Enhanced Solubility and One-Step Purification of Functional Dimeric Carboxypeptidase G2
Atefeh Khodakarami1, Bahareh Dabirmanesh1,a*, Sedigheh Asad2, and Mohammad Khaledi1
1Department of Biochemistry, Faculty of Biological Sciences, Tarbiat Modares University, 14115 Tehran, Iran2Department of Biotechnology, College of Science, University of Tehran, 14155 Tehran, Iran
Received April 2, 2020; Revised September 22, 2020; Accepted October 13, 2020
Carboxypeptidase G2 is a bacterial enzyme that catalyzes methotrexate conversion to its inactive forms which are then eliminated via a non-renal pathway in patients with renal disorders during a high-dose methotrexate administration. Due to the increasing demand of this enzyme, it was of interest to simplify its production process. For this reason, we developed a method for production and one-step purification of this enzyme using an intein-mediated system with a chitin-binding affinity tag. The carboxypeptidase G2 gene from Pseudomonas RS16 was optimized, synthesized, cloned into the pTXB1 expression vector and finally transformed into Escherichia coli BL21 (DE3) cells. The optimal condition for the enzyme soluble expression was achieved in 2×YT medium containing 1% glucose at 25°C for 30 h with 0.5 mM IPTG. The enzyme without intein was expressed as inclusion bodies indicating the importance of intein for the protein solubility. The expressed homodimer protein was purified to homogeneity on a chitin affinity column. The Km and kcat values of 6.5 µM and 4.57 s–1, respectively, were obtained for the purified enzyme. Gel filtration analysis indicated that the resulting recombinant protein was a dimer of 83 kDa. Fluorescence and circular dichroism spectroscopy confirmed the enzyme tertiary and secondary structures, respectively. The use of intein-mediated system provided the possibility of the one-step carboxypeptidase G2 purification, paving the way to the application of this enzyme in pharmaceutics.
KEY WORDS: carboxypeptidase G2, intein, methotrexateDOI: 10.1134/S0006297921020073
INTRODUCTION
Carboxypeptidase G2 (CPG2) is a dimeric zinc-dependent metalloenzyme, a 83-kDa exopeptidase, which converts methotrexate to its inactive form, 4-deoxy-4-amino-N10-methylpteroic acid and glutamate, by removing the C-terminal glutamate from folic acid and its analogs [1, 2]. CPG2 enables a non-renal removal of methotrexate in patients with renal failure. This activity has made CPG2 an enzyme of medical interest. CPG2 is also known as glucarpidase and is commercialized under the trade name of Voraxaze [3, 4].
Due to the CPG2 importance, it is crucial to obtain this enzyme in a highly purified form while maintaining its activity and structure. Purification is usually the main step and a challenging process in the production of recombinant pharmaceutical proteins in the biomedical and biotechnological studies [5]. Conventional methods commonly require several costly and time-consuming isolation steps, including various chromatographic procedures. Therefore, the necessity for a simple and cost-effective purification scheme is obvious. Currently, various methods have been used to enrich proteins of interest from crude biological extracts using simple, inexpensive, and one-step methods for commercial purposes [6, 7].
Reversible affinity interaction between a ligand and its binding target has been widely used in the purification of biomolecules. The presence of an affinity tag often affects the characteristics and functions of the target protein, so it might be necessary to remove the tag, especially in the case of therapeutic proteins. Enzymes or chemicals are then required to separate the tag, which adds to the purification steps [8, 9]. These enzymatic methods can also increase the cost of the manufacturing process and in some cases, will lead to the addition of amino acid residues to the fusion protein at the cleavage site [10, 11]. Therefore, self-cleaving intein-fused affinity tags have been created for convenient tag removal via a variety of ways, such as adding reducing agents and changing pH or temperature, depending on the construct used. However, cleavage induction by temperature or pH change in vivo cannot be tightly controlled and may lead to the loss of the product, while thiol reduction can disrupt the structure of proteins containing disulfide bonds [12-14].
The objective of the present study was to simplify the production of CPG2. For this reason, the CPG2 gene sequence from Pseudomonas sp. (strain RS-16) was optimized, synthesized, and cloned into the expression vector, pTXB1. The recombinant construct was then transformed into Escherichia coli BL21 (DE3) cells, and the optimum condition for its expression was selected. The protein encoded by the pTXB1-CPG2 construct contained intein and chitin-binding domain (CBD) affinity tag at its C-terminus. The produced tagged enzyme was significantly more soluble than the native enzyme without the tag. Since the resulting recombinant protein had no disulfide bonds, the thiol-induced cleavage activity of the intein was used to release the CPG2 from the chitin column, while the intein and the CBD remained attached to the resin (one-step purification). The kinetic parameters of the released enzyme were determined. The secondary and the tertiary structures of the purified CPG2 were analyzed using circular dichroism (CD) and fluorescence spectroscopies.
MATERIALS AND METHODS
Chemicals, bacterial strains, and plasmids. Yeast extract and tryptone were from Liofilchem (Roseto Degli Abruzzi, Italy). Isopropyl-β-D-1-thiogalactopyranoside (IPTG) was from Cinnagen (Cinnagen, Karaj, Iran). Methotrexate (MTX) and CPG2 were from Sigma-Aldrich (USA). Restriction endonucleases, Pfu DNA polymerase, and T4 DNA ligase were obtained from Thermo- Scientific (USA). Molecular biology kits were from GeneAll (South Korea). Primers were supplied by Macrogen (South Korea). The intein-mediated purification affinity chitin-binding tag system was from New England Biolab (United Kingdom). Q-Sepharose was provided by GE Healthcare (United Kingdom). Unless stated differently, other chemicals were from Merck (Germany). All chemicals were of analytical grade purity.
Cloning and construction of pTXB1-CPG2 recombinant plasmid. The gene coding for CPG2 was synthesized (GeneCust, France) based on the E. coli codon preference. The primers (forward primer: 5′-GGTGGTCATATGGCACTGGCACAAAAGCGTGAC-3′; reverse primer: 5′-GTGGTTGCTCTTCCGCATTTGCCAGCACCCAGATCCATAATCAGG-3′) were designed to amplify and to clone the CPG2 gene into pTXB1 by the NdeI and SapI sites. The PCR reaction for the gene amplification included the following steps: 95°C for 5 min, 30 cycles of 95°C for 1 min, 59°C for 60 s and 72°C for 1 min, with a final extension at 72°C for 10 min in a Bio-Rad thermocycler (Bio-Rad, USA). The PCR product was then loaded on 1% (w/v) agarose gel and purified. The purified fragment was digested with NdeI and SapI and cloned into the double-digested pTXB1 plasmid (New England Biolabs). DNA sequencing (Macrogen) confirmed the sequence inserted. The resulting construct (pTXB1-CPG2) was used for the transformation of E. coli BL21 (DE3) cells.
Expression of CPG2 in E. coli. In order to select the optimum expression condition, various concentrations of IPTG (0.1, 0.5, and 1 mM), different culture media [Luria Bertani (LB), Minimal medium (M9) and Yeast Extract Tryptone (2×YT)], incubation times (10, 20, and 30 h), and temperatures (25, 30, and 37°C) were tested. 2×YT medium (1 liter) containing ampicillin (100 µg/ml) was inoculated with the transformed bacterial cells and incubated at 37°C at 180 rpm until OD600 of 0.5-0.6. Protein expression was then induced with 0.5 mM IPTG at 25°C for 30 h in the presence of 1% glucose. The cells were harvested by centrifugation (8000g, 15 min, 4°C); the bacterial pellets were resuspended in 20 mM Tris-HCl containing 300 mM NaCl (pH 8) and sonicated on ice with 8 pulses of 10 s. The soluble and insoluble protein fractions were separated by centrifugation (8000g, 30 min, 4°C).
Purification. Chitin column chromatography. To purify the enzyme and to remove its chitin-binding tag and intein, affinity chromatography was carried on a chitin-coated resin as recommended by the manufacturer. Briefly, the chitin column was equilibrated with 20 mM Tris-HCl (pH 8) containing 500 mM NaCl. The protein was loaded onto the column and washed with 20-bed volumes of the same buffer. The bound protein was incubated in 20 mM Tris-HCl (pH 8) and 500 mM NaCl containing 50 mM dithiothreitol (DTT) at 4°C for 16 to 40 h. The eluted samples were collected as 0.5 mL fractions. SDS-PAGE (12% polyacrylamide) was used to confirm the purity of CPG2 and complete removal of the intein and CBD.
Q-Sepharose column chromatography. The CPG2 fusion protein with the intein and intact CBD tag was purified on Q-Sepharose. For this, the supernatant of the crude bacterial cell extract was loaded onto a Q-Sepharose column equilibrated with 20 mM Tris-HCl buffer (pH 8).
After washing the column with the same buffer, a linear gradient of 0-2 M NaCl in 20 mM Tris-HCl (pH 8) was applied. The fusion enzyme was eluted in 20 mM Tris-HCl containing 0.8 M NaCl. The buffer was then exchanged for 50 mM Tris-HCl (pH 7.5) using an Amicon Ultra-15 centrifugal filter unit (Millipore, Billerica, MA, USA; UFC 910024; cut-off, 10 kDa). All steps were carried out at 4°C. After SDS-PAGE, the proteins were stained with Coomassie brilliant blue R-250 [15]. Protein concentration was estimated by the Bradford method [16].
Enzyme activity and biochemical characterization. The activity of CPG2 was measured using MTX as a substrate. Different concentrations of the substrate (1-200 µM) were made by diluting a stock solution (50 mM) with 50 mM Tris-HCl (pH 7.5) containing 50 mM NaCl and 0.2 mM ZnSO4. The reaction was started by adding the enzyme to the reaction mixture (final volume of 300 µl): 50 µl of the purified recombinant fusion enzyme (1 mg/ml) or 10 µl of either recombinant or commercial enzyme with no intein (0.4 mg/ml).
The absorbance at 320 nm (for MTX) was recorded continuously with a Perkin Elmer spectrophotometer and the initial reaction rate was determined using linear regression. The data were fitted into the Michaelis–Menten equation, and the kinetic parameters were calculated using the GraphPad Prism 7 software. One unit of the enzyme was determined as the amount of CPG2 required to convert 1 µmol of the substrate to the product in 1 min [17, 18].
Before activity determination, the lyophilized commercial enzyme (0.4 mg/ml) was dissolved in 50 mM Tris-HCl (pH 7.5) and dialyzed (cut off, 30 kDa) against the same buffer (50 mM Tris-HCl) to remove possible unknown impurities.
Gel filtration. Lysozyme (14 kDa), ovalbumin (43 kDa), bovine serum albumin (66 kDa), and aldolase (150 kDa) were used to generate a standard curve (log MW versus volume) using Sephadex G100 gel filtration. Then, the purified CPG2 was loaded onto the column in 50 mM Tris-HCl containing 150 mM NaCl and eluted with the same buffer to determine its molecular mass.
Structural studies. Fluorescence spectroscopy. Fluorescence emission of aromatic residues of CPG2 was measured in 50 mM Tris-HCl buffer (pH 7.5) with a Perkin Elmer luminescence spectrometer LS 55 (USA). The samples were excited at 280 nm and the emission spectra were recorded from 300 to 400 nm. Both the excitation and the emission slits were set to 10 nm. The enzyme concentration was 30 µg/ml.
CD spectroscopy. Far-UV CD spectra (200-250 nm) were recorded using a JASCO (Japan) J-715 spectropolarimeter. The CPG2 concentration was 0.2 mg/ml in 50 mM Tris-HCl buffer (pH 7.5). The results were expressed as molar ellipticity [θ] (deg·cm2·dmol-1), that was calculated according to the formula [θ]λ = (θ × 100 MRW)/(cl), where c is the protein concentration in mg/ml, l is the optic path length (cm), and θ is the measured ellipticity (degrees) at wavelength λ.
RESULTS AND DISCUSSION
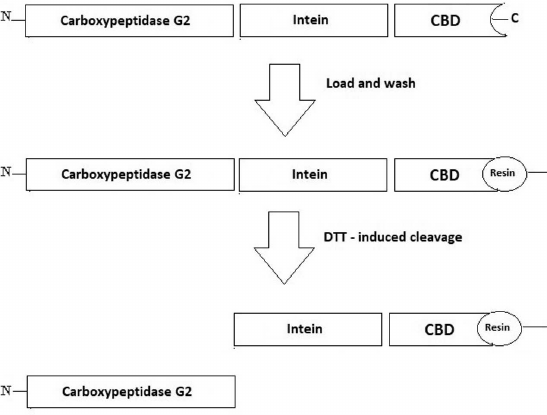
The used pTXB1 vector codes for the intein sequence from Mycobacterium xenopi and the CBD at the C-terminus that can be used to purify the target proteins on a chitin column. CBD is one of the better alternatives to the histidine tags because it is cheaper and can be regenerated more times [19, 20]. The genetically engineered M. xenopi intein can undergo self-cleavage in the presence of thiol reagents resulting in the target protein release (Fig. 1). Here, the CPG2 gene was codon-optimized, synthesized, and then inserted into NdeI and SapI restriction sites of the pTXB1 vector to prevent an addition of any extra amino acid to the purified enzyme after its self-cleavage (Fig. 1). The correct cloning of the full gene was confirmed by DNA sequencing of the resulting construct (pTXB-CPG2).
Fig. 1. Schematic representation of the fusion CPG2 protein. The CPG2 coding sequence was inserted into the pTXB1 vector. The fusion protein consisted of three parts: enzyme itself, intein, and C-terminal CBD to bind to the chitin-coated resin and to facilitate the one-step purification.
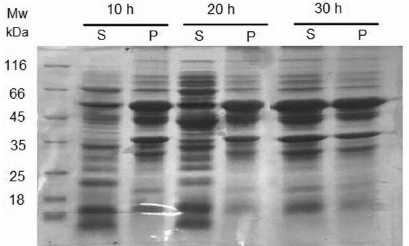
A variety of expression conditions including different culture media (LB, 2×YT, and M9), IPTG concentrations (0.1, 0.5, and 1 mM), temperatures (16, 25, and 37°C), and incubation times (10, 20, and 30 h) were tested. The highest amount of soluble protein (about 50%) was achieved in 2×YT medium containing 1% glucose after induction with 0.5 mM IPTG for 30 h at 25°C (Fig. 2). Under other conditions, protein expression led to a very low amount of soluble protein or to the formation of inclusion bodies. The relative content of soluble CPG2 at the optimal expression condition was quantified using the ImageJ software (Fig. 2), which demonstrated equal CPG2 amounts of in the supernatant and the pellet. Soluble CPG2 comprised about 35% of the total proteins in the supernatant.
Fig. 2. SDS-PAGE analysis of CPG2 expression under the optimal condition. Expression in 2×YT medium containing 1% glucose; induction with 0.5 mM IPTG at 25°C for 10, 20, and 30 h. The optimal expression was observed after 30 h at 25°C; S, supernatant; P, pellet.
Soluble expressed protein was loaded onto the chitin-binding column. The intein cleavage was induced with 50 mM DTT for 10, 20, and 40 h at 4°C. The incubation period of 20 h led to a better cleavage; while incubation for 40 h did not affect significantly the extent of cleavage but dramatically reduced the activity of the enzyme. The yield of active CPG2 was 305 mg CPG2 per liter of culture (Table 1). The purified protein was observed as a 42-kDa band on SDS-PAGE (corresponding to one CPG2 subunit), thus confirming complete removal of intein and CBD from CPG2 (Fig. 3a).
Table 1. Purification of soluble CPG2 from 1
liter of culture
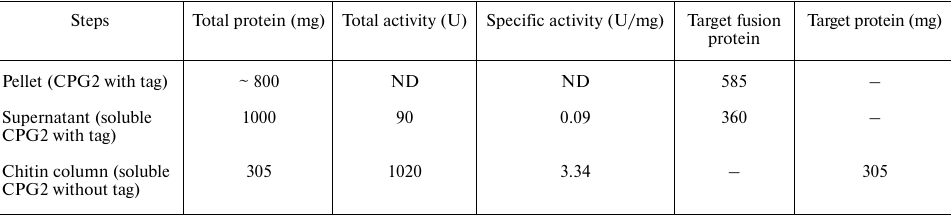
Fig. 3. a) SDS-PAGE for analyzing the on-column cleavage of the CPG2 fusion protein in the presence of DTT at different time points for 40 h. The optimal cleavage time was 20 h. The enzyme was purified to homogeneity after the cleavage. b) Molecular weight determination by gel filtration on Sephadex G100.
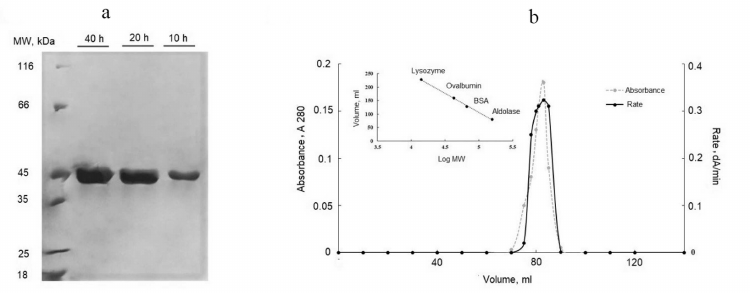
The molecular mass of the enzyme was determined by gel filtration on Sephadex G100. Protein standards were used to provide a calibration curve (log molecular mass versus elution volume). The enzyme was eluted as an active fraction in the volume of 110 ml, which corresponded to the molecular mass of 83 kDa, indicating the presence of a dimeric protein (Fig. 3b).
The kinetic parameters of the purified enzyme with and without the CBD tag were calculated using the Michaelis-Menten equation. The Km and kcat values for the purified enzyme were 6.5 ± 0.3 µM and 4.57 ± 0.23 s–1, respectively, which did not differ significantly from the kcat and Km values of commercially available from Pseudomonas sp. CPG2 (Sigma), used as a control (Table 2). Despite the use of the large CBD tag and DTT treatment for the one-step purification of CPG2, the enzyme activity assay and the gel filtration revealed that the obtained CPG2 retained its dimeric structure and its activity after the on-column cleavage. At the same time, the Km value for the purified enzyme with intein and CBD was about 47 mM and the kcat was 0.47 s–1. It appeared that the presence of the tag significantly reduced the catalytic efficiency of the enzyme due to its large size.
Table 2. Kinetic parameters of CPG2
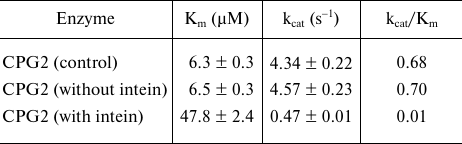
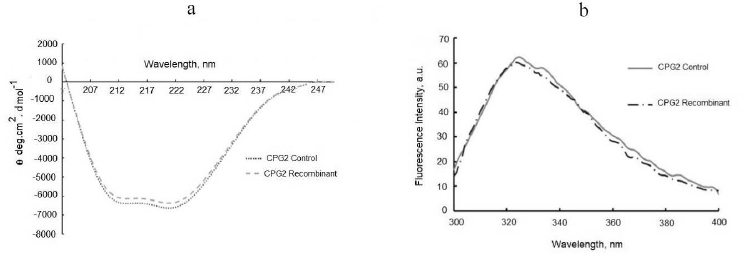
The secondary and the tertiary structures of the soluble CPG2 were studied using far-UV CD and fluorescence spectroscopies to assure that no structural changes had happened. The far-UV CD spectrum of the obtained enzyme was similar to that of the control CPG2 showing negative peaks corresponding to alpha-helical and beta-sheet structures (Fig. 4a) that have been previously reported for CPG2 [21]. The fluorescence spectrum of the recombinant CPG2 did not differ from the control (Fig. 4b).
Fig. 4. Structural analysis of CPG2. a) Far-UV CD spectra of CPG2 enzymes (0.2 mg/ml) in 50 mM Tris-HCl buffer, pH 7.5. b) Intrinsic fluorescence of the purified and control CPG2 enzymes. The excitation wavelength was 280 nm and the emission was recorded from 300 to 400 nm. Enzyme concentration was 60 µg/ml in 50 mM Tris-HCl buffer, pH 7.5.
Our previous research showed that intein could be used for the purification of a tetrameric enzyme (uricase) [22]. Similar to the current results, no functional and structural differences were observed when compared with the control [22]. Human acidic and basic fibroblast growth factors, human α-1 anti-trypsin, and cecropin (antimicrobial) were also expressed using this intein system [23-25].
Previously, CPG2 was purified from its native Pseudomonas sp. RS 16 strain using three purification steps involving chromatographies on SP-Sephadex, DEAE-Sepharose, and finally Sepharose-immobilized Procion Red H-8BN [26]. In another study by Minton et al. [27], this enzyme was expressed in E. coli with a very low yield [27]. To improve its expression, Goda et al. expressed the codon-optimized synthetic gene as inclusion bodies that required refolding, which might have reduced the yield of the active protein. In the same study, the His-tag was used for a single-step purification on a Ni-NTA column, but the tag remained attached to the protein [2]. Recently, Jeyharan et al. reported production of the soluble protein (250 mg/liter) using the His-tag for a simple purification [28]. In their study, the tag was removed by TEV protease, which required an extra purification step on a Ni-NTA column followed by Superdex 75GL chromatography. TEV protease is an expensive enzyme; besides, the cleavage with TEV leaves an extra residue, which may not be suitable for therapeutic proteins [28].
In the current study, GPG2 purification was simplified to a single step using the self-cleaving intein with no added amino acids. So far, several studies have reported purification of proteins using inteins, but the unique characteristics of each protein may result in different outcomes. Therefore, each protein requires to be independently assessed. Here, an appropriate yield of the soluble enzyme (305 mg/liter) was achieved at the optimal cultivation condition with no structural and functional changes occurring to the enzyme. Another key aspect is the ability of this tag to promote the solubility of CPG2. The ccSOL omics prediction was used to calculate the solubility propensity score and to identify soluble fragments within the protein. This program predicts the results of endogenous and heterologous expression of proteins in E. coli based on their hydrophobicity, hydrophilicity, as well as propensity to form β-sheet and α-helical structures [29, 30]. The probability that CPG2 fused with intein and CBD will be expressed in a soluble form was calculated as ~60%, which was more than the overall score obtained for the enzyme alone (35%). The ccSOL omics also identified more insoluble regions in the CPG2 primary sequence when compared to the intein fragment. We have previously cloned the gene encoding CPG2 (with no tag) into the pET28 vector and examined its expression under various conditions; however, only a small amount of the protein was in the soluble fraction (unpublished data). In agreement with our other experimental data, this result also confirmed the ability of this construct to increase the solubility of CPG2.
Although this commercial self-cleaving intein system has been used before for purifying several proteins, to our best knowledge, our investigation is the first one to assess its effect on the protein solubility. Overall, we were able to increase the solubility of the expressed CPG2 and to purify it in a one-step intein-mediated procedure. Notably, the enzyme retained its activity and homodimer structure after expression and purification. Our result brings about the hope that using inteins for purifying multimeric proteins could be an option in the future. Nevertheless, more research is required to improve the intein system alone or in a combination with other systems for the large-scale purification of therapeutic enzymes, especially the multimeric ones.
Acknowledgments. We gratefully appreciate the Research Council of Tarbiat Modares University, Prof. Khosro Khajeh, and Iran National Institute for Medical Research Development (NIMAD, project 940711) for their financial support through this investigation.
Ethics declarations. The authors declare no conflict of interest. This article does not contain description of studies with the involvement of humans or animal subjects.
REFERENCES
1.Rowsell, S., Pauptit, R. A., Tucker, A. D., Melton,
R. G., Blow, D. M., and Brick, P. (1997) Crystal structure of
carboxypeptidase G2, a bacterial enzyme with applications in cancer
therapy, Structure, 5, 337-347, doi:
10.1016/S0969-2126(97)00191-3.
2.Goda, S. K., Rashidi, F. A. B., Fakharo, A. A., and
Al-Obaidli, A. (2009) Functional overexpression and purification of a
codon optimized synthetic glucarpidase (Carboxypeptidase G2) in
Escherichia coli, Protein J., 28, 435-442, doi:
10.1016/J.ENZMICTEC.2016.08.001.
3.Ramsey, L. B., Balis, F. M., O’Brien, M. M.,
Schmiegelow, K., Pauley, J. L., et al. (2018) Consensus guideline for
use of glucarpidase in patients with high-dose methotrexate induced
acute kidney injury and delayed methotrexate clearance,
Oncologist, 23, 52-61.
4.Rattu, M. A., Shah, N., Lee, J. M., Pham, A. Q.,
and Marzella, N. (2013) Glucarpidase (voraxaze), a carboxypeptidase
enzyme for methotrexate toxicity, P T, 38, 732-744.
5.Wingfield, P. T. (2015) Overview of the
purification of recombinant proteins, Curr. Protoc. Protein
Sci., 80, 6.1.1-6.1.35, doi:
10.1002/0471140864.ps0601s80.
6.Rosano, G. L., and Ceccarelli, E. A. (2014)
Recombinant protein expression in Escherichia coli: advances and
challenges, Front. Microbiol., 5, 172, doi:
10.3389/fmicb.2014.00172.
7.Goh, H. C., Sobota, R. M., Ghadessy, F. J., and
Nirantar, S. (2017) Going native: Complete removal of protein
purification affinity tags by simple modification of existing tags and
proteases, Protein Expr. Purif., 129, 18-24, doi:
10.1016/J.PEP.2016.09.001.
8.Li, Y. (2011) Self-cleaving fusion tags for
recombinant protein production, Biotechnol. Lett., 33,
869-881, doi: 10.1007/s10529-011-0533-8.
9.Fan, Y., Miozzi, J. M., Stimple, S. D., Han, T. C.,
and Wood, D. W. (2018) Column-free purification methods for recombinant
proteins using self-cleaving aggregating tags, Polymers (Basel),
10, 468, doi: 10.3390/polym10050468.
10.Wu, W. Y., Mee, C., Califano, F., Banki, R., and
Wood, D. W. (2006) Recombinant protein purification by self-cleaving
aggregation tag, Nat. Protoc., 1, 2257-2262, doi:
10.1038/nprot.2006.314.
11.Arnau, J., Lauritzen, C., Petersen, G. E., and
Pedersen, J. (2006) Current strategies for the use of affinity tags and
tag removal for the purification of recombinant proteins, Protein
Expr. Purif., 48, 1-13, doi: 10.1016/J.PEP.2005.12.002.
12.Belfort, M., Stoddard, B. L., Wood, D. W., and
Derbyshire, V. (2006). Homing endonucleases and inteins, Springer
Science & Business Media.
13.Banki, R., and Wood, D. W. (2005) Inteins and
affinity resin substitutes for protein purification and scale up,
Microb. Cell Fact., 4, 1-6, doi:
10.1186/1475-2859-4-32.
14.Lahiry, A., Fan, Y., Stimple, S. D., Raith, M.,
and Wood, D. W. (2018) Inteins as tools for tagless and traceless
protein purification, J. Chem. Technol. Biotechnol., 93,
1827-1835, doi: 10.1002/jctb.5415.
15.Laemmli, U. K. (1970) Cleavage of structural
proteins during the assembly of the head of bacteriophage T4,
Nature, 227, 680.
16.Bradford, M. M. (1976) A rapid and sensitive
method for the quantitation of microgram quantities of protein
utilizing the principle of protein-dye binding, Anal. Biochem.,
72, 248-254.
17.Rashidi, F. B., AlQhatani, A. D., Bashraheel, S.
S., Shaabani, S., Groves, M. R., et al. (2018) Isolation and molecular
characterization of novel glucarpidases: enzymes to improve the
antibody directed enzyme pro-drug therapy for cancer treatment, PLoS
One, 13, e0196254, doi: 10.1371/journal.pone.0196254.
18.AlQahtani, A. D, Al-Mansoori, L., Bashraheel, S.
S., Rashidi, F. B., Al-Yafei, A., et al. (2019) Production of
“biobetter” glucarpidase variants to improve drug
detoxification and antibody directed enzyme prodrug therapy for cancer
treatment, Eur. J. Pharm. Sci., 127, 79-91, doi:
10.1016/J.EJPS.2018.10.014.
19.Wang, L, Kang, J. H., Kim, K. H., and Leeb, E. K.
(2009) Expression of intein-tagged fusion protein and its applications
in downstream processing, J. Chem. Technol. Biotechnol.,
85, 11-18, doi: 10.1002/jctb.2277.
20.Fong, B. A., Wu, W. Y., and Wood, D. W. (2010)
The potential role of self-cleaving purification tags in
commercial-scale processes, Trends Biotechnol., 28,
272-279, doi: 10.1016/j.tibtech.2010.02.003.
21.Yachnin, B. J., and Khare, S. D. (2017)
Engineering carboxypeptidase G2 circular permutations for the design of
an autoinhibited enzyme, Protein Eng. Des., 30, 321-331,
doi: 10.1093/protein/gzx005.
22.Alishah, K., Asad, S., Khajeh, K., and Akbari, N.
(2016) Utilizing intein-mediated protein cleaving for purification of
uricase, a multimeric enzyme, Enzyme Microb. Technol.,
93-94, 92-98, doi: 10.1016/J.Enzmictec.2016.08.001.
23.Wood, D. W., Derbyshire, V., Wu, W., Chartrain,
M., Belfort, M., and Belfort, G. (2000) Optimized single-step affinity
purification with a self-cleaving intein applied to human acidic
fibroblast growth factor, Biotechnol. Prog., 16,
1055-1063, doi: 10.1021/bp0000858.
24.Sharma, S. S., Chong, S., and Harcum, S. W.
(2006) Intein-mediated protein purification of fusion proteins
expressed under high-cell density conditions in E. coli, J.
Biotechnol., 125, 48-56, doi:
10.1016/j.jbiotec.2006.01.018.
25.Díaz, M., Venturini, E., Marchetti, S.,
Arenas, G., and Marshall, S. H. (2012) Intein-mediated expression of
cecropin in Escherichia coli, Electron. J. Biotechnol.,
15, 1-10.
26.Sherwood, R. F, Melton, R. G., Alwan, S. M., and
Hughes, P. (1985) Purification and properties of carboxypeptidase G2
from Pseudomonas sp. strain RS-16, Eur. J. Biochem.,
148, 447-453, doi: 10.1111/j.1432-1033.1985.tb08860.x.
27.Minton, N. P., Atkinson, T., and Sherwood, R. F.
(1983) Molecular cloning of the Pseudomonas carboxypeptidase G2
gene and its expression in Escherichia coli and Pseudomonas
putida, J. Bacteriol., 156, 1222-1227.
28.Jeyaharan, D., Aston, P., Garcia-Perez, A.,
Schouten, J., Davis, P., and Dixon, A. M. (2016) Soluble expression,
purification and functional characterisation of carboxypeptidase G2 and
its individual domains, Protein Expr. Purif., 127, 44-52,
doi: 10.1016/J.PEP.2016.06.015.
29.Agostini, F., Cirillo, D., Livi, C. M.,
DelliPonti, R., and Tartaglia, G. G. (2014) ccSOL omics: a webserver
for solubility prediction of endogenous and heterologous expression in
Escherichia coli, Bioinformatics, 30, 2975-2977,
doi: 10.1093/bioinformatics/btu420.
30.Hebditch, M., Carballo-Amador, M. A., Charonis,
S., Curtis, R., and Warwicker, J. (2017) Protein-Sol: a web tool for
predicting protein solubility from sequence, Bioinformatics,
33, 3098-3100, doi: 10.1093/bioinformatics/btx345.