REVIEW: Glucocorticoids: Dr. Jekyll and Mr. Hyde of Hippocampal Neuroinflammation
Alexey P. Bolshakov1, Liya V. Tret’yakova1, Alexey A. Kvichansky1, and Natalia V. Gulyaeva1,2,a*
1Institute of Higher Nervous Activity and Neurophysiology, Russian Academy of Sciences, 117485 Moscow, Russia2Research and Clinical Center for Neuropsychiatry of Moscow Healthcare Department, 115419 Moscow, Russia
* To whom correspondence should be addressed.
Received August 9, 2020; Revised October 13, 2020; Accepted October 13, 2020
Glucocorticoids (GCs) are an important component of adaptive response of an organism to stressogenic stimuli, a typical stress response being accompanied by elevation of GC levels in blood. Anti-inflammatory effects of GCs are widely used in clinical practice, while pro-inflammatory effects of GCs are believed to underlie neurodegeneration. This is particularly critical for the hippocampus, brain region controlling both cognitive function and emotions/affective behavior, and selectively vulnerable to neuroinflammation and neurodegeneration. The hippocampus is believed to be the main target of GCs since it has the highest density of GC receptors potentially underlying high sensitivity of hippocampal cells to severe stress. In this review, we analyzed the results of studies on pro- and anti-inflammatory effects of GCs in the hippocampus in different models of stress and stress-related pathologies. The available data form a sophisticated, though often quite phenomenological, picture of a modulatory role of GCs in hippocampal neuroinflammation. Understanding the dual nature of GC-mediated effects as well as causes and mechanisms of switching can provide us with effective approaches and tools to avert hippocampal neuroinflammatory events and as a result to prevent and treat brain diseases, both neurological and psychiatric. In the framework of a mechanistic view, we propose a new hypothesis describing how the anti-inflammatory effects of GCs may transform into the pro-inflammatory ones. According to it, long-term elevation of GC level or preliminary treatment with GC triggers accumulation of FKBP51 protein that suppresses activity of GC receptors and activates pro-inflammatory cascades, which, finally, leads to enhanced neuroinflammation.
KEY WORDS: glucocorticoids, stress response, hippocampus, neuroinflammation, cytokines, corticosterone, FKBP51DOI: 10.1134/S0006297921020048
Abbreviations: ACTH, adrenocorticotropic hormone; AP1, activator protein-1; CRH, corticotropin-releasing hormone; DEX, dexamethasone; GC, glucocorticoid; GR, glucocorticoid receptor; GRE, glucocorticoid response element; HPA, hypothalamic-pituitary-adrenal; HRE, hormone response element; IL, interleukin; LPS, lipopolysaccharide; MCAO, middle cerebral artery occlusion; MR, mineralocorticoid receptor; NF-κB, nuclear factor kappa B; TNF-α, tumor necrosis factor-α.
INTRODUCTION
The hypothalamic-pituitary-adrenal (HPA) axis is the major stress-reactive system of the organism providing an efficient adaptation to environmental conditions when exposed to physical or psychosocial stressogenic factors. The HPA axis is a complex of positive and negative feedbacks between the brain and endocrine system, in particular hypothalamus, anterior pituitary, and adrenal glands. The paraventricular nucleus of the hypothalamus contains stress-responsive neuroendocrine cells synthesizing and releasing the corticotropin-releasing hormone (CRH) recognized by the anterior lobe of the pituitary gland. CRH stimulates secretion of the adrenocorticotropic hormone (ACTH) from the pituitary to blood. ACTH, in turn, is transported by blood to the adrenal glands, where it triggers synthesis of glucocorticoids (GCs) in the adrenal cortex and their release into blood. Thus, GCs, hormones of corticosteroid family, are messengers of the HPA axis, its effector molecules. Cortisol is the most functionally active GC in humans and corticosterone, in rodents [1]. The HPA axis contains several negative feedback loops, their activation leads to the HPA activity suppression. Cortisol/corticosterone produced by the adrenal cortex inhibits both the hypothalamus and the pituitary, which reduces the synthesis of CRH and ACTH and, subsequently, results in the decreased production of GCs. These negative feedbacks decrease or terminate adaptation response induced by stress factors. In this review, we will analyze the role of GCs in the stress-related regulation of neuroinflammation. Since the majority of experimental data have been obtained in rodents, corticosterone will be the main object of the discussion.
In addition to the contribution to the development of stress response, GCs are recognized as modulators of inflammation. Inflammation is a typical tissue response to injury or infection aiming at elimination of pro-inflammatory factors and/or repair of the affected tissue. Neuroinflammation is a specific reaction of the nervous system including the following cellular and molecular elements: (i) changes in the microglial shape and gene expression pattern; (ii) activation of cytokine synthesis (IL-1β, IL-6, TNF-α) and of other paracrine inflammatory mediators production (prostaglandins, active forms of nitrogen and oxygen); (iii) attraction of cells of the immune system from the peripheral bloodstream due to altered functioning of the blood-brain barrier. Neuroinflammatory reaction can be induced by various internal and external pathological stimuli such as injuries, infections, toxic substances, etc. However, limited and controlled local neuroinflammation is important for optimal neuroplasticity, including neurogenesis and synaptic functions [2]. This may be especially important for the hippocampus, a particularly vulnerable to stress key limbic structure involved in both cognitive and emotional functions. It is generally accepted that uncontrolled neuroinflammatory process in hippocampus is one of the key links in psychiatric and neurological diseases such as depressive spectrum disorders, epilepsy, and post-stroke and post-traumatic affective and cognitive impairments [3]. A major hypothesis that explains development of the uncontrolled neuroinflammation specifically in hippocampus is that the exceptionally high density of GC receptors in the hippocampal cells makes this part of the brain highly sensitive to the stress-induced GC challenges, which, depending on conditions, could produce either pro- or anti-inflammatory effect in the hippocampus. Experimental data shaping current views on the mechanisms of the corticosterone-dependent regulation of neuroinflammatory processes are extremely controversial. These contradictions cause the necessity of systemic analysis of the information on the GC-dependent mechanisms modulating neuroinflammatory processes in the hippocampus.
RECEPTORS OF GLUCOCORTICOIDS
Two main types of receptors binding cortisol/corticosterone are glucocorticoid (GR) and mineralocorticoid (MR) receptors belonging to the nuclear receptor subfamily 3 group C, NR3C. GR is encoded by the NR3C1 gene, while MR by NR3C2 gene. Two isoforms of GR represent an active (GRα) and inactive (GRβ) receptor forms. Expression of the GRβ isoform could be increased in response to the pro-inflammatory cytokines, resulting in the decreased sensitivity to GCs underlying the phenomenon of the so-called glucocorticoid resistance [4]. Therefore, response of cells to GCs depends on the balance between these isoforms and could play an important role in shaping of the cellular response to elevated levels of GCs. Importantly, binding of corticosterone to GR does not necessarily result in the GR activation. It was shown that chaperones Hsp70 and Hsp90 interact with GR and either inactivate or activate GR [5, 6]. FKBP51 is a protein encoded by the fkbp5 gene, which interacts with GR-Hsp90 complex and decreases its affinity to GCs [7]. The FKBP51 expression is GR-dependent and transient activation of GR could elevate the FKBP51 level; its binding to GR subsequently suppresses the GR-dependent response either via inhibition of the GR protein complex translocation to the nucleus or via decrease of the affinity of HSP90-GR complex to GCs [8]. Characteristics of the interaction between GR and FKBP51, as well as the factors affecting these interactions, are described in more detail in reviews [9-11]. Since FKBP51 can act as a regulator of GR function, it has been actively studied due to its potential involvement in the mechanisms of depression and post-traumatic stress disorder. In addition, it was shown that expression of this protein in the hippocampal neurons could be increased after stress [12-14], as well as after injection of dexamethasone (10 mg/kg) [12], which may be due to a decrease in methylation of the fkbp5 gene promoter [15]. Below we will demonstrate that the FKBP51-mediated suppression of GR function may be a good candidate for explanation of the corticosterone-induced priming of neuroinflammation.
Glucocorticoids, in addition to the binding to GR, interact with the MR with higher affinity. The isoforms and transcript variants of MRs are not studied well, however, two MR splice variants have been found in human and rats [16]. Functional significance of these MR transcripts and their expression in different parts of the brain are still poorly understood. Like GRs, MRs can bind corticosterone, however, binding affinity of corticosterone to MR is several times higher than that to GR [17]. Occurrence of these receptors in the brain structures is different. While initially MR was found mainly in the limbic areas, especially in the hippocampus; later the single cell RNAseq studies showed that the majority of cells in all brain areas contain MR mRNA [18-20]. The only exception is microglia which does not express MRs. Unlike MR, GR is abundant in the entire brain, in all areas and cell types studied.
GR interact with endogenous GCs, such as corticosterone or cortisol, or with synthetic GC analogues [dexamethasone (DEX), prednisolone, etc]. GR is located in the cytoplasm as a monomer in the complex with heat shock proteins. The ligand-receptor binding triggers conformational changes in the receptor leading to formation of the homodimer and its translocation from the cytoplasm to the nucleus. In the nucleus, the activated GR binds to the glucocorticoid response elements (GREs) and negative GREs that have a palindromic structure. Subunits of the homodimer interact with the half of their sequence providing high binding affinity of the GR dimer. GR binding with DNA triggers assembly of the transcriptional regulatory complexes serving as co-activators or co-repressors of transcription [21]. Activation of MR stimulates similar mechanisms: after binding with an agonist, they form protein complexes and reach the nucleus, where they interact with the hormone response elements (HREs) and regulate expression of genes. A stress-induced increase in the corticosterone level results in activation of MRs and GRs and accumulation of these receptors in the nuclei of brain cells of the stressed animals [22]. Notably, the time course of nuclear accumulation of GRs and MRs after acute stress varies between brain areas: the rapid (in 10 min) accumulation of MRs is observed in the prefrontal cortex, while GRs are translocated to the nuclei in the cells of the dorsal hippocampus. A delayed (in 1 h) elevation in the MR level was observed in both parts of hippocampus, while GRs remained at high level in the nuclei of the dorsal hippocampus and accumulated in the frontal cortex. Importantly, the blood level of corticosterone remained high for at least 60 min after a 30-min stress induced by placing an animal on the elevated platform [22]. These data have several important outcomes. Firstly, translocation of MRs and GRs to the nucleus does not directly follow blood corticosterone level and depends on some additional factors such as local corticosterone concentration and modifications of MRs and GRs. Secondly, mechanisms regulating the level of MR and GR activation depend on the brain structure [23] suggesting that the balance between the pro- and anti-inflammatory activity of GCs may also vary between the brain structures. Thirdly, the stress induces long-term changes in the activity of both MRs and GRs preventing reliable prediction of the local pattern of gene expression after stress.
A membrane form of MR has been identified that mediates rapid non-genomic effects [24]. These receptors were firstly found in the hippocampal pyramidal neurons. Later the short-term activation of MRs by aldosterone was shown to enhance the long-term potentiation in the ventral but not dorsal hippocampus [25]. So far, it is not clear whether these membrane MRs are present in other cell types except neurons and whether they may be involved in the regulation of neuroinflammation.
It also remains unknown whether MRs in the brain cells are involved in the regulation of neuroinflammation. Current literature contains no studies where the role of these receptors was directly investigated. The role of MR is most studied in the regulation of neuroinflammation under conditions of arterial hypertension and associated stroke. It is known that neuroinflammation during arterial hypertension is associated with MR activation. Rats with spontaneous hypertension are characterized by the increased level of MR expression and neuroinflammation in the hippocampus [26]. At the same time, it is known that the development of inflammation in these rats is mediated by MR of the vascular endothelium, which can be activated by aldosterone [27]. It can be assumed that, in this model, neuroinflammation is primed by the vascular endothelium sensitive to aldosterone, and not by the cells of the central nervous system, and an increase in MR expression is a consequence of already developed neuroinflammation. This hypothesis is supported by the work of Bay-Richter, C. et al. demonstrating that the systemic administration of aldosterone facilitates development of neuroinflammation in the frontal cortex after systemic administration of bacterial lipopolysaccharide [28]. This MR agonist is inactive in this region of the brain [29] but can activate MR in the endothelial cells or in the periphery. It is assumed that the protective role of spironolactone in stroke is due to its action on the walls of blood vessels and prevention of the migration of cells of the immune system into the parenchyma of the central nervous system [30].
It was found using the models of brain damage in mice that the disruption of MR functioning, including in cells of the immune system outside the brain, has a protective effect. It has been shown that the deletion of MR in the LysM-positive cells (myeloid cells) leads to a decrease in the size of the infarction zone and to a decrease in the intensity of neuroinflammation in the model of middle cerebral artery occlusion [31]. Microglia are the main population of the healthy mouse brain cells that can be affected by this deletion, but microgliocytes in the brain of adult mice do not express MR [20]. Montes-Cobos et al. have shown that the similar deletion facilitates the course of experimental autoimmune encephalitis in mice. This study also showed that this effect may not be specifically associated with the tissues of the central nervous system since functionality of the entire immune system was impaired in these mice [32]. Thus, currently, there are no data in the literature that allow us to unambiguously state that MR expressed by the CNS cells regulates the processes of neuroinflammation.
It should also be noted that GCs are secreted in individual peaks, forming an ultradian rhythm. In rats, there is an approximately hourly cycle of the corticosterone release regulated by activation and inhibition of the HPA axis. At the signal transduction level, the response to corticosterone release peaks depends on its dynamic interaction with GR and MR [33]. The effects of low basal concentrations of endogenous GCs seem to be mediated by MRs because of their higher affinity. Low affinity GRs play a key role at high concentrations of endogenous glucocorticoids (under stress or daily peaks) [34]. It seems that the optimal function of GCs is realized at their moderate concentration, while hyper- and hyposecretion cause negative consequences [35]. Cushing’s syndrome is the typical clinical example of GCs hypersecretion resulting from different pathologies [36]. Remarkably, Cushing’s syndrome is frequently associated with psychopathology, the spectrum of behavioral abnormalities observed in the patients ranging from severe depression to mania [37]. Importantly, the long-term elevation in GC levels could induce reduced sensitivity or resistance of the receptors to GCs [38]. Overall, GC resistance can be caused by disruptions at various levels of the GC signaling cascade: impaired expression of GR, impaired binding of GC to its receptor, impaired translocation to the nucleus, impaired cofactor activity [39]. Since one of the major effects of GCs is related to the suppression of inflammation, the development of GC resistance may result in the enhancement of pro-inflammatory response.
ANTI-INFLAMMATORY ACTION OF GCs
On the periphery, GCs are known to act as anti-inflammatory agents. In most cases, activation of GR and MR leads to inhibition of the activity of immune cells and induction of apoptosis in lymphocytes [40]. However, among immune cells, subpopulation of Th17 lymphocytes demonstrates a significantly greater resistance to the GC-induced apoptosis [41]. This lymphocyte subpopulation is known to be responsible for chronic inflammatory airway diseases [42]. The subpopulation of T-suppressors may be significantly more sensitive to the inhibitory effect of low concentrations of GCs than the subpopulations of T-helpers (including Th17) and T-killers, as well as B-cells. Thus, at the relatively low concentrations, GCs rather exhibits an immunostimulating (pro-inflammatory) effect, shifting the T-helper/T-suppressor ratio towards predominance of the T-helper activity. At the higher concentrations, GCs could have an immunosuppressive effect with intensity of immunosuppression being directly proportional to the GC concentration in blood and growing almost linearly up to the levels 100 times higher than the physiological one [43]. In addition to modulation of the activity of lymphocytes, GCs could inhibit inflammation via several other ways: (i) GCs inhibit tissue infiltration of blood immune cells due to suppression of their chemotaxis [44]; (ii) GCs inhibit cytokine gene expression [45]; (iii) GCs inhibit expression of adhesion molecules [46]; (iv) GCs inhibit various functions of leukocytes related to inflammation (epithelial adhesion, emigration, chemotaxis, phagocytosis, respiratory burst, etc.) [47]; (v) GCs suppress phospholipase A2 expression [48]; (vi) GCs inhibit prostaglandin synthesis at the level of cyclooxygenase/PGE isomerase (COX-1 and COX-2) [49]; (vii) GCs induce expression of angiotensin-converting enzyme and endothelin, sensitize endothelial cells to vasoconstrictors, and inhibit production of vasodilators that cause decrease in the blood flow to the inflammatory areas [50].
Most of the long-term anti-inflammatory effects of GCs could be caused by the important negative regulatory mechanism called transrepression. The ligand-bound GR is recruited to chromatin through protein-protein interactions with the DNA-bound transcription factors, in particular, with the nuclear factor NF-κB and activator protein-1 (AP1), a complex of c-Fos and c-Jun proteins. GR directly binds to the Jun subunit of AP1 and the p65 subunit of NF-κB interfering with the transcriptional activation of these two proteins [51]. Eventually, this results in suppression of the pro-inflammatory cytokine synthesis.
In addition to the above mechanisms, there are other ways for GCs to affect inflammation indirectly. Elevation of GCs induces an increase in the level of endocannabinoids [52], which are known to have complex effect on inflammation. First of all, elevation of the endocannabinoid levels leads to suppression of HPA activation and suppression of GC release. Second, endocannabinoids induce direct anti-inflammatory effects, either dependent or independent of activation of their receptors. However, the endocannabinoid-mediated suppression of the release of some set of pro-inflammatory cytokines (TNF-α, IL-1β) may be simultaneously associated with activation of release of pro- (IL-6) and anti-inflammatory (IL-10) cytokines [53]. Therefore, GC elevation not only triggers direct anti-inflammatory action but also activates endocannabinoid system, which also helps to control the inflammatory response and suppress acute inflammation.
The anti-inflammatory action of GCs in the brain was confirmed in several studies. In the majority of these studies, systemic inflammatory response was induced by injection of bacterial lipopolysaccharide (LPS). The LPS-induced production of prostaglandins in the dentate gyrus of rats is suppressed by corticosterone released in response to LPS [54]. This finding supports previous results about the anti-inflammatory role of GCs after LPS injection. The acute restraint stress immediately after systemic LPS injection resulted in the suppression of LPS-induced inflammatory response in 1-2 h both at the periphery and in the brain [55]. In the adrenalectomized mice, the LPS-induced increase in gene expression and concentration of the pro-inflammatory cytokines after 2-4 h was higher in both the peripheral and brain tissues as compared with the sham operated mice [56]. These results suggest that the endogenous GCs regulate the components of the host response to inflammation by inhibiting cytokine expression in the peripheral organs and in the brain when the impact is acute and brief. GCs also suppress the pro-inflammatory response to LPS in both peripheral macrophages and microglia, when they are administered after the systemic LPS exposure [57]. The anti-inflammatory response was also observed after the long-term (36 h) exposure of rats to an extremely high dose of corticosterone (100 mg/pellet, sc). In this study, corticosterone suppressed expression of Il-1β and Il-1α mRNA in the hypothalamus and hippocampus, however, the expression of TNF-α in these structures remained unchanged. Paradoxically, these anti-inflammatory changes were accompanied by elevation of Il-6 mRNA in both the hypothalamus and hippocampus [58]. The reasons for the discrepancy between the effects of corticosterone on cytokine mRNA and protein still remain obscure.
PRO-INFLAMMATORY ACTION OF GCs
The pro-inflammatory effects of GCs were described in several experimental paradigms with acute and chronic GC administration prior to the treatment provoking inflammation. Acute systemic exposure to exogenous corticosterone potentiated a pro-inflammatory response to LPS administration in both the peripheral macrophages (liver) and hippocampal microglia if corticosterone injection preceded (2 or 24 h) LPS administration [57]. Hippocampal cells were primed by corticosterone since the LPS-induced increase in mRNA expression and protein concentrations of TNF-α, IL-1β, and IL-6 was considerably higher after pre-treatment with corticosterone [57]. Priming of hippocampal microglia may be induced by acute stress [59]. The microglia isolated from the rat brain 24 h after induction of the acute inescapable stress demonstrated stronger pro-inflammatory cytokine response (e.g., IL-1β gene expression) to LPS; pre-treatment with GC receptor antagonist mifepristone or adrenalectomy eliminated this sensitization of microglia. Thus, the microglial pro-inflammatory response (IL-1β, IL-6, NFκBIα) to immunologic challenges may be sensitized by the stress-induced GCs, similarly to peripheral macrophages. Priming of the neuroinflammatory response was also observed in the experiments with local administration of LPS after stress. Intrahippocampal LPS injection to the stressed animals increased the number of reactive microglial cells and expression of IL-1β and TNF-α mRNA [60]. These results suggest that the preliminary elevation of GCs alters the inflammatory system acting as an endogenous danger signal and preparing the body to cope with the subsequent immunological threats.
The majority of studies discussed in this review frequently analyzed only the levels of some pro-inflammatory cytokines in the hippocampus, while the GC-induced increase in the level of these cytokines could be associated with the reduction in expression of the receptors of these cytokines. It was shown that the increase in the serum GC concentration following immobilization stress or DEX treatment (short- or long-term) did not modify density of IL-1 receptors in the murine hippocampus under basal conditions or after peripheral LPS injection [61]. Importantly, IL-1 or IL-6 administration did not affect expression of GRs and MRs either. Expression of the hippocampal GRs was dramatically decreased after the TNF-α injection [62]. Taken together, these results suggest that the levels of IL-1 receptors in the hippocampus are relatively resistant to modulation by varying levels of the circulating GCs and TNF-α could modulate activity of the HPA axis during immune/inflammatory processes.
Signs of neuroinflammation are frequently observed in two qualitatively different situations of long-term or chronic pathological conditions. The first one includes states not accompanied by brain injury, for example, depression or infection modeled by LPS administration. The second situation is associated with brain injury and subsequent neurodegeneration (stroke, traumatic brain injury, seizures). The important difference between these situations is that the cell death induced by brain injury provokes a release of the damage-associated molecular species activating and maintaining inflammatory cascades.
Neuroinflammation as a consequence of a chronic stress has been reported by several groups; however, involvement of GCs in this process is still undetermined. Positive correlation between the development of neuroinflammation in the depressive states and increased concentration of GCs in the blood of patients with depressive disorders and in the brain of animals in depression models is well known [63]. Systemic administration of DEX for 21 consecutive days induced depressive-like behavior in mice and glucocorticoid resistance. In this case, hyperactivation of GR led to the decrease in corticosterone levels, which was accompanied by the reduced expression of the GR mRNA in the hippocampus, striatum, and prefrontal cortex, and increased expression of FKBP51 suppressing functional activity of the GR [64]. Notably, DEX has much higher affinity to the GR than corticosterone, however, in most studies it is used at similar concentration range as corticosterone (1-10 mg/kg) [65, 66]. Therefore, it is very likely that chronic treatment with DEX at these concentrations would cause over-activation of the GR-mediated signaling and induce glucocorticoid resistance. Glucocorticoid resistance may serve as a priming factor enhancing inflammatory response due to attenuation of the immunosuppressive cascades related to GRs. Unfortunately, dose-dependences of the DEX effects on neuroinflammation have not been systematically studied making interpretation of the data obtained using DEX unreliable.
Both acute and chronic severe stress resulted in accumulation of corticosterone in the rat hippocampus accompanied by the increased expression of the IL-1β mRNA [23]. A milder chronic unpredictable stress also induced an increase in the IL-1β mRNA expression in the rat hippocampus [67]. Although these studies confirm involvement of GCs in the development of stress response under these conditions, they are phenomenological with respect to revealing direct mechanistic association between the elevation of corticosterone and enhancement of synthesis of pro-inflammatory cytokines in the hippocampus.
An attempt to reveal relationship between the GC system and neuroinflammation was performed by R. Sapolsky group [68, 69]. The authors used systemic administration of LPS after chronic stress to examine the role of GCs in the regulation of expression of pro-inflammatory cytokines IL-1β and TNF-α. As expected, in unstressed rats LPS induced an increase in the level of these cytokines in the hippocampus and blockage of GRs by mifepristone potentiated this increase. However, mifepristone had the opposite effect in the rats subjected to stress suppressing the LPS-induced increase in the cytokine level. The latter finding clearly suggests that activity of the GC system is pro-inflammatory in the chronically stressed animals [68]. Later, the authors extended these findings and performed further analysis of association between the chronic elevation of corticosterone and response to LPS [69]. In this study, the blood level of corticosterone in adrenalectomized animals was chronically maintained at three levels: basal, moderately elevated, and high by implantation of the corticosterone-releasing pellets. It was expected that the maintenance of low basal corticosterone level during LPS challenge should be associated with enhanced neuroinflammation due to the loss of the corticosterone-mediated suppression of inflammation. However, the expression of pro-inflammatory cytokines (IL-1β and TNF-α) was similar to the intact animals treated with LPS. Paradoxically, chronic moderate elevation of corticosterone enhanced LPS-induced inflammatory response in the hippocampus as compared to the intact animals, and chronic high level of corticosterone had no effect on the LPS-induced inflammatory response.
The shift to pro-inflammatory action of GCs was also described by Frank et al. (2014) [70]. The rats were subjected to adrenalectomy to tightly control the level of corticosterone, and, next, supplemented with different concentrations of corticosterone in drinking water. Noteworthy, the “high” concentration of corticosterone in this study corresponded to the “moderate” concentration in the study reported by Munhoz et al. (2010) [69]. Supplementation with high doses of corticosterone increased expression of the genes associated with inflammation (Iba1, MHCII, NLRP3) in the hippocampus. Moreover, hippocampal microglia isolated from the brain of rats subjected to high corticosterone levels showed enhanced expression of the pro-inflammatory cytokines (Il-1β, Il-6, TNF-α) and inflammasome component (NLRP3) in response to the challenge with high dose of LPS [70].
Taken together, analysis of the GCs effects in the animal models of pathologies not associated with direct brain injury suggests that the high levels of corticosterone observed after either stress or LPS administration, may change the GCs action towards pro-inflammatory. These data suggest that regulation of the pro-inflammatory response may have other contours in addition to GCs and that long-term moderate elevation of corticosterone may in some way enhance inflammatory response. Remarkably, the long-term exposure to corticosterone at high doses did not have pro-inflammatory effect suggesting that a “pro-inflammatory” concentration range of corticosterone may exist.
The pro-inflammatory action of GCs was also revealed in the animal models of brain pathologies associated with direct injuries of the nervous tissue. The endogenous GCs increase damage to the hippocampus and provoke development of neuroinflammation after injection of kainate. Dinkel et al. (2003) found that injection of kainate to rats with chronically elevated corticosterone level induced accumulation of the inflammatory cells (granulocytes, monocytes/macrophages, and microglia) in the hippocampus. The inflammatory response in the hippocampus, which was evaluated using the level of cytokines IL-1β and TNF-α, was also enhanced in the rats with chronically high GC profile [71]. Further analysis of the GCs effects on the kainate-induced inflammatory response showed that the chronically high level of corticosterone augmented IL-1β expression [72] and accelerated accumulation of the pro-inflammatory cytokine CCL2 [73] in the hippocampus. In the model of lithium-pilocarpine induced epilepsy two injections of DEX 24 and 36 h after the status epilepticus suppressed neuroinflammatory response in the hippocampus [74]. Most likely the extremely high dose of DEX used in this study was sufficient to overcome its expected pro-inflammatory effect, similarly to the effect of corticosterone shown by Munhoz et al. in 2010 [69].
The stress-induced elevation of corticosterone concentration depends on the type of brain injury; the middle cerebral artery occlusion may induce higher corticosterone elevation as compared to the kainate-induced injury [75]. Focal ischemic damage of neocortex in the model of middle cerebral artery occlusion (MCAO) induced accumulation of corticosterone and IL-1β in the hippocampus, this effect was more expressed in the ventral hippocampus [76]. The neonatal pro-inflammatory stress (LPS administration to rat pups) resulted in accumulation of corticosterone and IL-6 in the hippocampus of adolescent animals, while IL-1β remained at the normal range [77]. However, so far, the mechanism(s) of GC-dependent cytokine accumulation in the hippocampus in the above models remains obscure. Based on the experimental data on the pro-inflammatory effect of GCs under several conditions, it has been hypothesized that the distant damage to the hippocampus after MCAO may occur as a result of pro-inflammatory action of GCs bound to their receptors in the hippocampus [3, 78].
HOW GLUCOCORTICOIDS BECOME PRO-INFLAMMATORY: A HYPOTHESIS
As mentioned above, activation of GRs leads to expression of FKBP51, a protein that suppresses nuclear translocation of GRs and decreases GR affinity to GCs; FKBP51 provides a negative feedback for the GR-dependent processes. The GC-induced activation of GRs after 1.5 h leads to FKBP51 elevation and removal of the GR-mediated suppression of pro-inflammatory response [7]. In addition, FKBP51 potentiates NF-κB signaling indirectly, thus enhancing pro-inflammatory response [8]; the detailed mechanisms of this enhancement are still under investigation [9]. This means that various inflammation-inducing factors would cause stronger pro-inflammatory response, when applied under conditions of elevated FKBP51 levels. Therefore, it may be hypothesized that the development of a pro-inflammatory effect of GCs strongly depends on accumulation of FKBP51 in the cells: the higher FKBP51 level should be associated with the stronger pro-inflammatory action of GCs.
The above description of FKBP51 features suggests that the effect of GCs strongly depends on the dose of GCs and moment of its application with respect to the inflammation-inducing factor. We can consider several variants of action of the inflammation-inducing factor action and GCs (to keep it simple, we use LPS as a universal pro-inflammatory stimulus): (i) LPS application followed by GC treatment; (ii) GC application followed by LPS several hours later (2-24 h); (iii) LPS application over the period of chronic GC elevation. In the first case, LPS induces corticosterone elevation exerting anti-inflammatory action [68] (figure, upper panel). Simultaneously, the corticosterone elevation has to induce slow accumulation of FKBP51, which should finally (in >1.5 h) remove the corticosterone-mediated suppression of the inflammation due to the decrease in GR affinity to GCs. Addition of the exogenous GC in this situation will have anti-inflammatory effect since this would help to overcome the FKBP51-induced low affinity of GRs. The second case is a situation of the so-called priming [57], when preliminary treatment with GCs enhances pro-inflammatory response caused by the following LPS injection. The most probable sequence of events is as follows: (i) GCs cause suppression of the pro-inflammatory response and enhancement of FKBP51 expression; (ii) accumulated FKBP51 removes the GC-dependent suppression of pro-inflammatory mechanisms; (iii) addition of LPS induces both GC elevation and expression of pro-inflammatory cytokines, however, due to suppressing effect of the accumulated FKBP51, the inflammatory response is stronger (figure, bottom panel). In this case, addition of GCs (CORT or DEX) at high doses will be anti-inflammatory because high doses should overcome the suppressive effect of FKBP51. The third case is very similar to the second one; the only difference is that GCs remain elevated for a long period of time during the long-term or repeated stress [23, 67], after MCAO [76] or during long-term administration of GCs [64, 69, 70]. It is highly probable that the elevated level of corticosterone results in accumulation of FKBP51 and, consequently, reduction of the GR sensitivity to corticosterone and removal of the GC-mediated suppression of pro-inflammatory response. Again, treatment with GCs at high doses should still have an anti-inflammatory effect under these conditions. The only exclusion here may be the situation when animals are chronically treated with DEX at high doses leading to glucocorticoid resistance, a general reduction of sensitivity of all cells to GCs. In his case, the GC-mediated suppression of inflammation is also absent, however, the mechanism may be not limited to FKBP51.
Hypothetical scheme of pro-inflammatory response priming by GCs. Upper panel illustrates a typical anti-inflammatory effect of GCs in response to LPS challenge: elevation of GC leads to activation of GR and suppression of activity of NF-kB and other pathways triggering expression of pro-inflammatory cytokines. Priming before the LPS treatment transforms GC-associated signaling to the state with high level of FKBP51, which promotes suppression of GR activity and potentiation of the NF-kB-dependent pathways. Administration of LPS in this case is associated with increased pro-inflammatory response (thick arrows) due to the strong FKBP51-mediated enhancement of the NF-kB pathway and inhibition of the GR-mediated immunosuppression.
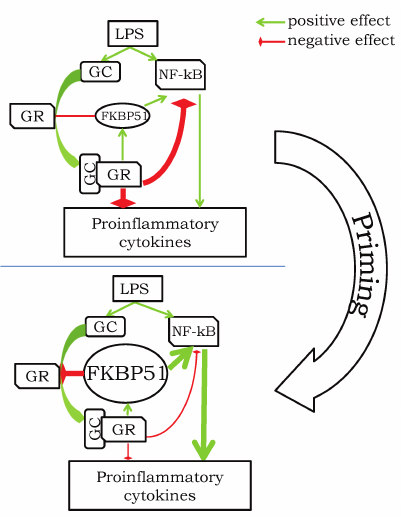
There are data apparently contradicting the proposed hypothesis in the case when adrenalectomized animals have constant “basal” level of corticosterone due to its supplementation from the implanted corticosterone-containing pellets [69]. Maintenance of the “basal” level does result in the pro-inflammatory action of corticosterone. Elimination of the important circadian and ultradian oscillations of the corticosterone levels characteristic for a healthy organism is a specific feature of this scenario. These oscillations may prevent accumulation of FKBP51 and suppression of the anti-inflammatory effect of GCs, whereas maintenance of the constant corticosterone level even at night, when it is normally low, apparently, leads to FKBP51 accumulation and suppression of the GR-mediated effects.
The above hypothesis strongly opposes to the hypothesis suggested by R. Sapolsky on the inverted-U dependence of GC effects [79]. Our hypothesis postulates that GCs generally have an anti-inflammatory effect, however, simultaneously with this effect, they also trigger processes decreasing the GR sensitivity to GCs and, this, inevitably, results in cancellation of the anti-inflammatory effect and activation of the pro-inflammatory cascades, which still could be suppressed by a pulse of high-dose GCs.
CONCLUDING REMARKS
The anti-inflammatory effects of GCs are widely used in clinical practice, while the pro-inflammatory effects of GCs are believed to underlie neurodegeneration. This is particularly critical for the hippocampus, brain region controlling both cognitive function and emotions/affective behavior, and selectively vulnerable to neuroinflammation and neurodegeneration. Abundance of GRs and MRs in the hippocampus underlies complex mechanisms of GC-mediated control of these processes. Our understanding of the dual effects of GCs really reminds the gothic novella “The Strange Case of Dr. Jekyll and Mr. Hyde” by Robert Louis Stevenson, first published in 1886. The good Dr. Jekyll was unpredictably turning into the evil Mr. Hyde and then restored his nature. An essential question is when and why the anti-inflammatory Dr. Jekyll becomes the pro-inflammatory Mr. Hyde and vice versa? Understanding the Jekyll/Hyde nature of GCs as well as reasons and mechanisms of this transition can provide us with effective approaches and tools to avert hippocampal neuroinflammatory events and as a result to prevent and treat brain diseases, both neurological and psychiatric.
The data summarized in our review form a sophisticated, though often quite phenomenological picture of a modulatory role of GCs in the hippocampal neuroinflammation. We suggested a hypothesis within the mechanistic framework describing how the anti-inflammatory effects of GCs may transform into the pro-inflammatory ones. So far, no experiments were performed that would support or reject the proposed mechanism. The majority of experiments concerning characteristics of FKBP51 and its role in the regulation of GR-mediated functions and inflammation were performed using either cell cultures or non-neural tissues. However, if our hypothesis is correct, it may provide an additional instrument to regulate the GC-mediated pathways especially in the cases of pathologies associated with high GC levels, where suppression of the FKBP51 function may help to restore the anti-inflammatory action of GCs.
Two issues remained out of the scope of our review though they may have an important impact on the GC-dependent modulation of inflammation. Firstly, the pro- and anti-inflammatory effects of GCs are frequently evaluated by measuring the levels of pro-inflammatory cytokines such as IL-1β or TNF-α. However, the cytokine network is extensive and complicated, and GCs may have multifaceted effects, e.g., potentiating expression of both pro- and anti-inflammatory cytokines. Yet, the effects of GCs on the expression of anti-inflammatory cytokines remain practically unstudied. The only study by Frank et al. (2010) showed that a single preliminary injection of corticosterone not only enhanced expression of the pro-inflammatory cytokines after LPS administration but also potentiated expression of the anti-inflammatory cytokines such as IL-10, IL-4, and CX3CL1 in the hippocampus [57]. This study points to the necessity of measuring levels of both pro- and anti-inflammatory cytokines as well as effect of GCs on both to evaluate the direction and intensity of the inflammatory process and the effects of GCs.
Another important issue usually ignored during analysis of the effects of stress and elevation of GCs on neuroinflammation is that stress may affect the cytokine system not necessarily directly through the GC-dependent mechanisms. For example, stress leads to elevation of IL-6 in blood [80-82] and, recently, it has been shown that this effect results from activation of the brown fat cells by the sympathetic system [83]. Moreover, the stress-induced accumulation of IL-6 decreases tolerance to the subsequent inflammatory challenge casting doubts on the exclusive role of GCs in the regulation of inflammatory processes [83]. This suggests that some factors, in addition to GCs, may be induced by stress and determine time course of the inflammatory response. Future studies in this field should help establishing and clarifying the role of each stress response component in the regulation of neuroinflammation.
Funding. This study was financially supported by the Russian Science Foundation [project no. 20-65-47029 (pro-inflammatory effects of GCs)] and by the Russian Foundation for Basic Research [project no. 19-315-90081 (anti-inflammatory effects of GCs)].
Ethics declarations. The authors declare no conflict of interest in financial or any other sphere. This article does not contain any studies with human participants or animals performed by any of the authors.
Open access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
REFERENCES
1.Hall, J. E. (2016) Guyton and Hall Textbook of
Medical Physiology, 13th Edn., Saunders, Philadelphia, PA.
2.Estes, M. L., and McAllister, A. K. (2014)
Alterations in immune cells and mediators in the brain: it’s not
always neuroinflammation! Brain Pathol., 24, 623-630,
doi: 10.1111/bpa.12198.
3.Gulyaeva, N. V. (2019) Functional neurochemistry of
the ventral and dorsal hippocampus: stress, depression, dementia and
remote hippocampal damage, Neurochem. Res., 44,
1306-1322, doi: 10.1007/s11064-018-2662-0.
4.Silverman, M. N., and Sternberg, E. M. (2012)
Glucocorticoid regulation of inflammation and its functional
correlates: from HPA axis to glucocorticoid receptor dysfunction,
Ann. N. Y. Acad. Sci., 1261, 55-63, doi:
10.1111/j.1749-6632.2012.06633.x.
5.Kirschke, E., Goswami, D., Southworth, D., Griffin,
P. R., and Agard, D. A. (2014) Glucocorticoid receptor function
regulated by coordinated action of the Hsp90 and Hsp70 chaperone
cycles, Cell, 157, 1685-1697, doi:
10.1016/j.cell.2014.04.038.
6.Baker, J. D., Ozsan, I., Ospina, S. R., Gulick, D.,
and Blair, L. J. (2019) Hsp90 heterocomplexes regulate steroid hormone
receptors: from stress response to psychiatric disease, Int. J. Mol.
Sci., 20, 79, doi: 10.3390/ijms20010079.
7.Paakinaho, V., Makkonen, H., Jaaskelainen, T., and
Palvimo, J. J. (2010) Glucocorticoid receptor activates poised FKBP51
locus through long‐distance interactions, Mol.
Endocrinol., 24, 511-525, doi: 10.1210/me.2009-0443.
8.Kästle, M., Kistler, B., Lamla, T.,
Bretschneider, T., Lamb, D., et al. (2018) FKBP51 modulates steroid
sensitivity and NF-κB signalling: A novel anti-inflammatory drug
target, Eur. J. Immunol., 48, 1904-1914, doi:
10.1002/eji.201847699.
9.Fries, G. R., Gassen, N. C., and Rein, T. (2017)
The FKBP51 glucocorticoid receptor co-chaperone: regulation, function,
and implications in health and disease, Int. J. Mol. Sci.,
18, 2614, doi: 10.3390/ijms18122614.
10.Liberman, A. C., Budziñski, M. L., Sokn,
C., Gobbini, R. P., Ugo, M. B., and Arzt, E. (2020) SUMO conjugation as
regulator of the glucocorticoid receptor-FKBP51 cellular response to
stress, Steroids, 153, 108520, doi:
10.1016/j.steroids.2019.108520.
11.Merkulov, V. M., Merkulova, T. I., and Bondar, N.
P. (2017) Mechanisms of brain glucocorticoid resistance in
stress-induced psychopathologies, Biochemistry (Moscow),
82, 351-365, doi: 10.1134/S0006297917030142.
12.Scharf, S. H., Liebl, C., Binder, E. B., Schmidt,
M. V., andMüller, M. B. (2011) Expression and regulation of the
Fkbp5 gene in the adult mouse brain, PLoS One, 6,
e16883, doi: 10.1371/journal.pone.0016883.
13.Bourke, C. H., Raees, M. Q., Malviya, S.,
Bradburn, C. A., Binder, E. B., and Neigh, G. N. (2013) Glucocorticoid
sensitizers Bag1 and Ppid are regulated by adolescent stress in a
sex-dependent manner, Psychoneuroendocrinology, 38,
84-93, doi: 10.1016/j.psyneuen.2012.05.001.
14.Green, M. R., Nottrodt, R. E., Simone, J. J., and
McCormick, C. M. (2016) Glucocorticoid receptor translocation and
expression of relevant genes in the hippocampus of adolescent and adult
male rats, Psychoneuroendocrinology, 37, 32-41, doi:
10.1016/j.psyneuen.2016.07.210.
15.Sawamura, T., Klengel, T., Armario, A.,
Jovanovic, T., Norrholm, S. D., et al. (2016) Dexamethasone treatment
leads to enhanced fear extinction and dynamic Fkbp5 regulation in
amygdale, Neuropsychopharmacology, 41, 832-846, doi:
10.1038/npp.2015.210.
16.Bloem, L. J., Guo, C., and Pratt, J. H. (1995)
Identification of a splice variant of the rat and human
mineralocorticoid receptor genes, J. Steroid Biochem. Mol.
Biol., 55, 159-162, doi: 10.1016/0960-0760(95)00162-s.
17.Young, E. A., Lopez, J. F., Murohy-Weinberg, V.,
Watson, S. J., and Akil, H. (2003) Mineralocorticoid receptor function
in major depression, Arch. Gen. Psychiatry, 60, 24-28,
doi: 10.1001/archpsyc.60.1.24.
18.Tasic, B., Yao, Z., Graybuck, L. T., Smith, K.
A., Nguyen, T. N., et al. (2018) Shared and distinct transcriptomic
cell types across neocortical areas, Nature, 563, 72-78,
doi: 10.1038/s41586-018-0654-5.
19.Saunders, A., Macosko, E. Z., Wysoker, A.,
Goldman, M., Krienen, F. M., et al. (2018) Molecular diversity and
specializations among the cells of the adult mouse brain, Cell,
174, 1015-1030, doi: 10.1016/j.cell.2018.07.028.
20.Zeisel, A., Hochgerner, H., Lonnerberg, P.,
Johnsson, A., Memic, F., et al. (2018) Molecular architecture of the
mouse nervous system, Cell, 174, 999-1014, doi:
10.1016/j.cell.2018.06.021.
21.Krauss, P. G. (2006) Biochemistry of Signal
Transduction and Regulation, 3rd Edn., Completely
Revised Edition, John Wiley & Sons.
22.Caudal, D., Jay, T. M., and Godsil, B. P. (2014)
Behavioral stress induces regionally-distinct shifts of brain
mineralocorticoid and glucocorticoid receptor levels, Front. Behav.
Neurosci., 8, 1-8, doi: 10.3389/fnbeh.2014.00019.
23.Piskunov, A., Stepanichev, M., Tishkina, A.,
Novikova, M., Levshina, I., and Gulyaeva, N. (2016) Chronic combined
stress induces selective and long-lasting inflammatory response evoked
by changes in corticosterone accumulation and signaling in rat
hippocampus, Metab. Br. Dis., 31, 445-454, doi:
10.1007/s11011-015-9785-7.
24.Karst, H., Derijk, R., and De Kloet, E. R. (2007)
The coming out of the brain mineralocorticoid receptor, Trends
Neurosci., 31, 1-7, doi: 10.1016/j.tins.2007.10.005.
25.Maggio, N., and Segal, M. (2010) Cellular basis
of a rapid effect of mineralocorticosteroid receptors activation on LTP
in ventral hippocampal slices, Hippocampus, 22, 267-275,
doi: 10.1002/hipo.20893.
26.Brocca, M. E., Pietranera, L., de Kloet, E. R.,
and De Nicola, A. F. (2019) Mineralocorticoid receptors,
neuroinflammation and hypertensive encephalopathy, Cell. Mol.
Neurobiol., 39, 483-492, doi: 10.1007/s10571-018-0610-9.
27.Dinh, Q. N., Young, M. J., Evans, M. A.,
Drummond, G. R., Sobey, C. G., and Sophocles, C. (2016)
Aldosterone-induced oxidative stress and inflammation in the brain are
mediated by the endothelial cell mineralocorticoid receptor, Brain
Res., 1637, 146-153, doi:
10.1016/j.brainres.2016.02.034.
28.Bay-Richter, C., Hallberg, L., Ventorp, F.,
Janelidze, S., and Brundin, L. (2012) Aldosterone synergizes with
peripheral inflammation to induce brain IL-1β expression and
depressive-like effects, Cytokine, 60, 749-754, doi:
10.1016/j.cyto.2012.08.016.
29.Joëls, M., and de Kloet, E. R. (2017) The
brain mineralocorticoid receptor: A saga in three episodes, J.
Endocrinol., 234, 49-66, doi: 10.1530/JOE-16-0660.
30.Dinh, Q. N., Drummond, G. R., Sobey, C. G., and
Chrissobolis, S. (2016) Cell-specific mineralocorticoid receptors:
future therapeutic targets for stroke? Neural Regen. Res.,
11, 1230-1231, doi: 10.4103/1673-5374.189173.
31.Frieler, R. A., Meng, H., Duan, S. Z, Berger, S.,
Schütz, G., et al. (2011) Myeloid-specific deletion of the
mineralocorticoid receptor reduces infarct volume and alters
inflammation during cerebral ischemia, Stroke, 42,
179-185, doi: 10.1161/STROKEAHA.110.598441.
32.Montes-Cobos, E., Schweingruber, N., Xiao, L.,
Fischer, H. J., Reichardt, H. M., Lühder, F. (2017) Deletion of
the mineralocorticoid receptor in myeloid cells attenuates central
nervous system autoimmunity, Front. Immunol., 8, 1319,
doi: 10.3389/fimmu.2017.01319.
33.Lightman, S. L., Wiles, C. C., Atkinson, H. C.,
Henley, D. E., Russell, G. M., et al. (2008) The significance of
glucocorticoid pulsatility, Eur. J. Pharmacol., 583,
255-262, doi: 10.1016/j.ejphar.2007.11.073.
34.Pariante, C. M., and Lightman, S. L. (2008) The
HPA axis in major depression: classical theories and new developments,
Trends Neurosci., 31, 464-468, doi:
10.1016/j.tins.2008.06.006.
35.De Kloet, E. R., Vreugdenhil, E., Oitzl, M. S.,
and Joels, M. (1998) Brain corticosteroid receptor balance in health
and disease, Endocr. Rev., 19, 269-301, doi:
10.1210/edrv.19.3.0331.
36.Sharma, S. T., and Nieman, L. K. (2011)
Cushing’s syndrome: all variants, detection, and treatment,
Endocrinol. Metab. Clin. N. Am., 40, 379-391, doi:
10.1016/j.ecl.2011.01.006.
37.Pereira, A. M., Tiemensma, J., and Romijn, J. A.
(2010) Neuropsychiatric disorders in Cushing’s syndrome,
Neuroendocrinology, 92, 65-70, doi:
10.1159/000314317.
38.Rodriguez, J. M., Monsalves-Alvareza, M.,
Henriquez, S., Llanos, M. N., and Troncoso, R. (2016) Glucocorticoid
resistance in chronic diseases, Steroids, 115, 182-192,
doi: 10.1016/j.steroids.2016.09.010.
39.Vandewalle, J., Luypaert, A., De Bosscher, K.,
and Libert, C. (2018) Therapeutic mechanisms of glucocorticoids,
Trends Endocrinol. Metab., 29, 42-54, doi:
10.1016/j.tem.2017.10.010.
40.Cain, D. W., and Cidlowski, J. A. (2017) Immune
regulation by glucocorticoids, Nat. Rev. Immunol., 17,
233-247, doi: 10.1038/nri.2017.1.
41.Banuelos, J., Shin, S., Cao, Y., Bochner, B. S.,
Morales-Nebreda, L., et al. (2016) BCL-2 protects human and mouse Th17
cells from glucocorticoid-induced apoptosis, Eur. J. Allergy Clin.
Immunol., 71, 640-650, doi: 10.1111/all.12840.
42.McKinley, L., Alcorn, J. F., Peterson, A.,
DuPont, R. B., Kapadia, S., et al. (2008) Th17 cells mediate
steroid-resistant airway inflammation and airway hyperresponsiveness in
mice, J. Immunol., 181, 4089-4097, doi:
10.4049/jimmunol.181.6.4089.
43.Banuelos, J., and Lu, N. Z. (2016) A gradient of
glucocorticoid sensitivity among helper T cell cytokines, Cytokine
Growth Factor Rev., 31, 27-35, doi:
10.1016/j.cytogfr.2016.05.002.
44.Spencer, R. L., Kalman, B. A., and Dhabhar, F. S.
(2011) Role of Endogenous Glucocorticoids in Immune System Function:
Regulation and Counterregulation, Comprehensive Physiology,
Handbook of Physiology, The Endocrine System, Coping with the
Environment: Neural and Endocrine Mechanisms, doi:
10.1002/cphy.cp070418.
45.Goujon, E., Layé, S., Parnet, P., and
Dantzer, R. (1997) Regulation of cytokine gene expression in the
central nervous system by glucocorticoids: Mechanisms and functional
consequences, Psychoneuroendocrinology, 22, S75-S80, doi:
10.1016/s0306-4530(97)00009-7.
46.Cronstein, B. N., Kimmel, S. C., Levin, R. I.,
Martiniuk, F., and Weissmann, G. (1992) A mechanism for the
anti-inflammatory effects of corticosterone: The glucocorticoid
receptor regulates leukocyte adhesion to endothelial cells and
expression of endothelial-leukocyte adhesion molecule 1 and
intercellular adhesion molecule 1, Proc. Natl. Acad. Sci.
USA, 89, 9991-9995, doi: 10.1073/pnas.89.21.9991.
47.Flower, R. J., and Rothwell, N. J. (1994)
Lipocortin-1: cellular mechanisms and clinical relevance, Trends
Pharmacol. Sci., 15, 71-76, doi:
10.1016/0165-6147(94)90281-X.
48.Dolan-O’Keefe, M., and Nick, H. S. (1999)
Inhibition of cytoplasmic phospholipase A2 expression by
glucocorticoids in rat intestinal epithelial cells,
Gastroenterology, 116, 855-864, doi:
10.1016/S0016-5085(99)70068-5.
49.Goppelt-Struebe, M., Wolter, D., and Resch, K.
(1989) Glucocorticoids inhibit prostaglandin synthesis not only at the
level of phospholipase A2 but also at the level of cyclo-oxygenase/PGE
isomerase, Br. J. Pharmacol., 98, 1287-1295, doi:
10.1111/j.1476-5381.1989.tb12676.x.
50.Perretti, M., and Ahluwalia, A. (2000) The
microcirculation and inflammation: site of action for glucocorticoids,
Microcirculation, 7, 147-161, doi:
10.1111/j.1549-8719.2000.tb00117.x.
51.Nissen, R. M., and Yamamoto, K. R. (2000) The
glucocorticoid receptor inhibits NFκB by interfering with
serine-2 phosphorylation of the RNA polymerase II carboxy-terminal
domain, Genes Dev., 14, 2314-2329, doi:
10.1101/gad.827900.
52.Hillard, C. J. (2018) Circulating
endocannabinoids: from whence do they come and where are they going?
Neuropsychopharmacology, 43, 155-172, doi:
10.1038/npp.2017.130.
53.Klein, T. W. (2005) Cannabinoid-based drugs as
anti-inflammatory therapeutics, Nat. Rev. Immunol., 5,
400-411, doi: 10.1038/nri1602.
54.Ma, Y., Matsuwaki, T., Yamanouchi, K., and
Nishihara, M. (2017) Glucocorticoids suppress the protective effect of
cyclooxygenase-2-related signaling on hippocampal neurogenesis under
acute immune stress, Mol. Neurobiol., 54, 1953-1966, doi:
10.1007/s12035-016-9766-9.
55.Goujon, E., Parnet, P., Laye, S., Combe, C.,
Kelley, K. W., and Dantzer, R. (1995) Stress downregulates
lipopolysaccharide-induced expression of proinflammatory cytokines in
the spleen, pituitary, and brain of mice, Brain Behav. Immun.,
9, 292-303, doi: 10.1006/brbi.1995.1028.
56.Goujon, E., Parnet, P., Layé, S., Combe,
C., and Dantzer, R. (1996) Adrenalectomy enhances pro-inflammatory
cytokines gene expression, in the spleen, pituitary and brain of mice
in response to lipopolysaccharide, Mol. Brain Res., 36,
53-62, doi: 10.1016/0169-328X(95)00242-K.
57.Frank, M. G., Miguel, Z. D., Watkins, L. R., and
Maier, S. F. (2010) Prior exposure to glucocorticoids sensitizes the
neuroinflammatory and peripheral inflammatory responses to E.
coli lipopolysaccharide, Brain Behav. Immun., 24,
19-30, doi: 10.1016/j.bbi.2009.07.008.
58.Chai, Z., Alheim, K., Lundkvist, J., Gatti, S.,
and Bartfai, T. (1996) Subchronic glucocorticoid pretreatment
reversibly attenuates IL-1β induced fever in rats; IL-6 mRNA is
elevated while IL-1α and IL-1β mRNAs are suppressed, in the
CNS, Cytokine, 8, 227-237, doi:
10.1006/cyto.1996.0032.
59.Frank, M. G., Thompson, B. M., Watkins, L. R.,
and Maier, S. F. (2012) Glucocorticoids mediate stress-induced priming
of microglial pro-inflammatory responses, Brain Behav. Immun.,
26, 337-345, doi: 10.1016/j.bbi.2011.10.005.
60.Espinosa-Oliva, A. M., De Pablos, R. M.,
Villarán, R. F., Argüelles, S., Venero, J. L., et al.
(2009) Stress is critical for LPS-induced activation of microglia and
damage in the rat hippocampus, Neurobiol. Aging, 32,
85-102, doi: 10.1016/j.neurobiolaging.2009.01.012.
61.Betancur, C., Lledo, A., Borrell, J., and Guaza,
C. (1994) Corticosteroid regulation of IL-1 receptors in the mouse
hippocampus: effects of glucocorticoid treatment, stress, and
adrenalectomy, Neuroendocrinology, 59, 120-128, doi:
10.1159/000126648.
62.Betancur, C., Borrell, J., and Guaza, C. (1995)
Cytokine regulation of corticosteroid receptors in the rat hippocampus:
effects of interleukin-1, interleukin-6, tumor necrosis factor and
lipopolysaccharide, Neuroendocrinology, 62, 47-54, doi:
10.1159/000126987.
63.Frank, M. G., Watkins, L. R., and Maier, S. F.
(2015) The permissive role of glucocorticoids in neuroinflammatory
priming: mechanisms and insights, Curr. Opin. Endocrinol. Diabetes
Obes., 22, 300-305, doi: 10.1097/MED.0000000000000168.
64.Skupio, U., Tertil, M., Sikora, M., Golda, S.,
Wawrzczak-Bargiela, A., and Przewlocki, R. (2015) Behavioral and
molecular alterations in mice resulting from chronic treatment with
dexamethasone: relevance to depression, Neuroscience,
286, 141-150, doi: 10.1016/j.neuroscience.2014.11.035.
65.Sigwalt, A. R., Budde, H., Helmich, I., Glaser,
V., Ghisoni, K., et al. (2011) Molecular aspects involved in swimming
exercise training reducing anhedonia in a rat model of depression,
Neuroscience, 192, 661-674, doi:
10.1016/j.neuroscience.2011.05.075.
66.Al-Shorbagy, M. Y., El Sayeh, B. M., and
Abdallah, D. M. (2012) Diverse effects of variant doses of
dexamethasone in lithium-pilocarpine induced seizures in rats, Can.
J. Physiol. Pharmacol., 90, 13-21, doi: 10.1139/y11-096.
67.Stepanichev, M. Yu., Peregud, D. I., Manolova, A.
O., Lazareva, N. A., Onufriev, M. V., and Gulyaeva, N. V., (2018) Mild
chronic stress increases expression of genes of proinflammatory
cytokines in the rat brain, Herald Russ. Acad. Sci. Biol. Ser.,
2, 209-215, doi: 10.7868/S0002332918020108.
68.Munhoz, C. D., Lepsch, L. B., Kawamoto, E. M.,
Malta, M. B., De Sa Lima, L., et al. (2006) Chronic unpredictable
stress exacerbates lipopolysaccharide-induced activation of nuclear
factor-κB in the frontal cortex and hippocampus via
glucocorticoid secretion, J. Neurosci., 26, 3813-3820,
doi: 10.1523/JNEUROSCI.4398-05.2006.
69.Munhoz, C. D., Sorrells, S. F., Caso, J. R.,
Scavone, C., and Sapolsky, R. M. (2010) Glucocorticoids exacerbate
lipopolysaccharide-induced signaling in the frontal cortex and
hippocampus in a dose-dependent manner, J. Neurosci., 30,
13690-13698, doi: 10.1523/JNEUROSCI.0303-09.2010.
70.Frank, M. G., Hershman, S. A., Weber, M. D.,
Watkins, L. R., and Maier, S. F. (2014) Chronic exposure to exogenous
glucocorticoids primes microglia to pro-inflammatory stimuli and
induces NLRP3 mRNA in the hippocampus, Psychoneuroendocrinology,
40, 191-200, doi: 10.1016/j.psyneuen.2013.11.006.
71.Dinkel, K., MacPherson, A., and Sapolsky, R. M.
(2003) Novel glucocorticoid effects on acute inflammation in the CNS,
J. Neurochem., 84, 705-716, doi:
10.1046/j.1471-4159.2003.01604.x.
72.Sorrells, S. F., Caso, J. R., Munhoz, C. D., Hu,
C. K., Tran, K. V., et al. (2013) Glucocorticoid signaling in myeloid
cells worsens acute CNS injury and inflammation, J. Neurosci.,
33, 7877-7889, doi: 10.1523/JNEUROSCI.4705-12.2013.
73.Sorrells, S. F., Munhoz, C. D., Manley, N. C.,
Yen, S., and Sapolsky, R. M. (2014) Glucocorticoids increase
excitotoxic injury and inflammation in the hippocampus of adult male
rats, Neuroendocrinology, 100, 129-140, doi:
10.1159/000367849.
74.Vizuete, A. F. K., Hansen, F., Negri, E., Leite,
M. C., De Oliveira, D. L., and Gonçalves, C. A. (2018) Effects
of dexamethasone on the Li-pilocarpine model of epilepsy: protection
against hippocampal inflammation and astrogliosis, J.
Neuroinflam., 15, 68, doi: 10.1186/s12974-018-1109-5.
75.Smith-Swintosky, V. L., Pettigrew, L. C.,
Sapolsky, R. M., Phares, C., Craddock, S. D., et al. (1996) Metyrapone,
an inhibitor of glucocorticoid production, reduces brain injury induced
by focal and global ischemia and seizures, J. Cerebr. Bl. Fl.
Metab., 16, 585-598, doi:
10.1097/00004647-199607000-00008.
76.Onufriev, M. V., Freiman, S. V., Moiseeva, Y. V.,
Stepanichev, M. Y., et al. (2017) Accumulation of corticosterone and
interleukin-1β in the hippocampus after focal ischemic damage of
the neocortex: selective vulnerability of the ventral hippocampus,
Neurochem. J., 11, 236-241, doi:
10.1134/S1819712417030084.
77.Onufriev, M. V., Freiman, S. V., Peregud, D. I.,
Kudryashova, I. V., Tishkina, A. O., et al. (2017) Neonatal
Proinflammatory stress induces accumulation of corticosterone and
Interleukin-6 in the hippocampus of juvenile rats: potential mechanism
of synaptic plasticity impairments, Biochemistry (Moscow),
82, 281, doi: 10.1134/S0006297917030051.
78.Gulyaeva, N. V. (2019) Biochemical mechanisms and
translational relevance of hippocampal vulnerability to distant focal
brain injury: the price of stress response, Biochemistry
(Moscow), 84, 1306-1328, doi: 10.1134/S0006297919110087.
79.Sapolsky, R. M. (2015) Stress and the brain:
individual variability and the inverted-U, Nat. Neurosci.,
18, 1344-1346, doi: 10.1038/nn.4109.
80.Cheng, Y., Jope, R. S., and Beurel, E. (2015) A
pre-conditioning stress accelerates increases in mouse plasma
inflammatory cytokines induced by stress, BMC Neurosci.,
16, 31, doi: 10.1186/s12868-015-0169-z.
81.LeMay, L. G., Vander, A. J., and Kluger, M J.
(1990) The effects of psychological stress on plasma interleukin-6
activity in rats, Physiol. Behav., 47, 957-961, doi:
10.1016/0031-9384(90)90024-X.
82.Maes, M., Song, C., Lin, A., De Jongh, R., Van
Gastel, A., et al. (1998) The effects of psychological stress on
humans: increased production of pro-inflammatory cytokines and a
Th1-like response in stress-induced anxiety, Cytokine,
10, 313-318, doi: 10.1006/cyto.1997.0290.
83.Qing, H., Desrouleaux, R., Israni-Wingeret, K.,
Mineur, Y. S., Fogelman, N., et al. (2020) Origin and function of
stress-induced IL-6 in murine models, Cell, 182, 372-387,
doi: 10.1016/j.cell.2020.05.054.