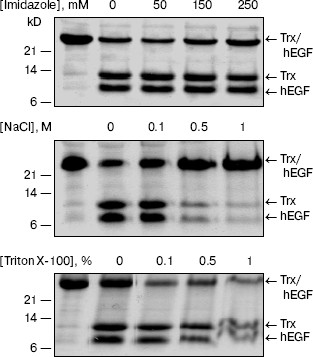
Table 5. Effects of protease inhibitors on
hydrolytic activity of L-HEP
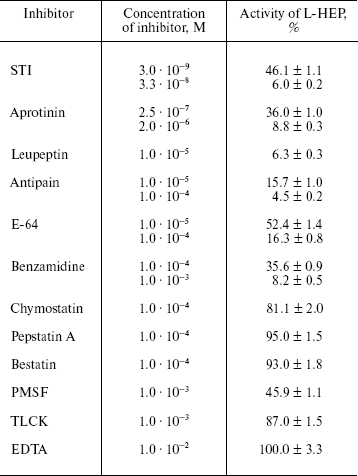
Note: Data are presented as mean ± S.D. from at least three
independent experiments.
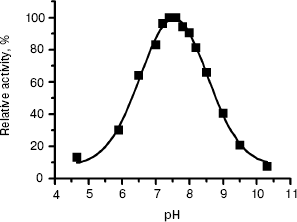
The pH dependence of L-HEP enzymatic activity was measured for hydrolysis of Z-Lys-SBzl as described in “Materials and Methods”. The enzyme cleaved it over a broad pH range with pH optimum 7.5 and 50% activity at pH 6.3 and 8.7 (Fig. 3).
Thus, we have obtained and characterized for the first time the human enteropeptidase catalytic subunit. Our preparation retained 100% activity during storage at -20°C for at least for 1.5 years. High activity, specificity, and stability of human enteropeptidase make it a very useful reagent in biotechnology for the cleavage of fusion proteins.Fig. 3. The pH dependence of Z-Lys-SBzl hydrolysis by L-HEP. Data (filled squares) were treated to fit a Gaussian curve with pH optimum 7.54 ± 0.02.
We thank Anna Gorozhanina, a student from the Moscow State University Department of Bioengineering, for skilful technical assistance.
The study was supported by the Russian Academy of Sciences Program on Molecular and Cellular Biology.
REFERENCES
1.Kunitz, M. (1939) J. Gen. Physiol.,
22, 429-446.
2.Baratti, J., Maroux, S., Louvard, D., and
Desnuelle, P. (1973) Biochim. Biophys. Acta, 315,
147-161.
3.Anderson, L. E., Walsh, K. A., and Neurath, H.
(1977) Biochemistry, 16, 3354-3360.
4.Liepnieks, J. J., and Light, A. (1979) J. Biol.
Chem., 254, 1677-1683.
5.Magee, A. I., Grant, D. A., and Taylor, J. H.
(1981) Clin. Chim. Acta, 115, 241-254.
6.Lojda, Z., and Gossrau, R. (1983)
Histochemistry, 78, 251-270.
7.Mitoshi, Y., Onishi, T., Sano, T., and Komi, N.
(1990) Gastroenterol. Jpn., 25, 320-327.
8.Hadorn, B., Tarlow, M. J., Lloyd, J. K., and Wolff,
O. H. (1969) Lancet, 1, 812-813.
9.Haworth, J. C., Gourley, B., Hadorn, B., and
Sumida, C. (1971) J. Pediatr., 78, 481-490.
10.Holzinger, A., Maier, E. M., Buck, C.,
Mayerhofer, P. U., Kappler, M., Haworth, J. C., Moroz, S. P., Hadorn,
H. B., Sadler, J. E., and Roscher, A. A. (2002) Am. J. Hum.
Genet., 70, 20-25.
11.Collins-Racie, L. A., McColgan, J. M., Grant, K.
L., DiBlasio-Smith, E. A., McCoy, J. M., and LaVallie, E. R. (1995)
Biotechnology (NY), 13, 982-987.
12.Vozza, L. A., Wittwer, L., Higgins, D. R.,
Purcell, T. J., Bergseid, M., Collins-Racie, L. A., LaVallie, E. R.,
and Hoeffler, J. P. (1996) Biotechnology (NY), 14,
77-81.
13.Svetina, M., Krasevec, N., Gaberc-Porekar, V.,
and Komel, R. (2000) J. Biotechnol., 76, 245-251.
14.Choi, S. I., Song, H. W., Moon, J. W., and Seong,
B. L. (2001) Biotechnol. Bioeng., 75, 718-724.
15.Lu, D., Futterer, K., Korolev, S., Zheng, X.,
Tan, K., Waksman, G., and Sadler, J. E. (1999) J. Mol. Biol.,
292, 361-373.
16.Gasparian, M. E., Ostapchenko, V. G., Schulga, A.
A., Dolgikh, D. A., and Kirpichnikov, M. P. (2003) Protein. Exp.
Purif., 31, 133-139.
17.Grant, D. A., and Hermon-Taylor, J. (1979)
Biochim. Biophys. Acta, 567, 207-215.
18.Green, G. D., and Shaw, E. (1979) Analyt.
Biochem., 93, 223-226.
19.Schagger, H., and von Jagow, G. (1987) Analyt.
Biochem., 166, 368-379.
20.Bradford, M. M. (1976) Analyt. Biochem.,
72, 248-254.
21.Lu, D., Yuan, X., Zheng, X., and Sadler, J. E.
(1997) J. Biol. Chem., 272, 31293-31300.
22.Rose, T., and Di Cera, E. (2002) J. Biol.
Chem., 277, 19243-19246.
23.Matsushima, M., Ichinose, M., Yahagi, N.,
Tsukada-Kato, S., Miki, K., Omata, M., Kim, Y. T., Ito, H., Takahashi,
T., Sakurai, Y., Tsuchiya, Y., Athauda, S. B., Inoue, H., and
Takahashi, K. (1999) J. Biochem. (Tokyo), 125,
947-951.
24.Chen, J. M., Kukor, Z., Le Marechal, C., Toth,
M., Tsakiris, L., Raguenes, O., Ferec, C., and Sahin-Toth, M. (2003)
Mol. Biol. Evol., 20, 1767-1777.
25.Sreedharan, S. K., Verma, C., Caves, L. S.,
Brocklehurst, S. M., Gharbia, S. E., Shah, H. N., and Brocklehurst, K.
(1996) Biochem. J., 316, 777-786.
26.Varughese, K. I., Ahmed, F. R., Carey, P. R.,
Hasnain, S., Huber, C. P., and Storer, A. C. (1989)
Biochemistry, 28, 1330-1332.