REVIEW: Circular RNAs: Biogenesis, Functions, and Role in Myocardial Hypertrophy
Natalia M. Baulina1,2,a*, Ivan S. Kiselev1,2, Olga S. Chumakova1, and Olga O. Favorova1,2
1Chazov National Medical Research Centre of Cardiology, Ministry of Health of the Russian Federation, 121552 Moscow, Russia2Pirogov Russian National Research Medical University, 117997 Moscow, Russia
* To whom correspondence should be addressed.
Received May 30, 2023; Revised July 14, 2023; Accepted July 16, 2023
Circular RNAs (circRNAs) are a large class of endogenous single-stranded covalently closed RNA molecules. High-throughput RNA sequencing and bioinformatic algorithms have identified thousands of eukaryotic circRNAs characterized by high stability and tissue-specific expression pattern. Recent studies have shown that circRNAs play an important role in the regulation of physiological processes in the norm and in various diseases, including cardiovascular disorders. The review presents current concepts of circRNA biogenesis, structural features, and biological functions, describes the methods of circRNA analysis, and summarizes the results of studies on the role of circRNAs in the pathogenesis of hypertrophic cardiomyopathy, the most common inherited heart disease.
KEY WORDS: circRNA, hypertrophic cardiomyopathy, backsplicing, competing endogenous RNADOI: 10.1134/S0006297924140013
Abbreviations: circRNA, circular RNA; HCM, hypertrophic cardiomyopathy; LVH, left ventricular hypertrophy.
INTRODUCTION
Circular RNAs (circRNAs) are a large class of single-stranded covalently closed RNA molecules. Viroids, exogenous infectious agents of plants, were the first circRNA molecules discovered more than 40 years ago [1]. Several years later, endogenous circRNAs were found in the cytoplasmic fraction of eukaryotic HeLa cells by electron microscopy [2]. For a long time, they had been considered as a “junk” resulting from aberrant RNA splicing [3], with the only exception for the circRNA transcript of the testis-determining gene Sry with a presumable specific function in the mouse testes [4].
However, a large body of evidence has been accumulated suggesting that circRNAs (traditionally classified as non-coding RNAs) serve as transcription and translation regulators [5] and play an important role in both healthy organism and various pathologies. Due to the stability of the closed ring structure, they have attracted attention as promising biomarkers of both diseases and therapy efficacy [6].
In recent years, thousands of eukaryotic circRNAs with tissue-specific gene expression patterns have been identified in fungi, protists, plants, worms, fish, insects, and mammals using high-throughput RNA sequencing (RNAseq) and bioinformatics algorithms [7]. According to the circAtlas 2.0 database (updated in March 2020), humans have more than 420 thousand circRNAs [8]. Unfortunately, current circRNAs databases remain incomplete and disconnected. Despite the efforts made by several research groups to streamline the information on circRNAs, these databases use different variants of nomenclature to annotate circRNAs. Typically, the identification number of a circRNA refers to a biological species and contains a numerical code unique to each database. Thus, the circRNA for the human CAMSAP1 gene encoded by the chr9:138773478-138799005 (hg19) locus is designated as hsa_circ_0001900 in the circBase (the first major database) [9] and as HSA_CIRCpedia_63397 in the CIRCpedia database [10]. Some databases, e.g., circBank [11], suggests a more convenient nomenclature that uses the name of a gene from which circRNA is transcribed (e.g., hsa_circCAMSAP1_006). The issue of circRNA nomenclature standardization remains unresolved, which hinders comparison of experimental data by different research groups.
It has been increasingly clear that circRNAs play an essential role in the pathogenesis of cardiovascular diseases in humans [6]. Left ventricular hypertrophy (LVH) is a common risk factor for developing heart failure, arrhythmia, and sudden cardiac death, i.e., diseases that hold the first place among the causes of morbidity and mortality worldwide. In most cases, the so-called secondary LVH emerges as an adaptive response to the long-term increased hemodynamic pressure or blood overload in the left ventricle (LV), e.g., in the case of hypertension or heart defect. Less commonly, primary LVH develops due to the intrinsic cardiomyocyte abnormality, namely, mutation in one of the genes coding for the sarcomere proteins. Hypertrophic cardiomyopathy (HCM) is a hereditary disease that develops because of the primary LVH. The mechanisms underlying the primary and secondary LVHs and the relevant treatments differ significantly despite some common elements in their pathogenesis. Studying the role of circRNAs as regulators of biological processes in LVH of various origin is an important and promising field of scientific research.
Here, we present current concepts on the biogenesis, degradation, and major functions of circRNA, as well as methods for their analysis. The data of yet not numerous studies on the role of circRNAs in pathological process are discussed in detail with regard to the LVH development, as well as a potential use of circRNAs as therapeutic targets and biomarkers in HCM.
BIOGENESIS AND DEGRADATION of circRNAs
Biogenesis of circRNAs. The majority of circRNAs are transcriptional products of thousands of protein-coding genes [12]. As of today, exonic circRNAs, intronic circRNAs, and exon–intron circRNAs are considered as the most common types of circRNAs, while circRNAs transcribed from the 3′- and 5′-untranslated regions are less common [5]. Exonic and exon–intron circRNAs can consist of either one or several exons, which indicates their generation by alternative splicing [13]. In some cases, circRNAs can be transcribed from the antisense DNA strand (antisense circRNAs) or intergenic regions (intergenic circRNAs) [14, 15]. The most studied among circRNAs are exonic circRNAs that account for >80% of all known circRNAs. The structural diversity of circRNAs is determined by different mechanisms of their formation (see below).
The canonical mechanism for the eukaryotic mRNA maturation is splicing of the precursor mRNA (pre-mRNA) with the involvement of the spliceosome, when the donor splicing site (5′-end of the intron) connects to the acceptor splicing site (3′-end of the same intron) followed by the lariat formation and its removal. Spliced mature mRNA can contain all exons of the pre-mRNA or only some of them in different combinations (in the case of alternative splicing), although the linear order of exons is always preserved. In contrast to canonical splicing, most circRNAs are formed via another mechanism (backsplicing), in which circRNA exons are linked in reverse order compared to the canonical mechanism [16].
Backsplicing is a type of pre-mRNA splicing in which the splicing donor site located closer to the pre-mRNA 3′-end (downstream) connects to the acceptor site located closer to the 5′-end (upstream) [12]. We will keep this, in our opinion, the most appropriate definition of backsplicing, although one can find other definitions of this process, often contradictory, in published articles. Backsplicing can result in the formation of a closed loop containing a single exon (exonic circRNAs) or several exons interspersed with intron(s) (exon–intron circRNAs). Exon–intron circRNAs can then lose introns via canonical splicing and become exonic circRNAs. The presence of long introns in the transcript increases the probability of backsplicing, because such introns often contain complementary inverted repeats, such as Alu sequences capable of forming antiparallel double-stranded RNA structures (“stalks”) that bring the backsplicing sites closer to each other [17-19]. There are also RNA-binding proteins, such as Quaking, Muscleblind, and Fused in sarcoma, whose monomers can bind to specific sequences in the upstream and downstream introns, respectively, dimerize, and bring the splice sites closer to each other, thus promoting circRNA formation [20].
Because backsplicing and canonical splicing are catalyzed by the same spliceosome-dependent mechanism [21], they directly compete with each other [22]. Depletion of the spliceosome components, such as SF3a and SF3b, can shift the balance toward the backsplicing [23]. The processing efficiency of the most circRNAs is very low; nevertheless, circRNAs can accumulate in the cells in relatively high quantities due to their resistance to degradation by exonucleases [24]. For some genes, circRNAs represent dominant transcription products [22]. Biogenesis of circRNA can start with splicing by the canonical mechanism. The resulting RNA fragment containing both exons and introns is excised with the formation of the lariat, which is then processed by backsplicing with the generation of exonic circRNAs and sometimes exon–intron circRNAs [25]. Moreover, some introns excised from the precursor mRNA during canonical splicing do not degrade, but instead close into intronic circRNAs [26]. Figure 1 shows the splicing events leading to the emergence of various circRNA types. As seen from the figure, the same gene locus can generate several different circRNAs due to the use of alternative backsplicing sites. The products of linear RNA splicing can become components of circRNAs; moreover, some circRNAs contain exons absent in the linear transcripts [27].
Fig. 1. Formation of mature mRNA and various circRNAs from the precursor mRNA. Backsplicing results in the generation of exonic circRNA bearing exon 2 via the head-to-tail junction between the 5′ splice site of intron 2 and 3′ splice site of intron 1. Alternative backsplicing can result in the excision of exons 2 and 3 and intron 2, while subsequent intron removal via canonical splicing generates exonic circRNAs consisting of exons 2 and 3. Canonical splicing can result in both linear mRNA and circRNAs (via removal of the “tail” from the lariat-intron 4 sequence). Alternative canonical splicing forms an exon–intron lariat that is processed by backsplicing with the generation of exon-intron circRNA. Excision of intron 3 from the exon-intron circRNA by the canonical pathway leads to the formation of exonic circRNA consisting of exons 3 and 4. Exons and introns are depicted as multi-colored rectangles and black lines, respectively; green dashed line, canonical splicing, green dash-dotted line, alternative canonical splicing; red dashed line, backsplicing; red dash-dotted line, alternative backsplicing. The convergence of the flanking introns due to the dimerization of RNA-binding proteins (RBPs) (purple pentagons) and interaction of inverted complementary sequences (blue rectangles) are shown on gray background.
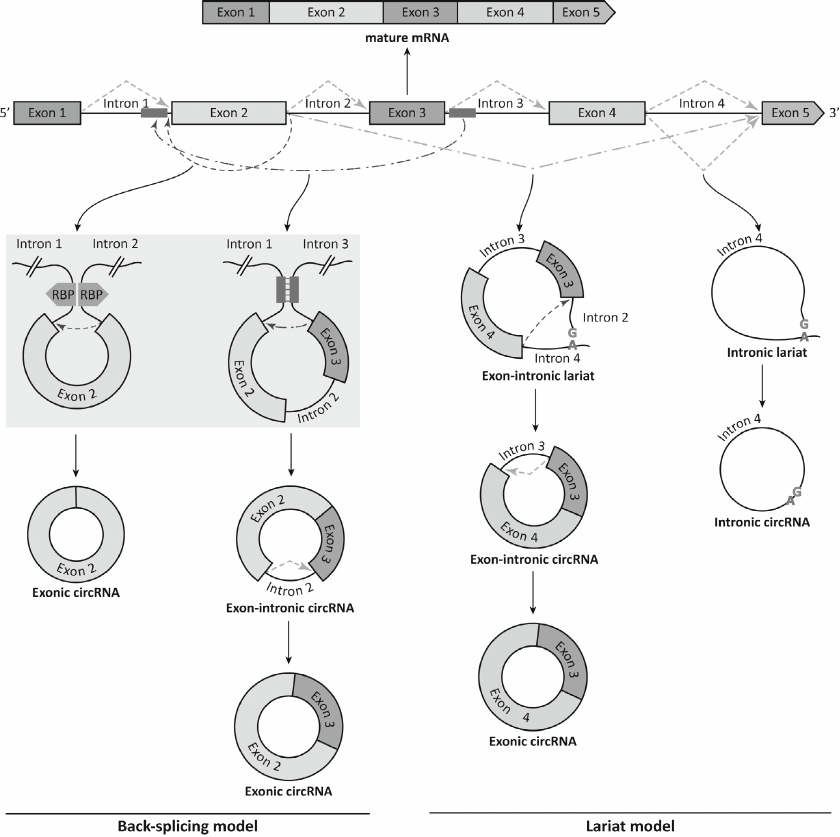
In general, biogenesis of circRNAs is a multi-stage process that depends on many factors, including position of inverted repeats and binding sites for proteins that mediate the looping or, conversely, prevent backsplicing.
Despite the lack of poly(A) tail and cap, the majority of emerging exonic circRNAs are transported to the cytosol, whereas intronic circRNAs and exon-intron circRNAs reside in the nucleus [7]. The mechanism of circRNA export from the nucleus to the cytosol depends on the sequence length: shorter circRNAs are transported with the help of ATP-dependent RNA helicase URH49, while the export of longer circRNAs is mediated by the spliceosomal RNA helicase UAP56 [28].
Degradation of circRNAs. Normal functioning of cellular circRNAs requires a balance between their formation and degradation [29, 30]. However, the mechanisms and underlying conditions of circRNA degradation have not yet been fully clarified. Because circRNAs are extremely stable and resistant to the cleavage by exonucleases due to the lack of free ends [5, 18, 31], the major role in the circRNA degradation belongs to endonucleases. For instance, AGO2 endonuclease cleaves circRNAs via a microRNA-regulated mechanism [32]. There is also an alternative N6-methyladenosine (m6A)-related pathway for the endonuclease-mediated circRNA cleavage: m6A-containing circRNAs are cleaved by endonuclease P in a complex with the multidrug resistance protein (RNase P/MRP). The complex interacts with the m6A-binding protein YTHDF2 through the adapter protein HRSP12 [33]. Several other proteins have been described that perform specific functions in the degradation of circRNAs [23, 34, 35].
circRNAs are often found in exosomes and extracellular vesicles, which presumably participate in their clearance or act as intermediaries in the circRNA-mediated cell–cell communication [36-38].
FUNCTIONS of circRNA
Currently, circRNAs are believed to perform diverse biological functions by acting as transcriptional regulators and templates for protein synthesis. Some circRNAs serve as protein decoys, scaffolds, and recruiters by recognizing specific amino acid sequences in proteins (Fig. 2).
Fig. 2. Main circRNA functions: microRNA binding (a), direct binding and modulation of host genes by the R-loop formation (b), binding to the transcriptional regulators (c), cap-independent circRNA translation followed by peptide production (d).
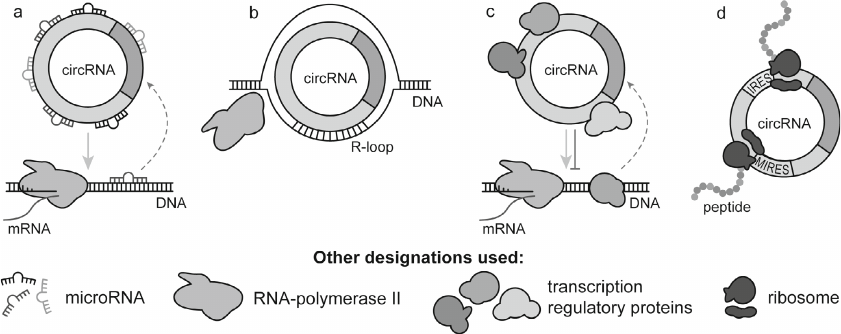
Recent studies have revealed that circRNAs are enriched in microRNA-binding sites and act as “sponges” by binding microRNAs and blocking their inhibitory effect on the target genes [39], resulting in the upregulated expression of the latter (Fig. 2a). This competition for microRNAs with the mRNAs of the target genes allows to consider circRNAs as the intracellular competitive endogenous RNAs (ceRNAs) that affect the functional activity of both microRNAs and target genes [40]. Because circRNAs are actively expressed in cells, show high stability, and bear many microRNA-binding sites, they might be as efficient microRNA sponges as lncRNAs (other non-coding molecules with the same function) [39, 41].
It was shown that nuclear circRNAs can directly modulate expression of the own (host) genes by forming a hybrid with the complementary DNA strand (R-loop structure) to pause or terminate their transcription (Fig. 2b). Formation of the circRNA–DNA hybrid loop can also contribute to the upregulated expression of truncated transcripts or transcripts with skipped exons resulting in the production of phenotypically defective gene products [42]. circRNAs can also promote expression of the host genes, as it was shown for some nuclear exon–intron circRNAs (e.g., circEIF3J and circPAIP2), by recruiting spliceosomal small nuclear ribonucleoprotein U1 and RNA polymerase II to their promoters [43]. Under certain physiological and pathological conditions, circRNAs act as protein decoys, scaffolds, and recruiters (Fig. 2c). Depending on the conditions, the same circRNA can bind to a particular protein or interact with multiple proteins [44].
Originally, circRNAs had been classified as non-coding RNAs; however, it was demonstrated that some of them encode peptides with yet unknown functions [45, 46] (Fig. 2d). Some circRNAs contain an internal ribosome entry site (IRES) ensuring a cap-independent initiation of translation and an open reading frame; they bind to polyribosomes and are translated into short peptides or proteins [47, 48]. Certain circRNAs are translated via another cap-independent mechanism mediated by the methylation of nitrogen at position 6 in adenosine (N6-methyladenosine, m6A). The presence of a single m6A-induced ribosome engagement site (MIRES) in the 5′-untranslated region promotes selective translation of circRNAs under stress conditions via direct binding of the translation initiation factor eIF3 [49, 50].
METHODS FOR circRNA ANALYSIS
The studies of circRNAs had been limited for a long time because of the difficulties in the identification of circRNAs caused by the size variability and lack of free ends in these molecules. Indeed, circRNAs cannot be separated from other RNAs by gel electrophoresis and detected with classical molecular biology techniques, such as reverse transcription of poly(A)-rich RNAs or rapid amplification of cDNA ends (RACE). Moreover, the sequences of circRNAs and mRNAs encoded by the host genes are identical, which further complicates their differentiation.
High-throughput circRNA analysis. The last decade has been characterized by a significant progress in the development of RNA sequencing techniques, including various ribosomal RNA depletion strategies, methods allowing increased read lengths and greater sequencing depth, and new algorithms for alignment of genomic reads. These advancements have resulted in the identification of splice variants of mRNAs and non-polyadenylated RNAs, including circRNAs. The fact that the ends of circRNAs molecules are joined in a head-to-tail manner provides a unique opportunity for their detection [51]. Thus, preparation of circRNA samples for sequencing includes various procedures for destruction of linear RNAs, such as the poly(A) RNA depletion, treatment with 3′→5′ exoribonuclease R (RNase R), and RPAD (RNase R Treatment, Polyadenylation, and Poly(A)+ RNA Depletion), aimed to enrich the total RNA pool with circRNAs [52, 53]. However, since circRNAs can be formed by alternative (other than backsplicing) mechanisms, such as trans-splicing, tandem duplication, or reverse transcriptase template switching [13, 52], complete elimination of linear RNA sequences, including the products of their cyclization, remains a serious issue.
RNAseq data are aligned to the genome sequences using standard tools, such as STAR and BWA [54, 55]. The algorithms for further circRNA identification are continuously advanced; the most common among them are ACSF, CIRCexplorer2, CIRI2, DCC, KNIFE, and Uroborus [7]. Unfortunately, one of the negative consequences of such diversity of the data processing tools is the difficulty in the interpretation and comparison of data provided by different research teams.
Due to the advances in the sequencing technologies and bioinformatics tools for the annotation and identification of circRNAs, the identified types of circRNAs and their biological functions have been continuously expanding, making circRNAs quite competitive candidates for novel disease biomarkers.
Another step in the studies of circRNAs was development of DNA microarray platforms for detection and quantification of validated circRNAs [51]. Unlike RNAseq, microarrays utilize probes for identifying the regions of the head-to-tail junctions in known circRNAs. The first commercial microchips were manufactured by Arraystar Inc. (https://www.arraystar.com/circular-rna-array-service/); they contained more than 10,000 circRNAs selected based on the published data. Because the sequences of the microarray probes are similar to the sequences of linear RNAs, RNase R pretreatment of the total RNA pool can improve the specificity of detection of circular molecules. In contrast to RNAseq, DNA microarrays are limited by the number of probes for the validated circRNAs and do not allow to identify mature (post-splicing) circRNAs or to determine the ratio between the circRNA and linear RNA for the same gene. However, compared to RNAseq, the microarray data are much easier to process.
Methods for circRNA analysis based on PCR and Northern blotting. Since both high-throughput RNAseq and DNA microarrays can generate false-positive results, alternative approaches, such as reverse transcription-polymerase chain reaction (RT-PCR), Sanger sequencing of PCR products, and Northern blotting, are recommended for verification and more accurate quantification of identified circRNAs. Thus, PCR-based identification and quantification of circRNAs have become widely used due to their speed and convenience.
Specific circRNAs can be amplified by PCR using divergent primers flanking the head-to-tail junction sequence [56]. Because such primers do not amplify linear RNAs, no RNase R treatment of the total RNA is required for the PCR-based detection and quantification of circRNAs. The backsplicing sequence in the obtained PCR products can be verified by Sanger sequencing with divergent primers. However, this method is unable to obtain the full-length sequence of a circRNA or to distinguish between multiple circRNAs derived from the same gene region.
Recently, a new approach to detecting the full-length circRNA sequences named circRNA-RCA (circRNA-rolling circle amplification) was developed based on the primers complementary to the head-to-tail junction [57]. The number of circRNA copies obtained in the RCA reaction is determined by the initial number of molecules in the sample and their length, which significantly complicates normalization of results and their comparison for different circRNAs and their splicing variants. Hence, circRNA-RCA can identify circRNA variants with the same backsplicing junction sequences qualitatively but not quantitatively [13, 52].
Recently, it was demonstrated that circRNAs can be successfully quantified by the droplet digital PCR. Compared to RT-PCR, droplet digital PCR produces more accurate data due to the absence of prolonged incubation step [58]. Several alternative approaches (SplintQuant, NanoString) have been developed as well for accurate quantification of endogenous circRNAs [59, 60].
Northern blotting is recognized as a gold standard for the analysis of circRNAs, although it requires additional sample treatment with RNase R and RNase H [53]. Northern blotting also has a number of drawbacks, such as the need for very large quantities of RNA and use of radioactively labeled probes [7].
Online databases for the analysis of circRNAs. Several specialized circRNA databases containing information on the circRNA expression in tissues, evolutionary conservatism, association with diseases, and interactions with RNA-binding proteins and microRNAs are available for in silico analysis. Most of them provide the data on human circRNAs, but some have information on monkey, mouse, rat, chicken, and yeast circRNAs. The data on the expression levels for various circRNAs in different types of cells and tissues can be found in TSCD [61], CSCD [62], CIRCpedia [10], circBase [9], circAtlas [8], circMine [63], PanCircBase [64]. Several databases, such as CSCD [62], circAtlas [8], circInteractome [65], circNet [66], starBase [67] and circFunBase [68], have been developed for predicting the functions of circRNAs and their interactions with microRNAs and RNA-binding proteins. Other databases, including circAtlas [8], circInteractome [65], circBank [11], circRNADb [46], and riboCIRC [69], contain the information on the circRNA translational potential. All these resources provide predictive data about circRNAs that have to be experimentally verified.
circRNAs IN MYOCARDIAL HYPERTROPHY
The studies of molecular mechanisms underlying LVH and identification of therapeutic targets in this disease have recently attracted an increasing attention of researchers. Several circRNAs involved in the regulation of the pathological processes associated with LVH of various origin have been identified that were considered as potential diagnostic or therapeutic targets.
The association between circRNAs and secondary myocardial hypertrophy was discovered by RNAseq in the heart tissue of mice subjected to transverse aortic constriction (TAC). Almost 700 deregulated circRNAs were identified compared with the control animals [70]. Other studies revealed that the cardiac-specific circRNAs mm9-circ-012559 (circ-HRCR) that acts as an endogenous sponge for miR-223, counteracted hypertrophy induced by isoproterenol and TAC. Using a cardiac-specific transgenic mouse model, it was shown that miR-223 promotes LVH development in heart failure, whereas one of its targets, apoptosis repressor with a CARD domain (ARC), alleviates myocardial hypertrophy. Therefore, circ-HRCR attenuates hypertrophy in vivo by suppressing the activity of miR-223 and promoting the action of ARC [71].
circSlc8a1 has attracted a lot of attention because of its high abundance in cardiomyocytes. This circRNA serves as a sponge for miR-133a [72], which inhibits myocardial hypertrophy [73]. The knockdown of circSlc8a1 in mice reduced the LVH caused by the TAC-induced pressure overload, whereas experimental upregulation of circSlc8a1 expression in cardiomyocytes resulted in heart failure [72]. Moreover, it was shown that circSlc8a1 also regulates the targets of miR-133a, including serum response factor (Srf), connective tissue growth factor (Ctgf), beta-1 adrenergic receptor (Adrb1), adenylate cyclase 6 (Adcy6), and transcription factors interacting with these proteins. Hence, this study provided a compelling evidence that the circSlc8a1-mediated inhibition of miR-133a could be used to treat myocardial hypertrophy.
Li et al. [74] found that the circ-000203 aggravated LVH in angiotensin II-infused mice. By acting as a ceRNA, circ-000203 blocked suppression of the target Gata4 gene via binding specifically to miR-26b-5p and miR-140-3p [74]. Previously, it was shown that upregulation of the GATA4 expression promoted development of hypertrophy both in vivo and in vitro [75]. It can be speculated that hsa-circ-0036167 (a homolog of mouse circ-000203) plays a similar role in humans, so that decreasing its level can result in the myocardial hypertrophy regression.
The profiling of cardiac circRNAs in isoproterenol-treated (myocardial hypertrophy model) vs. control mice revealed 401 differentially expressed circRNAs [76]. Based on the Gene Ontology and Kyoto Encyclopedia of Genes and Genomes pathways, the host genes of differentially expressed circRNAs belong to the pathways involved in the circulatory system activity and development of cardiovascular diseases. Moreover, it was also shown that the circRNAs wwp1 decreased myocardial hypertrophy by regulating the levels of atrial natriuretic factor (ANF) and miR-23a.
Using the mouse models of TAC-induced myocardial hypertrophy and angiotensin II-stimulated mouse cardiomyocytes to mimic this pathological process, it was found that myocardial hypertrophy is regulated by the circNfix and circHIPK3 [77, 78]. The used myocardial hypertrophy models were characterized by the reduced circNfix expression, while an increase in the circNfix content attenuated LVH induced by the pressure overload by regulating the miR-145-5p/ATF3 axis [77]. In contrast, expression of circHIPK3 in the heart was significantly increased in cardiac hypertrophy [78]. The knockout of circHIPK3 inhibited development of the TAC-induced myocardial hypertrophy and improved the echocardiography parameters. The silencing of circHIPK3 in cardiomyocytes markedly reduced the size of these cells and decreased the content of hypertrophy markers, such as the ANF, brain natriuretic peptide, and β-myosin heavy chain. It was shown that circHIPK3 regulates cardiac hypertrophy via binding to miR-185-3p followed by the modulation of expression of the target calcium-sensing receptor (CaSR) gene [78]. On the other hand, circ_0001052 transcribed from the Hipk3 gene also exhibited the prohypertrophic effect in angiotensin II-stimulated cardiomyocytes [79] by sponging miR-148a-3p and miR-124-3p and thereby restraining their suppressive effect on the Hipk3 gene expression, as well as by recruiting Srsf1 to stabilize Hipk3 [79].
In the LVH cell model, angiotensin II-induced LVH was found to be associated with the increased expression of circ-TLR4 and its host gene TLR4 [80] coding for the Toll-like receptor 4 (TLR4). TLR4 is critical in the regulation of inflammatory response and cardiac hypertrophy. The knockdown of circ-TLR4 attenuated angiotensin II-induced hypertrophy of cardiomyocytes. Moreover, circ-TLR4 upregulated expression of the TLR4 gene by recruiting the DNA/RNA binding protein FUS to stabilize the TLR4 mRNA. In a similar LVH cell model, expression of another circRNA, circ_0018553, was upregulated, and this circRNA was highly abundant in the exosomes derived from endothelial progenitor cells [81]. It was shown that circ_0018553 acts as a sponge for miR-4731, which, in turn, represses expression of the longevity protein sirtuin 2 (SIRT2), one of the essential regulators of the cell response to DNA damage. Therefore, exosomal circ_0018553 derived from endothelial progenitor cells protects against angiotensin II-induced LVH by modulating the miR-4731/SIRT2 signaling pathway. Another member of the sirtuin family regulated by circRNAs, SIRT1, is also involved in the LVH development. Thus, circ-SIRT1/circ-Sirt1 transcribed from the SIRT1 gene alleviated angiotensin II-induced heart failure and induced autophagy in H9c2 cells and human cardiomyocytes derived from induced pluripotent stem cells [82]. It was found that circ-SIRT1 upregulated SIRT1 expression at the post-transcriptional level by lowering the content of cellular miR-3681-3p/miR-5195-3p and stabilizing SIRT1 protein by recruiting ubiquitin-specific peptidase USP22 for its deubiquitination. Hence, circ-SIRT1 restrains the development of heart failure via activation of the SIRT1-dependent autophagy.
The synthesis of circRNAs for the capture of prohypertrophic microRNAs has become one of the topic research areas in the therapy of myocardial hypertrophy. For instance, the circmiR sponge was created to target miR-132 and miR-212 [83] and stimulate cardiomyocyte growth [84]. CircmiRs containing 12 microRNA-binding sites were found to be the most efficient; when delivered in vivo into cardiomyocytes, they reduced the LVH and preserved the cardiac function in the mouse model of TAC-induced HCM. Compared with endogenous circRNAs, circmiRs have more advantages due to the flexibility of their design, long half-life, and possibility of application in low doses. These data evidence a high potential of engineered circRNAs as new therapeutic agents for HCM.
circRNAs IN HCM
HCM is the most common hereditary heart disease with the incidence rate of at least 1 : 500 in general population [85, 86]. HCM is defined as the LV hypertrophy unexplained by other hemodynamic causes [87]. In young people, HCM is related mainly to the risk of sudden cardiac death; in older individuals, it is associated with progressive heart failure, atrial fibrillation, and embolic strokes. HCM is a socially highly significant diseases because of the difficulties with its early diagnostics due to the asymptomatic onset and concomitant diseases that might result in the development of secondary LVH, as well as the absence (until recently) of treatment. HCM has a poorly predictable clinical course: a large portion of patients live a normal or almost normal life, whereas some persons develop complications for yet-unknown reasons. Individual clinical features of HCM can be explained by a significant genetic heterogeneity of the disease. Currently, pathogenetic variants (mutations) in various genes encoding sarcomere proteins or other related proteins are verified only in a half of HCM patients (genotype-positive patients). However, a substantial number of familial HCM cases are observed among genotype-negative individuals, which promotes further search for new disease-related genes and inheritance mechanisms. In recent years, a new hypothesis has emerged stating that some patients have a non-Mendelian polygenic inheritance of HCM [88]. External factors can also contribute to the development and clinical picture of primary LVH. Finally, many studies aimed at unraveling the mechanisms of HCM clinical heterogeneity investigate the impact of epigenetics and non-coding RNAs. The remaining large gap in the understanding of the mechanisms implicated in manifestation of various genetic defects and formation of the HCM phenotype hinders timely diagnostics, prediction of the disease course, and development of personalized treatment.
One of the most urgent tasks in modern medicine is the search for available biomarkers for the differential diagnostics between the primary and secondary LVH, as well as biomarkers of the HCM complications. Currently verified biochemical markers that are universal for all cardiovascular diseases and can be assessed in the peripheral blood (e.g., troponin and N-terminal fragment of the brain natriuretic peptide) may be of prognostic value in HCM [89] because they indicate progressive structural alterations in the myocardium (however, they do not reflect molecular events typical to this genetically determined disease). circRNAs might become effective and convenient HCM biomarkers due to their stability and ability to be released into the bloodstream where they can freely circulate. The level of circRNAs in the circulation changes in HCM, reflecting differential gene expression and subsequent splicing of the corresponding pre-mRNAs in the myocardium, might expand our understanding of the underlying pathological mechanisms. However, the active studies in this research field have just started.
One of the first works investigating the role of circRNAs in HCM showed that the levels of circDNAJC6, circMBOAT2, and circTMEM56 were significantly reduced in the serum from HCM patients vs. control subjects [90]. When assessed individually or together, these circRNAs demonstrated a capacity to discriminate between the HCM patients (including patient subgroups with obstructive and non-obstructive HCM) and control subjects (AUC, 0.722 to 0.949). The levels of two circRNAs, circTMEM56 and circDNAJC6, showed a strong correlation with the extent of LV obstruction and interventricular septum thickness and, therefore, can serve as indicators of disease severity in patients with obstructive HCM.
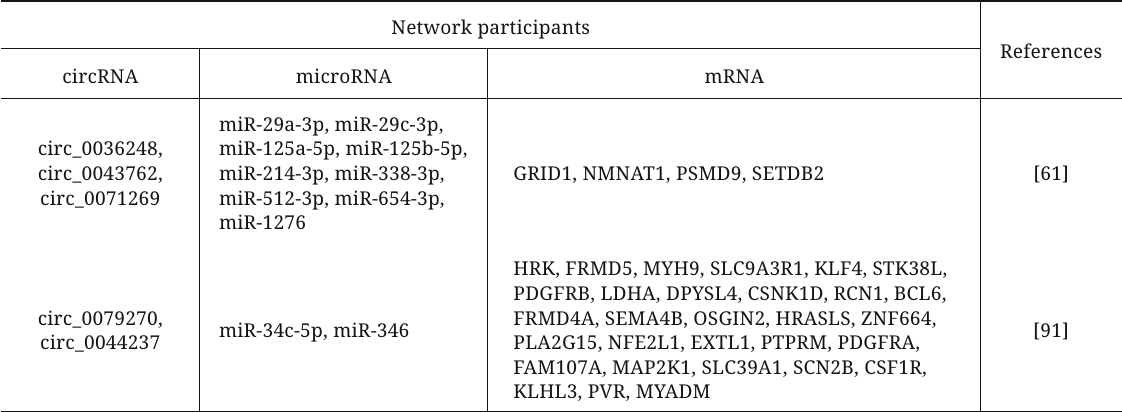
Various bioinformatics algorithms have been used to construct circRNA–microRNA–mRNA interaction networks and to reveal the key endogenous RNAs among circRNAs involved in the development of primary LVH by competing with mRNAs for the binding to the complementary region in microRNAs (table). For example, the authors of [61] used open data sets to analyze the plasma levels of circRNAs in HCM patients and volunteers and to build a network of ceRNAs, included three circRNAs (circ_0036248, circ_0043762, and circ_0071269). Functional analysis revealed that these circRNAs participate in the modulation of the calcium release channel activity and muscle filament sliding and, therefore, may be the key regulators involved in HCM pathogenesis. Another study analyzed open datasets on the levels of circRNAs, microRNAs, and mRNA in HCM to construct a similar network with circ_0079270 and circ_0044237 as its key components [91]. The authors confirmed downregulation of the circ_0079270 expression and changes in the expression of identified microRNAs and 18 mRNAs in the cell model of HCM. It was also shown that circ_0079270 is generated by reverse transcription and partial splicing of the ACTB gene-encoded sequence with the formation of a circular structure containing exons 2 and 5. Another mechanism of the ACTB-mediated development of HCM involving circRNAs was proposed by Feng et al. [92]. Based on comprehensive bioinformatics analysis, it was found that expression of circFN1 is decreased in HCM. According to the data of in vitro experiments, this circRNA can competitively bind to miR-206, thereby upregulating expression of the target ACTB gene, which promotes HCM development.
CircRNAs–microRNA–mRNA networks participating in the
development of myocardial hypertrophy
CONCLUSION
circRNAs has been well-established as RNA regulators of transcription and translation with essential in vivo functions. Recent studies suggest that circRNAs can be used as diagnostic markers and therapeutic agents in various pathologies, including myocardial hypertrophy, the cause of the common hereditary disease HCM. However, regardless of the methods used (experimental or bioinformatics), the data on the role of circRNAs in myocardium hypertrophy remain scattered and often controversial. This is not surprising, as the studies on the involvement of circRNAs in this pathology has only started, while the small size and heterogeneity of samples, different methods used for sample collection and isolation, and different algorithms and approaches for circRNA detection complicate interpretation of the obtained data. Nevertheless, the studies described in this review undoubtedly will lay the foundation for future advances in this research field.
Contributions. N.M.B. conceptualization, writing – original draft, review and editing; I.S.K. visualization and writing – review and editing; O.S.C. writing – medical issue; O.O.F. writing – review and editing.
Funding. The study was supported by the Russian Science Foundation (project no. 20-15-00353).
Ethics declarations. This work does not contain studies involving human and animal subjects. The authors of this work declare no conflict of interest.
REFERENCES
1.Sanger, H. L., Klotz, G., Riesner, D., Gross, H.
J., and Kleinschmidt, A. K. (1976) Viroids are single-stranded
covalently closed circular RNA molecules existing as highly base-paired
rod-like structures, Proc. Natl. Acad. Sci. USA, 73,
3852-3856, doi: 10.1073/pnas.73.11.3852.
2.Hsu, M. T., and Coca-Prados, M. (1979) Electron
microscopic evidence for the circular form of RNA in the cytoplasm of
eukaryotic cells, Nature, 280, 339-340, doi:
10.1038/280339a0.
3.Cocquerelle, C., Mascrez, B., Hétuin, D.,
and Bailleul, B. (1993) Mis-splicing yields circular RNA molecules,
FASEB J., 7, 155-160, doi:
10.1096/fasebj.7.1.7678559.
4.Capel, B., Swain, A., Nicolis, S., Hacker, A.,
Walter, M., Koopman, P., Goodfellow, P., and Lovell-Badge, R. (1993)
Circular transcripts of the testis-determining gene Sry in adult mouse
testis, Cell, 73, 1019-1030, doi:
10.1016/0092-8674(93)90279-y.
5.Memczak, S., Jens, M., Elefsinioti, A., Torti, F.,
Krueger, J., Rybak, A., Maier, L., Mackowiak, S. D., Gregersen, L. H.,
Munschauer, M., Loewer, A., Ziebold, U., Landthaler, M., Kocks, C., le
Noble, F., and Rajewsky, N. (2013) Circular RNAs are a large class of
animal RNAs with regulatory potency, Nature, 495,
333-338, doi: 10.1038/nature11928.
6.Verduci, L., Tarcitano, E., Strano, S., Yarden, Y.,
and Blandino, G. (2021) CircRNAs: Role in human diseases and potential
use as biomarkers, Cell Death Disease, 12, 468, doi:
10.1038/s41419-021-03743-3.
7.Kristensen, L. S., Andersen, M. S., Stagsted, L. V.
W., Ebbesen, K. K., Hansen, T. B., and Kjems, J. (2019) The biogenesis,
biology and characterization of circular RNAs, Nat. Rev. Genet.,
20, 675-691, doi: 10.1038/s41576-019-0158-7.
8.Wu, W., Ji, P., and Zhao, F. (2020) CircAtlas: An
integrated resource of one million highly accurate circular RNAs from
1070 vertebrate transcriptomes, Genome Biol., 21, 101,
doi: 10.1186/s13059-020-02018-y.
9.Glažar, P., Papavasileiou, P., and Rajewsky,
N. (2014) circBase: A database for circular RNAs, RNA (New York,
N.Y.), 20, 1666-1670, doi: 10.1261/rna.043687.113.
10.Dong, R., Ma, X.-K., Li, G.-W., and Yang, L.
(2018) CIRCpedia v2: an updated database for comprehensive circular RNA
annotation and expression comparison, Genom. Proteom.
Bioinform., 16, 226-233, doi: 10.1016/j.gpb.2018.08.001.
11.Liu, M., Wang, Q., Shen, J., Yang, B. B., and
Ding, X. (2019) Circbank: a comprehensive database for circRNA with
standard nomenclature, RNA Biol., 16, 899-905, doi:
10.1080/15476286.2019.1600395.
12.Barrett, S. P., and Salzman, J. (2016) Circular
RNAs: Analysis, expression and potential functions, Development,
143, 1838-1847, doi: 10.1242/dev.128074.
13.Szabo, L., and Salzman, J. (2016) Detecting
circular RNAs: Bioinformatic and experimental challenges, Nat. Rev.
Genet., 17, 679-692, doi: 10.1038/nrg.2016.114.
14.Ma, J., Du, W. W., Zeng, K., Wu, N., Fang, L.,
Lyu, J., Yee, A. J., and Yang, B. B. (2021) An antisense circular RNA
circSCRIB enhances cancer progression by suppressing parental gene
splicing and translation, Mol. Ther., 29, 2754-2768, doi:
10.1016/j.ymthe.2021.08.002.
15.Maass, P. G., Glažar, P., Memczak, S.,
Dittmar, G., Hollfinger, I., Schreyer, L., Sauer, A. V., Toka, O.,
Aiuti, A., Luft, F. C., and Rajewsky, N. (2017) A map of human circular
RNAs in clinically relevant tissues, J. Mol. Med., 95,
1179-1189, doi: 10.1007/s00109-017-1582-9.
16.Chen, I., Chen, C.-Y., and Chuang, T.-J. (2015)
Biogenesis, identification, and function of exonic circular RNAs,
Wiley Interdiscip. Rev. RNA, 6, 563-579, doi:
10.1002/wrna.1294.
17.Zhang, X.-O., Wang, H.-B., Zhang, Y., Lu, X.,
Chen, L.-L., and Yang, L. (2014) Complementary sequence-mediated exon
circularization, Cell, 159, 134-147, doi:
10.1016/j.cell.2014.09.001.
18.Jeck, W. R., Sorrentino, J. A., Wang, K., Slevin,
M. K., Burd, C. E., Liu, J., Marzluff, W. F., and Sharpless, N. E.
(2013) Circular RNAs are abundant, conserved, and associated with ALU
repeats, RNA, 19, 141-157, doi:
10.1261/rna.035667.112.
19.Liang, D., and Wilusz, J. E. (2014) Short
intronic repeat sequences facilitate circular RNA production, Genes
Dev., 28, 2233-2247, doi: 10.1101/gad.251926.114.
20.Ivanov, A., Memczak, S., Wyler, E., Torti, F.,
Porath, H. T., Orejuela, M. R., Piechotta, M., Levanon, E. Y.,
Landthaler, M., Dieterich, C., and Rajewsky, N. (2015) Analysis of
intron sequences reveals hallmarks of circular RNA biogenesis in
animals, Cell Rep., 10, 170-177, doi:
10.1016/j.celrep.2014.12.019.
21.Chen, L.-L., and Yang, L. (2015) Regulation of
circRNA biogenesis, RNA Biol., 12, 381-388, doi:
10.1080/15476286.2015.1020271.
22.Salzman, J., Gawad, C., Wang, P. L., Lacayo, N.,
and Brown, P. O. (2012) Circular RNAs are the predominant transcript
isoform from hundreds of human genes in diverse cell types, PLoS
One, 7, e30733, doi: 10.1371/journal.pone.0030733.
23.Liang, D., Tatomer, D. C., Luo, Z., Wu, H., Yang,
L., Chen, L.-L., Cherry, S., and Wilusz, J. E. (2017) The output of
protein-coding genes shifts to circular RNAs when the pre-mRNA processi
ng machinery is limiting, Mol. Cell, 68, 940-954.e3, doi:
10.1016/j.molcel.2017.10.034.
24.Zhang, Y., Xue, W., Li, X., Zhang, J., Chen, S.,
Zhang, J.-L., Yang, L., and Chen, L.-L. (2016) The biogenesis of
nascent circular RNAs, Cell Rep., 15, 611-624, doi:
10.1016/j.celrep.2016.03.058.
25.Barrett, S. P., Wang, P. L., and Salzman, J.
(2015) Circular RNA biogenesis can proceed through an exon-containing
lariat precursor, ELife, 4, e07540, doi:
10.7554/eLife.07540.
26.Zhang, Y., Zhang, X.-O., Chen, T., Xiang, J.-F.,
Yin, Q.-F., Xing, Y.-H., Zhu, S., Yang, L., and Chen, L.-L. (2013)
Circular intronic long noncoding RNAs, Mol. Cell, 51,
792-806, doi: 10.1016/j.molcel.2013.08.017.
27.Zhang, X.-O., Dong, R., Zhang, Y., Zhang, J.-L.,
Luo, Z., Zhang, J., Chen, L.-L., and Yang, L. (2016) Diverse
alternative back-splicing and alternative splicing landscape of
circular RNAs, Genome Res., 26, 1277-1287, doi:
10.1101/gr.202895.115.
28.Huang, C., Liang, D., Tatomer, D. C., and Wilusz,
J. E. (2018) A length-dependent evolutionarily conserved pathway
controls nuclear export of circular RNAs, Genes Dev., 32,
639-644, doi: 10.1101/gad.314856.118.
29.Wang, K., Gao, X.-Q., Wang, T., and Zhou, L.-Y.
(2023) The function and therapeutic potential of circular RNA in
cardiovascular diseases, Cardiovasc. Drugs Ther., 37,
181-198, doi: 10.1007/s10557-021-07228-5.
30.Huang, A., Zheng, H., Wu, Z., Chen, M., and
Huang, Y. (2020) Circular RNA-protein interactions: Functions,
mechanisms, and identification, Theranostics, 10,
3503-3517, doi: 10.7150/thno.42174.
31.Enuka, Y., Lauriola, M., Feldman, M. E.,
Sas-Chen, A., Ulitsky, I., and Yarden, Y. (2016) Circular RNAs are
long-lived and display only minimal early alterations in response to a
growth factor, Nucleic Acids Res., 44, 1370-1383, doi:
10.1093/nar/gkv1367.
32.Hansen, T. B., Wiklund, E. D., Bramsen, J. B.,
Villadsen, S. B., Statham, A. L., Clark, S. J., and Kjems, J. (2011)
MiRNA-dependent gene silencing involving Ago2-mediated cleavage of a
circular antisense RNA, EMBO J., 30, 4414-4422, doi:
10.1038/emboj.2011.359.
33.Park, O. H., Ha, H., Lee, Y., Boo, S. H., Kwon,
D. H., Song, H. K., and Kim, Y. K. (2019) Endoribonucleolytic cleavage
of m6A-containing RNAs by RNase P/MRP complex, Mol.
Cell, 74, 494-507.e8, doi: 10.1016/j.molcel.2019.02.034.
34.Jia, R., Xiao, M.-S., Li, Z., Shan, G., and
Huang, C. (2019) Defining an evolutionarily conserved role of GW182 in
circular RNA degradation, Cell Discov., 5, 45, doi:
10.1038/s41421-019-0113-y.
35.Liu, C.-X., Li, X., Nan, F., Jiang, S., Gao, X.,
Guo, S.-K., Xue, W., Cui, Y., Dong, K., Ding, H., Qu, B., Zhou, Z.,
Shen, N., Yang, L., and Chen, L.-L. (2019) Structure and degradation of
circular RNAs regulate PKR activation in innate immunity, Cell,
177, 865-880.e21, doi: 10.1016/j.cell.2019.03.046.
36.Dou, Y., Cha, D. J., Franklin, J. L.,
Higginbotham, J. N., Jeppesen, D. K., Weaver, A. M., Prasad, N., Levy,
S., Coffey, R. J., Patton, J. G., and Zhang, B. (2016) Circular RNAs
are down-regulated in KRAS mutant colon cancer cells and can be
transferred to exosomes, Sci. Rep., 6, 37982, doi:
10.1038/srep37982.
37.Lasda, E., and Parker, R. (2016) Circular RNAs
co-precipitate with extracellular vesicles: a possible mechanism for
circRNA clearance, PLoS One, 11, e0148407, doi:
10.1371/journal.pone.0148407.
38.Preußer, C., Hung, L.-H., Schneider, T.,
Schreiner, S., Hardt, M., Moebus, A., Santoso, S., and Bindereif, A.
(2018) Selective release of circRNAs in platelet-derived extracellular
vesicles, J. Extracell. Vesicles, 7, 1424473, doi:
10.1080/20013078.2018.1424473.
39.Hansen, T. B., Jensen, T. I., Clausen, B. H.,
Bramsen, J. B., Finsen, B., Damgaard, C. K., and Kjems, J. (2013)
Natural RNA circles function as efficient microRNA sponges,
Nature, 495, 384-388, doi: 10.1038/nature11993.
40.Thomson, D. W., and Dinger, M. E. (2016)
Endogenous microRNA sponges: Evidence and controversy, Nat. Rev.
Genet., 17, 272-283, doi: 10.1038/nrg.2016.20.
41.Cesana, M., Cacchiarelli, D., Legnini, I.,
Santini, T., Sthandier, O., Chinappi, M., Tramontano, A., and Bozzoni,
I. (2011) A long noncoding RNA controls muscle differentiation by
functioning as a competing endogenous RNA, Cell, 147,
358-369, doi: 10.1016/j.cell.2011.09.028.
42.Conn, V. M., Hugouvieux, V., Nayak, A., Conos, S.
A., Capovilla, G., Cildir, G., Jourdain, A., Tergaonkar, V., Schmid,
M., Zubieta, C., and Conn, S. J. (2017) A circRNA from SEPALLATA3
regulates splicing of its cognate mRNA through R-loop formation,
Nat. Plants, 3, 17053, doi: 10.1038/nplants.2017.53.
43.Li, Z., Huang, C., Bao, C., Chen, L., Lin, M.,
Wang, X., Zhong, G., Yu, B., Hu, W., Dai, L., Zhu, P., Chang, Z., Wu,
Q., Zhao, Y., Jia, Y., Xu, P., Liu, H., and Shan, G. (2015) Exon-intron
circular RNAs regulate transcription in the nucleus, Nat. Struct.
Mol. Biol., 22, 256-264, doi: 10.1038/nsmb.2959.
44.Zhou, W.-Y., Cai, Z.-R., Liu, J., Wang, D.-S.,
Ju, H.-Q., and Xu, R.-H. (2020) Circular RNA: Metabolism, functions and
interactions with proteins, Mol. Cancer, 19, 172, doi:
10.1186/s12943-020-01286-3.
45.Van Heesch, S., Witte, F., Schneider-Lunitz, V.,
Schulz, J. F., Adami, E., Faber, A. B., Kirchner, M., Maatz, H.,
Blachut, S., Sandmann, C.-L., Kanda, M., Worth, C. L., Schafer, S.,
Calviello, L., Merriott, R., Patone, G., Hummel, O., Wyler, E.,
Obermayer, B., et al. (2019) The translational landscape of the human
heart, Cell, 178, 242-260.e29, doi:
10.1016/j.cell.2019.05.010.
46.Chen, X., Han, P., Zhou, T., Guo, X., Song, X.,
and Li, Y. (2016) circRNADb: A comprehensive database for human
circular RNAs with protein-coding annotations, Sci. Rep.,
6, 34985, doi: 10.1038/srep34985.
47.Pamudurti, N. R., Bartok, O., Jens, M.,
Ashwal-Fluss, R., Stottmeister, C., Ruhe, L., Hanan, M., Wyler, E.,
Perez-Hernandez, D., Ramberger, E., Shenzis, S., Samson, M., Dittmar,
G., Landthaler, M., Chekulaeva, M., Rajewsky, N., and Kadener, S.
(2017) Translation of circRNAs, Mol. Cell, 66, 9-21.e7,
doi: 10.1016/j.molcel.2017.02.021.
48.Yang, Y., Fan, X., Mao, M., Song, X., Wu, P.,
Zhang, Y., Jin, Y., Yang, Y., Chen, L.-L., Wang, Y., Wong, C. C., Xiao,
X., and Wang, Z. (2017) Extensive translation of circular RNAs driven
by N6-methyladenosine, Cell Res., 27, 626-641,
doi: 10.1038/cr.2017.31.
49.Qin, S., Zhang, Q., Xu, Y., Ma, S., Wang, T.,
Huang, Y., and Ju, S. (2022) m6A-modified circRNAs:
detections, mechanisms, and prospects in cancers, Mol. Med.,
28, 79, doi: 10.1186/s10020-022-00505-5.
50.Prats, A.-C., David, F., Diallo, L. H., Roussel,
E., Tatin, F., Garmy-Susini, B., and Lacazette, E. (2020) Circular RNA,
the key for translation, Int. J. Mol. Sci., 21, 8591,
doi: 10.3390/ijms21228591.
51.Li, S., Teng, S., Xu, J., Su, G., Zhang, Y.,
Zhao, J., Zhang, S., Wang, H., Qin, W., Lu, Z. J., Guo, Y., Zhu, Q.,
and Wang, D. (2019) Microarray is an efficient tool for circRNA
profiling, Briefings Bioinform., 20, 1420-1433, doi:
10.1093/bib/bby006.
52.Jeck, W. R., and Sharpless, N. E. (2014)
Detecting and characterizing circular RNAs, Nat. Biotechnol.,
32, 453-461, doi: 10.1038/nbt.2890.
53.Pandey, P. R., Rout, P. K., Das, A., Gorospe, M.,
and Panda, A. C. (2019) RPAD (RNase R treatment, polyadenylation, and
poly(A)+ RNA depletion) method to isolate highly pure circular RNA,
Methods, 155, 41-48, doi:
10.1016/j.ymeth.2018.10.022.
54.Dobin, A., and Gingeras, T. R. (2015) Mapping
RNA-seq reads with STAR, Curr. Protoc. Bioinform., 51,
11.14.1-11.14.19, doi: 10.1002/0471250953.bi1114s51.
55.Hansen, T. B., Venø, M. T., Damgaard, C.
K., and Kjems, J. (2016) Comparison of circular RNA prediction tools,
Nucleic Acids Res., 44, e58, doi:
10.1093/nar/gkv1458.
56.Panda, A. C., and Gorospe, M. (2018) Detection
and analysis of circular RNAs by RT-PCR, BioProtocol, 8,
e2775, doi: 10.21769/BioProtoc.2775.
57.Das, A., Rout, P. K., Gorospe, M., and Panda, A.
C. (2019) Rolling circle cDNA synthesis uncovers circular RNA splice
variants, Int. J. Mol. Sci., 20, 3988, doi:
10.3390/ijms20163988.
58.Chen, D.-F., Zhang, L.-J., Tan, K., and Jing, Q.
(2018) Application of droplet digital PCR in quantitative detection of
the cell-free circulating circRNAs, Biotechnol. Biotechnol.
Equip., 32, 116-123, doi: 10.1080/13102818.2017.1398596.
59.Conn, V., and Conn, S. J. (2019) SplintQuant: A
method for accurately quantifying circular RNA transcript abundance
without reverse transcription bias, RNA, 25, 1202-1210,
doi: 10.1261/rna.070953.119.
60.Dahl, M., Daugaard, I., Andersen, M. S., Hansen,
T. B., Grønbæk, K., Kjems, J., and Kristensen, L. S.
(2018) Enzyme-free digital counting of endogenous circular RNA
molecules in B-cell malignancies, Lab. Invest., 98,
1657-1669, doi: 10.1038/s41374-018-0108-6.
61.Guo, Q., Wang, J., Sun, R., He, Z., Chen, Q.,
Liu, W., Wu, M., Bao, J., Liu, Z., Wang, J., and Zhang, Y. (2020)
Comprehensive construction of a circular RNA-associated competing
endogenous RNA network identified novel circular RNAs in hypertrophic
cardiomyopathy by integrated analysis, Front. Genet., 11,
764, doi: 10.3389/fgene.2020.00764.
62.Xia, S., Feng, J., Chen, K., Ma, Y., Gong, J.,
Cai, F., Jin, Y., Gao, Y., Xia, L., Chang, H., Wei, L., Han, L., and
He, C. (2018) CSCD: a database for cancer-specific circular RNAs,
Nucleic Acids Res., 46, D925-D929, doi:
10.1093/nar/gkx863.
63.Zhang, W., Liu, Y., Min, Z., Liang, G., Mo, J.,
Ju, Z., Zeng, B., Guan, W., Zhang, Y., Chen, J., Zhang, Q., Li, H.,
Zeng, C., Wei, Y., and Chan, G. C.-F. (2022) circMine: A comprehensive
database to integrate, analyze and visualize human disease-related
circRNA transcriptome, Nucleic Acids Res., 50, D83-D92,
doi: 10.1093/nar/gkab809.
64.Sinha, T., Mishra, S. S., Singh, S., and Panda,
A. C. (2022) PanCircBase: an online resource for the exploration of
circular RNAs in pancreatic islets, Front. Cell Dev. Biol.,
10, 942762, doi: 10.3389/fcell.2022.942762.
65.Dudekula, D. B., Panda, A. C., Grammatikakis, I.,
De, S., Abdelmohsen, K., and Gorospe, M. (2016) CircInteractome: a web
tool for exploring circular RNAs and their interacting proteins and
microRNAs, RNA Biol., 13, 34-42, doi:
10.1080/15476286.2015.1128065.
66.Liu, Y. C., Li, J. R., Sun, C. H., Andrews, E.,
Chao, R. F., Lin, F. M., Weng, S. L., Hsu, S. D., Huang, C. C., Cheng,
C., Liu, C. C., and Huang, H. D. (2016) CircNet: a database of circular
RNAs derived from transcriptome sequencing data, Nucleic Acids
Res., 44, D209-D215, doi: 10.1093/nar/gkv940.
67.Li, J.-H., Liu, S., Zhou, H., Qu, L.-H., and
Yang, J.-H. (2014) starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and
protein-RNA interaction networks from large-scale CLIP-Seq data,
Nucleic Acids Res., 42, D92-D97, doi:
10.1093/nar/gkt1248.
68.Meng, X., Hu, D., Zhang, P., Chen, Q., and Chen,
M. (2019) CircFunBase: a database for functional circular RNAs,
Database, 2019, baz003, doi: 10.1093/database/baz003.
69.Li, H., Xie, M., Wang, Y., Yang, L., Xie, Z., and
Wang, H. (2021) riboCIRC: a comprehensive database of translatable
circRNAs, Genome Biol., 22, 79, doi:
10.1186/s13059-021-02300-7.
70.Werfel, S., Nothjunge, S., Schwarzmayr, T.,
Strom, T.-M., Meitinger, T., and Engelhardt, S. (2016) Characterization
of circular RNAs in human, mouse and rat hearts, J. Mol. Cell.
Cardiol., 98, 103-107, doi: 10.1016/j.yjmcc.2016.07.007.
71.Wang, K., Long, B., Liu, F., Wang, J. X., Liu, C.
Y., Zhao, B., Zhou, L. Y., Sun, T., Wang, M., Yu, T., Gong, Y., Liu,
J., Dong, Y. H., Li, N., and Li, P. F. (2016) A circular RNA protects
the heart from pathological hypertrophy and heart failure by targeting
miR-223, Eur. Heart J., 37, 2602-2611, doi:
10.1093/eurheartj/ehv713.
72.Lim, T. B., Aliwarga, E., Luu, T. D. A., Li, Y.
P., Ng, S. L., Annadoray, L., Sian, S., Ackers-Johnson, M. A., and Foo,
R. S.-Y. (2019) Targeting the highly abundant circular RNA circSlc8a1
in cardiomyocytes attenuates pressure overload induced hypertrophy,
Cardiovasc. Res., 115, 1998-2007, doi:
10.1093/cvr/cvz130.
73.Carè, A., Catalucci, D., Felicetti, F.,
Bonci, D., Addario, A., Gallo, P., Bang, M.-L., Segnalini, P., Gu, Y.,
Dalton, N. D., Elia, L., Latronico, M. V. G., Høydal, M.,
Autore, C., Russo, M. A., Dorn, G. W., Ellingsen, O., Ruiz-Lozano, P.,
Peterson, K. L., et al. (2007) MicroRNA-133 controls cardiac
hypertrophy, Nat. Med., 13, 613-618, doi:
10.1038/nm1582.
74.Li, H., Xu, J.-D., Fang, X.-H., Zhu, J.-N., Yang,
J., Pan, R., Yuan, S.-J., Zeng, N., Yang, Z.-Z., Yang, H., Wang, X.-P.,
Duan, J.-Z., Wang, S., Luo, J.-F., Wu, S.-L., and Shan, Z.-X. (2020)
Circular RNA circRNA_000203 aggravates cardiac hypertrophy via
suppressing miR-26b-5p and miR-140-3p binding to Gata4, Cardiovasc.
Res., 116, 1323-1334, doi: 10.1093/cvr/cvz215.
75.Pikkarainen, S., Tokola, H., Kerkelä, R.,
and Ruskoaho, H. (2004) GATA transcription factors in the developing
and adult heart, Cardiovasc. Res., 63, 196-207, doi:
10.1016/j.cardiores.2004.03.025.
76.Yang, M.-H., Wang, H., Han, S.-N., Jia, X.,
Zhang, S., Dai, F.-F., Zhou, M.-J., Yin, Z., Wang, T.-Q., Zang, M.-X.,
and Xue, L.-X. (2020) Circular RNA expression in isoproterenol
hydrochloride-induced cardiac hypertrophy, Aging, 12,
2530-2544, doi: 10.18632/aging.102761.
77.Pan, J., Xu, Z., Guo, G., Xu, C., Song, Z., Li,
K., Zhong, K., and Wang, D. (2021) Circ_nuclear factor I X (circNfix)
attenuates pressure overload-induced cardiac hypertrophy via regulating
miR-145-5p/ATF3 axis, Bioengineered, 12, 5373-5385, doi:
10.1080/21655979.2021.1960462.
78.Xu, X., Wang, J., and Wang, X. (2020) Silencing
of circHIPK3 inhibits pressure overload-induced cardiac hypertrophy and
dysfunction by sponging miR-185-3p, Drug Design Dev. Ther.,
14, 5699-5710, doi: 10.2147/DDDT.S245199.
79.Yang, M., Wang, W., Wang, L., and Li, Y. (2023)
Circ_0001052 promotes cardiac hypertrophy via elevating Hipk3,
Aging, 15, 1025-1038, doi: 10.18632/aging.204521.
80.Li, C., Wang, J., Feng, J., Zhou, J., Hou, L.,
Gao, Y., and Cheng, Z. (2022) Circ-TLR4 promotes cardiac hypertrophy
through recruiting FUS to stabilize TLR4 mRNA, J. Intervent. Cardiac
Electrophysiol., 65, 153-163, doi:
10.1007/s10840-022-01209-w.
81.Zuo, H., Li, L., Wang, X., Chen, S., Liao, Z.,
Wei, S., Ruan, H., Li, T., and Chen, J. (2023) A novel circ_0018553
protects against angiotensin-induced cardiac hypertrophy in
cardiomyocytes by modulating the miR-4731/SIRT2 signaling pathway,
Hypertens. Res., 46, 421-436, doi:
10.1038/s41440-022-01111-y.
82.Wang, W., Wang, L., Yang, M., Wu, C., Lan, R.,
Wang, W., and Li, Y. (2021) Circ-SIRT1 inhibits cardiac hypertrophy via
activating SIRT1 to promote autophagy, Cell Death Disease,
12, 1069, doi: 10.1038/s41419-021-04059-y.
83.Lavenniah, A., Luu, T. D. A., Li, Y. P., Lim, T.
B., Jiang, J., Ackers-Johnson, M., and Foo, R. S. (2020) Engineered
circular RNA sponges act as miRNA inhibitors to attenuate pressure
overload-induced cardiac hypertrophy, Mol. Ther., 28,
1506-1517, doi: 10.1016/j.ymthe.2020.04.006.
84.Ucar, A., Gupta, S. K., Fiedler, J., Erikci, E.,
Kardasinski, M., Batkai, S., Dangwal, S., Kumarswamy, R., Bang, C.,
Holzmann, A., Remke, J., Caprio, M., Jentzsch, C., Engelhardt, S.,
Geisendorf, S., Glas, C., Hofmann, T. G., Nessling, M., Richter, K., et
al. (2012) The miRNA-212/132 family regulates both cardiac hypertrophy
and cardiomyocyte autophagy, Nat. Commun., 3, 1078, doi:
10.1038/ncomms2090.
85.Semsarian, C., Ingles, J., Maron, M. S., and
Maron, B. J. (2015) New perspectives on the prevalence of hypertrophic
cardiomyopathy, J. Am. College Cardiol., 65, 1249-1254,
doi: 10.1016/j.jacc.2015.01.019.
86.Maron, B. J., Gardin, J. M., Flack, J. M.,
Gidding, S. S., Kurosaki, T. T., and Bild, D. E. (1995) Prevalence of
hypertrophic cardiomyopathy in a general population of young adults.
Echocardiographic analysis of 4111 subjects in the CARDIA study.
Coronary artery risk development in (young) adults, Circulation,
92, 785-789, doi: 10.1161/01.cir.92.4.785.
87.The Task Force for the Diagnosis and Management
of Hypertrophic Cardiomyopathy of the European Society of Cardiology
(ESC) (2014) 2014 ESC guidelines on diagnosis and management of
hypertrophic cardiomyopathy, Eur. Heart J., 35,
2733-2779, doi: 10.1093/eurheartj/ehu284.
88.Harper, A. R., Goel, A., Grace, C., Thomson, K.
L., Petersen, S. E., Xu, X., Waring, A., Ormondroyd, E., Kramer, C. M.,
Ho, C. Y., Neubauer, S., HCMR Investigators, Tadros, R., Ware, J. S.,
Bezzina, C. R., Farrall, M., and Watkins, H. (2021) Common genetic
variants and modifiable risk factors underpin hypertrophic
cardiomyopathy susceptibility and expressivity, Nat. Genet.,
53, 135-142, doi: 10.1038/s41588-020-00764-0.
89.Jansen, M., Algül, S., Bosman, L. P.,
Michels, M., van der Velden, J., de Boer, R. A., van Tintelen, J. P.,
Asselbergs, F. W., and Baas, A. F. (2022) Blood-based biomarkers for
the prediction of hypertrophic cardiomyopathy prognosis: A systematic
review and meta-analysis, ESC Heart Failure, 9,
3418-3434, doi: 10.1002/ehf2.14073.
90.Sonnenschein, K., Wilczek, A. L., de
Gonzalo-Calvo, D., Pfanne, A., Derda, A. A., Zwadlo, C., Bavendiek, U.,
Bauersachs, J., Fiedler, J., and Thum, T. (2019) Serum circular RNAs
act as blood-based biomarkers for hypertrophic obstructive
cardiomyopathy, Sci. Rep., 9, 20350, doi:
10.1038/s41598-019-56617-2.
91.Gong, K., Yang, K., Xie, T., Luo, Y., Guo, H.,
Tan, Z., Chen, J., Wu, Q., Gong, Y., Wei, L., Luo, J., Yao, Y., Yang,
Y., and Xie, L. (2023) Identification of circRNA-miRNA-mRNA regulatory
network and its role in cardiac hypertrophy, PLoS One,
18, e0279638, doi: 10.1371/journal.pone.0279638.
92.Feng, W., and Han, S. (2022) LncRNA
ADAMTS9-AS1/circFN1 competitively binds to miR-206 to elevate the
expression of ACTB, thus inducing hypertrophic cardiomyopathy, Oxid.
Med. Cell. Longev., 2022, 1450610, doi:
10.1155/2022/1450610.