Role of Hsp70 (DnaK-DnaJ-GrpE) and Hsp100 (ClpA and ClpB) Chaperones in Refolding and Increased Thermal Stability of Bacterial Luciferases in Escherichia coli Cells
G. B. Zavilgelsky1*, V. Yu. Kotova1, M. M. Mazhul'2, and I. V. Manukhov1
1State Scientific Center of the Russian Federation GosNIIgenetika, 1-yi Dorozhnyi Proezd 1, Moscow, 117545 Russia; fax: (095) 315-0501; E-mail: zavilgel@genetika.ru2Department of Microbiology, School of Biology, Lomonosov Moscow State University, Moscow, 119899 Russia
* To whom correspondence should be addressed.
Received April 5, 2002; Revision received April 17, 2002
The role of chaperones Hsp70 (DnaK-DnaJ-GrpE) and Hsp100 (ClpA-ClpB-ClpX) in refolding of thermoinactivated luciferase from the marine bacterium Photobacterium fischeri and the terrestrial bacterium Photorhabdus luminescens has been studied. These luciferases are homologous, but differ greatly in the rate of thermal inactivation and the rate constant for the luminescence reaction. It was shown that refolding of thermoinactivated luciferases is completely determined by the DnaK-DnaJ-GrpE system. However these luciferases markedly differ in the rate and degree of refolding. The degree of refolding of thermolabile “quick” Ph. fischeri luciferase reaches 80% of the initial level over several minutes, whereas renaturation of thermostable “slow” Ph. luminescens luciferase proceeds substantially slower (the degree of renaturation reaches only ~7-8% of the initial level over tens of minutes). The measurement of the rate of thermal inactivation of luciferases in vivo in the cells of Escherichia coli wild strain and strains containing mutations in genes clpA, clpB, clpX showed that Ph. luminescens luciferase revealed reduced thermostability in mutant strain E. coli clpA-. It was shown that this effect was not connected with DnaK-dependent refolding. In the case of thermolabile Ph. fischeri luciferase, mutation in gene clpA has no effect on the shape of the curve of thermal inactivation. These data suggest that denatured Ph. luminescens luciferase has enhanced affinity with respect to chaperone ClpA in comparison with DnaK, whereas thermolabile Ph. fischeri luciferase is characterized by enhanced affinity with respect to chaperone DnaK. Denatured luciferase bound to ClpA does not aggregate and following refolding proceeds probably spontaneously and very quickly (over 1-2 min). It is evident that the process under discussion requires ATP, since the addition of uncoupler of oxidative phosphorylation carbonyl cyanide 3-chlorophenylhydrazone results in a sharp decrease in thermal stability of luciferase to the level typical of the enzyme in vitro. The enhanced thermosensitivity of luciferases was observed also in E. coli containing mutations in gene clpB. However, this effect, which takes place for Ph. fischeri luciferase as well as for Ph. luminescens luciferase, is determined by DnaK-dependent refolding and probably connected with the ability of chaperone ClpB to provide disaggregation of the proteins, resulting in their interaction with chaperones of the Hsp70 family (DnaK-DnaJ-GrpE).
KEY WORDS: chaperones, DnaK, ClpA, ClpB, refolding, luciferase
In a bacterial cell molecular chaperones participate in many processes connected with correct folding of nascent polypeptide chains during and after translation, with recovery of the native structure of partially denatured proteins (refolding), with disaggregation of protein supramolecular structures formed in the cell during heat shock, with translocation through membranes, and so on [1-5]. Firefly luciferase is widely used as a model protein for study of the role of various chaperone systems in folding and refolding of proteins [6-8]. In particular, it was shown that refolding of denatured firefly luciferase requires a combined action of chaperones belonging to the Hsp70 family [8, 9].
In the present paper the influence of bacterial chaperones of Hsp70 family (DnaK-DnaJ-GrpE) and Hsp100 family (ClpA, ClpB, ClpX) on thermal stability and refolding of bacterial luciferases has been studied. Bacterial luciferases are among the group of thermolabile proteins, which undergo inactivation at 37-42°C. This circumstance allows the peculiarities of thermal inactivation and refolding of these enzymes immediately in a bacterial cell to be studied. In contrast to monomeric firefly luciferase, bacterial luciferases are heterodimers consisting of alpha- and beta-subunits with molecular masses of ~40 and 35 kD, respectively. Subunits of luciferases are homologous. The active site of the enzyme is located on the alpha-subunit. The role of the beta-subunit remains unclear, but its presence in the complex enhances greatly the quantum yield of the reaction [10, 11]. In the present paper thermolabile luciferase from the marine bacterium Photobacterium fischeri and thermostable luciferase from the bacterium Photorhabdus luminescens were used. Earlier we showed that DnaK-dependent refolding is more efficient in the case of thermolabile luciferase from Ph. fischeri [12]. In the present work we have studied the role of chaperones ClpA and ClpB in refolding of Ph. fischeri and Ph. luminescens luciferases as well as the influence of these chaperones on thermal stability of luciferases in the cells of Escherichia coli. ClpA and ClpB ATPases belong to the family of chaperones Hsp100 and play an important role in unfolding and proteolysis of some bacterial proteins [13]. It was shown also that chaperone ClpB participates in refolding of proteins, providing disaggregation of denatured proteins in vitro as well as in vivo under the action of Hsp70 chaperones (DnaK-DnaJ-GrpE) [14-16]. As for ClpA, it was shown in vitro that this chaperone forms a complex with denatured polypeptides irrespective of whether or not these proteins are the substrates of ClpAP-proteinase. However ClpA does not participate in DnaK-dependent refolding of proteins [17, 18].
MATERIALS AND METHODS
Bacterial strains and plasmids. E. coli K12 SG20250 DeltalacU169 araD flbB relA clpA+ clpB+ and its insertion mutants SG22099 clpA::kan clpB+ and SG22100 clpA+ clpB::kan were obtained from S. Gottesman and Y. Zhou (USA) [19]. E. coli K12 MG1655 and its deletion mutant PK202 DeltadnaK14 DeltadnaJ14 dksA::kan were obtained from E. Craig (USA) [20]. E. coli K12 AB1157 and its deletion mutants NK113 DeltaclpP::cat and NK114 DeltaclpX::kan were obtained from N. E. Murray [21]. E. coli K12 TG1 was obtained from the collection of GosNIIgenetika.
Hybrid plasmid pF2 with genes luxAB Ph. fischeri placed under promotor Plac was constructed by recloning of DNA fragment from plasmid pF1 containing full lux-operon of Ph. fischeri [22]. Hybrid plasmid pXen4 contains fragment of DNA with genes luxAB recloned from hybrid plasmid pXen7 with full lux-operon of Ph. luminescens Zm1 [23].
Isolation and purification of bacterial luciferases. Bacterial cells containing hybrid plasmids with genes luxAB were grown in L-broth with ampicillin (100 µg/ml) at 28°C on a shaker till absorbance reached 2.5-3.0. After centrifugation the cells were resuspended in a tenth of the solution volume containing 50 mM phosphate buffer, pH 7.0, and 20 mM beta-mercaptoethanol. Protein extract was obtained by treatment of the suspension of the bacterial cells (portion-wise in 30 ml) by ultrasound (44 kHz; 6 times for 20 sec each time) with cooling in ice. The sonicated suspension was centrifuged for 30 min at 30,000g to remove cell fragments. Further purification was achieved by passing the protein through the column with Sephadex G-25; 0.1 M potassium phosphate buffer, pH 7.0, was used for elution. To detect the fractions containing luciferase, the bioluminescence method was applied. The content of protein in a tube after purification on the column with Sephadex G-25 was ~100 µg/ml.
Measurement of the intensity of bioluminescence and the rate constant of the enzymatic reaction. Bacterial luciferases (E) catalyze oxidation of long-chain aldehyde (RCHO) by oxygen (O2) of air with the participation of reduced flavin mononucleotide (FMNH2): FMNH2 + RCHO + O2 --> FMN + RCOOH + H2O + a quantum of light (490 nm). The reaction proceeds through several stages:
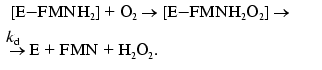 (2)
(2)Complex [E-FMNH2O2] breaks down through the dark pathway (2) with the rate constant kd or interacts with aliphatic aldehyde with the formation of ternary complex (3) which breaks down with generation of a quantum of light (k1 is the rate constant of this reaction) (4):
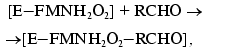 (3)
(3)
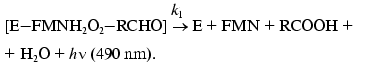 (4)
(4)The fact that the reaction is accompanied by generation of a quantum of blue-green light (490 nm) provides a possibility of simple and quick measuring the enzymatic activity of luciferase in the model systems in vitro as well as in vivo without destroying the bacterial cell.
The rate constant of luminescence decay (kt) is determined by the following expression: kt = (klA + kdKa)/(Ka + A), where Ka is the dissociation constant for the ternary complex and A is the concentration of aliphatic aldehyde [24]. We used high concentrations of aliphatic aldehyde n-decanal (A >> Ka), where the following relationship was valid: kt ~= kl.
Bioluminescence of the bacterial cells and cell-free extracts was measured with the aid of a model 1251 luminometer (Finland) at high concentration of n-decanal (6.8 µM) at 20°C. The reaction mixture in the in vitro measurements contained 0.1 M potassium phosphate buffer, pH 7.0, 20 mM beta-mercaptoethanol, 20 µM FMN (Sigma, USA), and 6.8 µM n-decanal (Sigma). The reaction was initiated by the addition of 20 µl of 0.1% solution of sodium dithionite (Fisher, Germany), which reduces FMN to FMNH2, to the cell with the reaction mixture placed immediately before the photomultiplier [25]. The final volume of the reaction mixture was 1 ml. Oxidation of FMNH2 proceeds quickly in comparison with the life time of intermediates of the luminescence reaction. Therefore, the enzyme exerts only one turnover, the intensity of emission reaching a maximum for the first several seconds. Further decrease in the reaction rate and the intensity of luminescence (I) proportional to the reaction rate is described by the first order kinetics: I = I0exp(-kt) [26]. The rate constant of the luminescence decay (k = kl) was determined by calculating the slope of the straight line in coordinates {lnI; t} (t is time). In this procedure we used only part of the decay curve corresponding to the interval of the I/Imax values from 0.8 to 0.2 (Imax is the maximum value of the intensity of luminescence).
Thermal inactivation and refolding of luciferases. Thermal inactivation of luciferases in vivo and in vitro was carried out in a water bath at a fixed temperature. In the experiments in vivo chloramphenicol (167 µg/ml) was added to the suspension of the cells to suppress the protein synthesis. To decrease ATP concentration in bacteria, we used uncoupler of oxidative phosphorylation carbonyl cyanide 3-chlorophenylhydrazone (CCCP; Sigma) as well as FCCP. ATP concentration in the bacterial cells was measured using firefly luciferase [27]. Refolding of luciferases was carried out at room temperature or at a temperature that is optimum for the given luciferase: 25°C for Ph. fischeri luciferase and 35°C for Ph. luminescens luciferase. Bioluminescence was measured in aliquots (200 µl) withdrawn at certain intervals. Preliminary “heat shock” was carried out by incubation of E. coli cells in the thermostat at 42°C in the absence of chloramphenicol.
RESULTS AND DISCUSSION
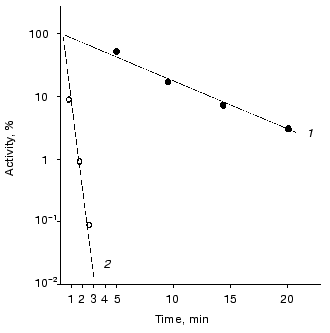
Thermal stability and rate constant for the luciferase reaction. Luciferases of Ph. fischeri and Ph. luminescens are highly homologous: 66% of the total number of the amino acid residues in the alpha-subunit (359 residues) and 52% of the total number of amino acid residues in beta-subunit (324 residues) are identical [28]. However, these luciferases differ greatly in the rate of thermal inactivation and the rate constant for the luciferase reaction. Figure 1 shows the kinetic curves of thermal inactivation of the purified preparations of luciferases from Ph. fischeri and Ph. luminescens at 43.5°C. The comparison of the slopes of the kinetic curves allows the following conclusion to be made: Ph. luminescens luciferase loses the activity ~15 times slower than Ph. fischeri luciferase. The values of the rate of the enzymatic reaction for these luciferases differ by a factor of ~10-15 (table). The table summarizes the average values of the rate constants for the luciferase reaction and the rate constants of thermal inactivation (averaging over the results of five experiments in each case). It should be noted that the above characteristics of the luciferases are determined by the alpha-subunit, where the active site is located [24].
Rate constants for the enzymatic reaction and rate constants of thermal inactivation at 43.5°C for bacterial luciferasesFig. 1. Kinetics of thermal inactivation of Ph. fischeri and Ph. luminescens luciferases (2 and 1, respectively) in vitro at 43.5°C. The ordinate axis shows the enzymatic activity of luciferase in percent of the initial level. The abscissa axis is the time of thermal inactivation.

Refolding of thermoinactivated luciferases. Registration of the kinetics and degree of refolding of thermoinactivated luciferases was carried out in vivo in the cells of E. coli K12 MG1655 and its deletion mutant PR202 DeltadnaKJ14 containing hybrid plasmid with genes luxAB (pF2 or pXen4). Bacteria were grown at 28°C to the middle of the logarithmic phase. Then chloramphenicol (167 µg/ml) was added to the suspension of the cells to suppress the following synthesis of protein and the culture was transferred into a water bath at elevated temperature for inactivation of luciferase. Temperature and time of incubation for each luciferase were selected so that the final level of the enzymatic activity was 0.01-0.1% of the initial level. Refolding of inactivated luciferases was carried out at optimum temperatures: at room temperature or at 25°C for Ph. fischeri luciferase or at 35°C for Ph. luminescens luciferase. Figure 2 shows the time course of renaturation of luciferases (in percent of the initial level). As can be seen from the figure, refolding is completely lacking in the cells of the mutant strain PK202 DeltadnaKJ. In the cells of E. coli MG1655 dnaK+ refolding takes place for both types of luciferases. However, in the case of thermolabile Ph. fischeri luciferase the degree of renaturation is markedly higher (80% of the initial level) than that for thermostable Ph. luminescens luciferase (6-8% of the initial level). The kinetics of refolding of these luciferases are different: renaturation of thermolabile luciferase proceeds very quickly and reaches 5% level over 1-2 min, whereas refolding of thermostable luciferase is a substantially slower process (the degree of renaturation reaches the same level over 10-20 min).
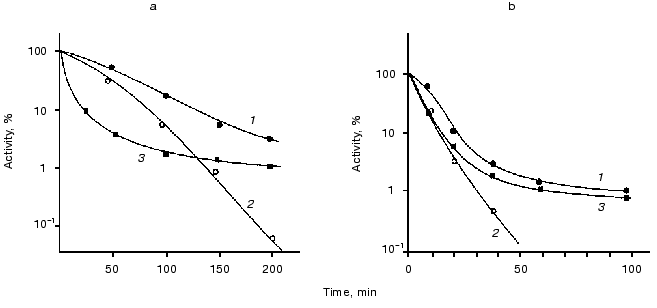
Influence of chaperones ClpA and ClpB on thermostability of luciferases. The goal of the next series of the experiments was to study the kinetics of thermal inactivation of luciferases in vivo in E. coli clpA+clpB+ and E. coli containing mutations in genes clpA and clpB. Figure 3 shows the kinetic curves of thermal inactivation of Ph. luminescens luciferase (Fig. 3a) and Ph. fischeri luciferase (Fig. 3b) in the cells of E. coli 20250 clpA+clpB+, 22099 clpA-clpB+ and 22100 clpA+clpB-. We discovered two characteristic features of the kinetics of thermal inactivation: 1) the shape of the kinetic curves obtained in strains clpB+ (clpA+clpB+ and clpA-clpB+) suggests the formation of a “thermoresistant fraction”, the appearance of this fraction becoming especially notable on long incubation of the cells at high temperature (Fig. 3, a and b, curves 1 and 3); 2) the thermostable luciferase from Ph. luminescens 22099 clpA-clpB+ reveals an enhanced thermosensitivity in the early stages of thermal inactivation (curve 3 in Fig. 3a); for thermolabile Ph. fischeri luciferase the effect of mutation in gene clpA on the shape of the inactivation curve is slight (curve 3 in Fig. 3b). In the case of E. coli mutant 22100 clpA+clpB- the “resistant fraction” is lacking (curves 2 in Fig. 3, a and b). ClpB-dependent “thermoresistant fraction” is connected with DnaKJ-refolding, because in strain PK202 DeltadnaKJ14 this fraction is also lacking (Fig. 4). Thus, one can conclude that the enhanced thermostability of Ph. luminescens luciferase observed on long incubation of the cells at high temperature is mainly determined by the combined action of chaperones DnaK-DnaJ-GrpE and ClpB and probably connected with refolding of aggregates of proteins [14-16]. It should be noted that in the case of thermolabile Ph. fischeri luciferase the portion of ClpB-dependent “resistant fraction” is a little higher. This is because refolding exerted by chaperones Hsp70 is more efficient (Fig. 2). As for the enhanced thermosensitivity of Ph. luminescens luciferase in the mutant strain 22099 clpA-clpB+, one can assume that chaperone ClpA has a protective effect during thermal inactivation. This effect coincides with the protective action of ClpA in the model system in vitro [29]. Mutation in gene clpX has no effect on the kinetics of thermal inactivation and refolding of luciferases (data not presented).Fig. 2. Kinetics of refolding of thermoinactivated Ph. fischeri luciferase (a) and Ph. luminescens luciferase (b) in cells of E. coli MG1655 dnaK+ (1) and E. coli PK202 DeltadnaKJ14 (2). The ordinate axis is the enzymatic activity of luciferase in percent of the initial level. The abscissa axis is the time of incubation at the temperature that is optimum for the particular luciferase (25°C for Ph. fischeri luciferase and 35°C for Ph. luminescens luciferase).
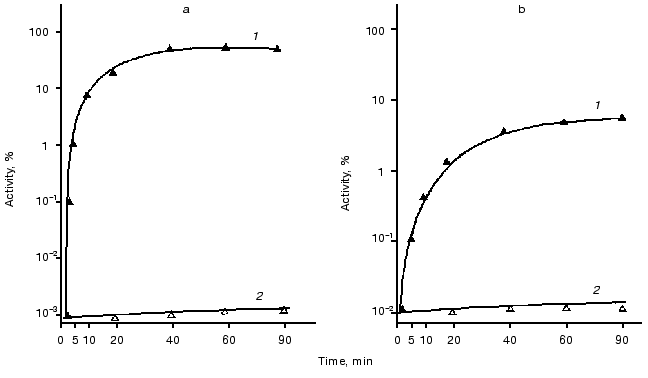
Fig. 3. Effect of mutations in genes clpA and clpB on the rate of thermal inactivation of Ph. luminescens (a) and Ph. fischeri (b) luciferases in the cells of E. coli 20250 clpA+clpB+ (1), E. coli 22100 clpA+clpB- (2), and E. coli 22099 clpA-clpB+ (3). The cells were incubated in a water bath at 45.5 (a) or 38°C (b). The ordinate axis corresponds to the luciferase activity (intensity of bioluminescence) in percent of the initial level. Chloramphenicol (167 µg/ml) was added in the suspension of the cells to suppress synthesis of proteins.
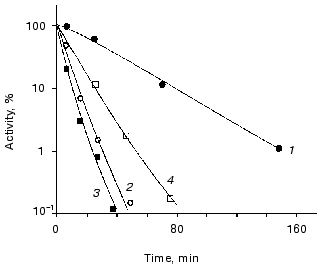
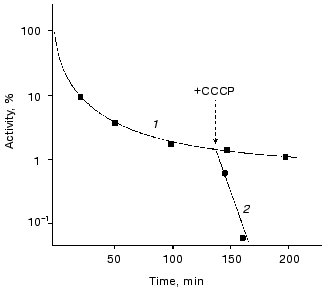
The role of ATP in enhancing thermostability of luciferases in vivo. As known, the effective action of chaperones ClpA and DnaKJE-ClpB requires large amounts of ATP [13-15, 30-33]. To reduce the content of ATP in the cells of E. coli, in the next series of the experiments we treated the cells with uncoupler of oxidative phosphorylation CCCP. As can be seen from the data shown in Fig. 5, the addition of CCCP to the suspension of the cells 22099 clpA-clpB+ (pXen4) at the instant of the formation of the “resistant fraction” on the kinetic curve of inactivation of luciferase (45°C) results in the rapid disappearance of the latter: the residual activity of Ph. luminescens luciferase decreased by several orders of magnitude over 7-10 min (the addition of CCCP is designated by the arrow). Thus, refolding of luciferase exerted by DnaKJE-ClpB requires substantial amounts of ATP. CCCP acts in analogous fashion when this uncoupler is added to the cells of E. coli clpA-clpB+ and clpA+clpB+ in the early stages of inactivation of the enzyme. To eliminate ATP-dependent refolding exerted by chaperones DnaKJE, in the next series of experiments CCCP was added to the cells PK202 DeltadnaKJ14 where refolding is lacking. Figure 4 shows the kinetic curves of thermal inactivation of Ph. luminescens luciferase in the strain of E. coli PK202 under standard conditions and in the case when CCCP was added in the early stages of thermal inactivation (43.5°C). Thermal inactivation shows a great increase in velocity and the kinetics of inactivation resemble those for the experiments in vitro. It should be noted that the addition of CCCP to the suspension of bacteria in L-broth at room temperature has no effect on the activity of luciferase and intensity of bioluminescence of the suspension throughout the experiment. The addition of CCCP results in the decrease in the concentration of ATP in the bacterial cells practically to the background level over some minutes at room and elevated temperatures. Thus, one can conclude that the low level of ATP in the cell is crucial for chaperone-dependent refolding of the protein, but not for the bioluminescence reaction. Analogously, uncoupler of oxidative phosphorylation FCCP enhances the rate of thermal inactivation of Ph. luminescens luciferase in the cells PK202 (data not presented). The addition of glucose to L-broth decreases the efficiency of action of CCCP. This result may be ascribed to the additional synthesis of ATP in the glycolytic process, which is independent of ATP-synthases (curve 4 in Fig. 4).Fig. 4. Kinetics of thermal inactivation of Ph. luminescens luciferase in the cells of E. coli P202 DeltadnaKJ at 43.5°C in the absence (1) and in the presence of 50 µM CCCP (2). Curve 3 corresponds to the isolated luciferase in vitro. Curve 4 was obtained in the presence of 50 µM CCCP and 0.6% glucose. The ordinate axis corresponds to the intensity of bioluminescence of the suspension of the cells in percent of the initial level.
As a working hypothesis, one can assume that there are two alternative pathways of renaturation of denatured Ph. luminescens and Ph. fischeri luciferases in the cells of E. coli. It is probable that thermolabile luciferase from Ph. fischeri has the higher affinity in respect to chaperone DnaK in comparison with ClpA. Therefore, the main pathway of renaturation of this protein should proceed with the participation of chaperones of the DnaK-DnaJ-GrpE system (direct refolding) or needs the additional participation of chaperone ClpB (disaggregation followed by refolding). It is likely that thermostable Ph. luminescens luciferase has enhanced affinity with respect to chaperone ClpA in comparison with DnaK. Therefore, for this enzyme DnaK-dependent refolding is of little importance. However the protein bound to chaperone ClpA does not aggregate. Subsequent hydrolysis of ATP provides release of denatured protein, the latter most probably spontaneously refolding into the native structure.Fig. 5. Effect of uncoupler of oxidative phosphorylation CCCP on the rate of thermal inactivation of Ph. luminescens luciferase in the cells of E. coli 22099 clpA-clpB+ in the region of the appearance of the “thermoresistant fraction” in the absence (1) and in the presence of 50 µM CCCP (2). The arrow indicates the point in time at which CCCP was added. Temperature of inactivation was 45.5°C. The ordinate axis corresponds to the intensity of bioluminescence of the suspension of the cells in percent of the initial level.
The authors are grateful to S. Gottesman and Y. Zhou (USA) for their gifts of clpAB mutants and fruitful discussion of the results of the present work, to E. Graig (USA) for the gift of strain P202, to N. Murray (Great Britain) for gifts of mutants clpXP, and to A. V. Bogachev for help in measuring the ATP content in the bacterial cells (Lomonosov Moscow State University) and for valuable advice and discussion of the results.
This study was supported by the Russian Foundation for Basic Research (grants 00-15-97855 and 00-04-48755).
REFERENCES
1.Hartl, F. U. (1996) Nature,381,
571-580.
2.Bukau, B., and Horwich, A. L. (1998)
Cell,92, 351-366.
3.Schroder, H., Langer, T., Hartl, F.-U., and Bukau,
B. (1993) EMBO J., 12, 4137-4144.
4.Lund, P. A. (2001) Adv. Microb. Physiol.,
44, 93-140.
5.Teter, S. A., Houry, W. A., Ang, D., Tradler, T.,
Rockabrandt, D., Fischer, G., Blum, P., Georgopoulos, C., and Hartl, F.
U. (1999) Cell,97, 755-765.
6.Frydman, J., Nimmesgern, E., Ohtsuka, K., and
Hartl, F.-U. (1994) Nature,370, 111-117.
7.Gamer, J., Multhaup, G., Tomayasa, T., McCarty, J.
S., Rudiger, S., Schonfeld, H. J., Schirra, C., Bujard, H., and Bukau,
B. (1996) EMBO J., 15, 607-617.
8.Hesterkamp, T., and Bukau, B. (1998) EMBO
J.,17, 4818-4828.
9.Goffin, L., and Georgopoulos, C. (1998) Mol.
Microbiol.,30, 329-340.
10.Fisher, A. J., Raushel, F. M., Baldwin, T. O.,
and Rayment, I. (1995) Biochemistry,34, 6581-6586.
11.Baldwin, T. O., Christopher, J. A., Raushel, F.
M., Sinclair, J. F., Ziegler, M. M., Fisher, A. J., and Rayment, I.
(1995) Curr. Opin. Struct. Biol., 5, 798-809.
12.Manukhov, I. V., Eroshnikov, G. E., Vyssokikh, M.
Yu., and Zavilgelsky, G. B. (1999) FEBS Lett.,448,
221-223.
13. Hoskins, J. R., Sharma, S., Sathyanarayana, B. K., and
Wickner, S. (2001) Adv. Protein Chem., 59, 413-429.
14.Goloubinoff, P., Mogk, A., Ben Zvi, A. P.,
Tomoyasu, T., and Bukau, B. (1999) Proc. Natl. Acad. Sci.
USA,96, 13732-13737.
15.Zolkiewski, M. (1999) J. Biol.
Chem.,274, 28083-28086.
16.Mogk, A., Tomoyasu, T., Goloubinoff, P., Rudiger,
S., Roder, D., Langen, H., and Bukau, B. (1999) EMBO
J.,18, 6934-6949.
17.Weber-Ban, E. U., Reid, B. G., Miranker, A. D.,
and Horwich, A. L. (1999) Nature, 401, 90-93.
18.Hoskins, J. R., Singh, S. K., Maurizi, M. R., and
Wickner, S. (2000) Proc. Natl. Acad. Sci. USA,97,
8892-8897.
19.Gottesman, S., Roche, E., Zhou, Y., and Sauer, R.
T. (1998) Genes Dev.,12, 1338-1347.
20.Kang, P.-J., and Craig, E. A. (1990) J.
Bacteriol., 172, 2055-2064.
21.Makovets, S., Titheradge, A. J. B., and Murray,
N. E. (1998) Mol. Microbiol., 28, 25-35.
22.Chernova, T. A., and Zavilgelsky, G. B. (1991)
Biotekhnologiya, No. 3, 17-22.
23.Manukhov, I. V., Rastorguev, S. M., Eroshnikov,
G. E., Zarubina, A. P., and Zavilgelsky, G. B. (2000) Genetika,
36, 322-330.
24.Valkova, N., Szittner, R., and Meighen, E. A.
(1999) Biochemistry,38, 13820-13828.
25.Meighen, E. A., and Hastings, J. W. (1971) J.
Biol. Chem., 246, 7666-7674.
26.Hastings, J. W., and Nealson, K. H. (1977)
Annu. Rev. Microbiol.,31, 549-595.
27.Lundin, A., and Thore, A. (1975) Appl.
Microbiol., 30, 713-721.
28.Zavigelsky, G. B., Zarubina, A. P., and Manukhov,
I. V. (2002) Mol Biol. (Moscow), 36, 848-856.
29.Wickner, S., Gottesman, S., Skowyra, D., Hoskins,
J., McKenney, K., and Maurizi, M. R. (1994) Proc. Natl. Acad. Sci.
USA,91, 12218-12222.
30.McCarty, J. S., Bucherberger, A., Reinstein, J.,
and Bukau, B. (1995) J. Mol. Biol.,249, 126-137.
31.Hoskins, J. R., Pak, M., Maurizi, M. R., and
Wickner, S. (1998) Proc. Natl. Acad. Sci. USA,95,
12135-12140.
32.Singh, S. K., Grimaud, R., Hoskins, J. R.,
Wickner, S., and Maurizi, M. R. (2000) Proc. Natl. Acad. Sci.
USA,97, 8898-8903.
33.Ben-Zvi, A. P., and Goloubinoff, P. (2001) J.
Struct. Biol.,135, 84-93.