REVIEW: Klotho Protein and Cardio-Vascular System
Ivan N. Tyurenkov1, Valentina N. Perfilova1,a*, Alla A. Nesterova2, and Yelena Glinka3
1Volgograd State Medical University, Ministry of Health of the Russian Federation, 400066 Volgograd, Russia2Pyatigorsk Medical and Pharmaceutical Institute, Branch of the Volgograd State Medical University, Ministry of Health of the Russian Federation, 357500 Pyatigorsk, Russia
3Keenan Research Centre, St. Michael’s Hospital, Canada, Toronto, ON M5B 1W8
* To whom correspondence should be addressed.
Received April 11, 2020; Revised August 20, 2020; Accepted September 8, 2020
Klotho protein affects a number of metabolic pathways essential for pathogenesis of cardio-vascular diseases and their prevention. It inhibits lipid peroxidation and inflammation, as well as prevents endothelial injury and calcification of blood vessels. Klotho decreases rigidity of blood vessels and suppresses development of the heart fibrosis. Low level of its expression is associated with a number of diseases. Cardioprotective effect of klotho is based on its ability to interact with multiple receptors and ion channels. Being a pleiotropic protein, klotho could be a useful target for therapeutic intervention in the treatment of cardio-vascular diseases. In this review we present data on pharmaceuticals that stimulate klotho expression and suggest some promising research directions.
KEY WORDS: Klotho protein, heart failure, heart ischemia, cardiomyopathy, arterial hypertensionDOI: 10.1134/S0006297921020024
Abbreviations: Akt, protein kinase B; CKD, chronic kidney disease; eNOS, endothelial nitric oxide synthase; FGF, fibroblast growth factor; FGFR, fibroblast growth factor receptor; HUVECs, human umbilical vein endothelial cells; IGF, insulin-like growth factor; IL, interleukin; SOD, superoxide dismutase; MAPK, mitogen-activated protein kinase; NF-κB, nuclear factor κB; PI3K, phosphatidylinositol-3-kinase; ROS, reactive oxygen species; TGF, transforming growth factor; TGFβRII, TGFβ1 receptor II; TNF-α, tumor necrosis factor α; VEGFR, vascular endothelial growth factor receptor.
INTRODUCTION
Cardio-vascular diseases remain a major cause of early disability and premature death. A number of pathogenic mechanisms lead to the development of these diseases, and the list of mechanisms continues to grow. Successful prophylactics and treatment of cardio-vascular diseases, improvement of the late outcomes depend on the identifying of new therapeutic targets, preferably those, which are involved with more than one pathogenic mechanism. Pleiotropic klotho protein participates in a number of metabolic processes. This makes it an attractive candidate for such a target. Here, we review the role of klotho in the regulation of physiology and pathology of cardio-vascular system, its effect on oxidative stress, endothelial injury, electric activity of the heart, vascular and heart remodeling, heart ischemia, arterial hypertension, heart failure, and more.
STRUCTURE, FUNCTION, AND LOCALIZATION OF KLOTHO PROTEIN
The Kl gene (known as α-klotho or klotho) has been first identified by Kuro-o, M. et al. (1997) [1] as a murine anti-aging gene. Human Kl is located on the 13q12 chromosome, it consists of 50 thousand base pairs and contains five exons. It is expressed in a number of organs and tissues: the highest level of Kl and its product is found in kidney, much less in prostate, lungs, liver, skeletal muscle, aorta, pancreatic islets, and brain. Its paralog, KlB, is located on the fourth chromosome and has the size and structure very similar to those of Kl. The main source of human β-klotho is adipose tissue. It is synthesized also in lungs and pancreas, but its mRNA is found in many tissues including skeletal muscle, aorta, and heart. Both genes encode for the membrane-bound and secretable proteins [2]. The third member of the klotho family is encoded by Lctl (Lactase-like gene). Its product is a transmembrane lactase-like protein with alternative names klotho/lactase-phlorizin hydrolase-related protein, KLPH or LCTL, or γ-klotho. The human gene is located on the chromosome 15. It is expressed at the same low level by the majority of organs. Testes express the highest level of this protein. Kl has the highest and Lctl the lowest level of expression [1-5]. Thus, mammalian genome encodes three members of the klotho family. They are transmembrane proteins of different size [6].
Alpha- and β-klotho consist of two extracellular subdomains (KL1 and KL2 of about 450 amino acids each), transmembrane, and C-end cytoplasmic domains. Gamma-Klotho, unlike its paralogs, contains only 567 amino acids and just one extracellular domain KL1. Extracellular domains are highly homologous to β-glucosidases, which hydrolyze bonds in saccharides, glycoproteins, and glycolipids. Klotho belongs to the glycoside hydrolase family 1, although the active site of klotho does not have glutamate residue essential for hydrolase activity. Very short intracellular domain (21 amino acid in α-klotho and 24 in β-klotho) is incapable of independent signal transduction and does not have PDZ domain or motifs that bind known adaptor proteins. This makes independent receptor activity of the membrane-bound klotho unlikely. To the best of our knowledge, the two-hybrid system has not been used to search for klotho binding partners. As to its extracellular domains, they interact with other receptor proteins and their ligands, thus modifying their function. The effect of this interaction on cell physiology depends on the type of the receptor and the level of its expression in the tissue.
It is proven experimentally that α-klotho directly binds to FGFR1c and FGFR4, IGF1/insulin receptors, and TGFβRII, as well as Wnt. Being bound to FGFR1c and FGFR4, klotho acts as a co-receptor, switches specificity of the receptor, and enhances its affinity to the endocrine FGF23 multifold, while decreasing its affinity to the canonical agonists FGF 1-20, which require heparin for their binding to the receptor. Structural analysis reveals that α-klotho interacts with both C-terminal of FGF23 and D3 domain of FGFR1Rc, acting as a chaperone and stabilizing the tertiary complex. C-terminal of FGF23 contains RXXR motif that binds to the FGFR1c-α-klotho dimer. Activity of the tertiary complex depends on heparan-sulphate, an obligatory cofactor of the paracrine signaling of FGF. Three aspartate and one cysteine residues in the klotho molecule coordinate zinc ion. Klotho conformation in this complex makes its enzymatic activity impossible [7]. Interaction with α-klotho radically changes physiological outcome of the FGFR1 stimulation. Canonical pathway activated by binding of FGF in the absence of α-klotho leads to dimerization and autophosphorylation of the receptor, activation of its substrate, ERK1/2, Akt, Ras/MAPK [8]. It participates in organogenesis, cell differentiation, induces cell proliferation and migration, cooperates with TGFβ1, Notch, and non-canonical Wnt pathways [9]. Unlike the described above canonical pathway, the ternary complex of endocrine FGF23 with FGFR1c and α-klotho controls calcium and inorganic phosphate homeostasis and regulates vitamin D metabolism. Signaling pathway activated by the formation of the ternary complex switches on the Ras/MAPK cascade typical for the klotho-expressing cells. Contrary to this, binding of FGF2 to cardiomyocytes not expressing this protein induces activation of phospholipase PLCγ, calcineurin, and NFAT [10].
In addition, α-klotho activates signaling pathways independent on FGFR. Interactions of α-klotho with Wnt and TGFβRII inhibit corresponding canonical signaling pathways of Wnt/β-catenin and TGFβ1/SMAD2 and fibrosis [11-13]. α-Klotho binding to insulin/IGF1 receptors prevents activation of its substrates IRS1 and IRS2 and downstream activation of the PI3K/Akt pathway [14, 15].
A number of α-klotho effects depend on the glucosidase or sialidase activity of its extracellular subdomains [16-18]. Structural studies reveal that the soluble klotho binds with α2-3-sialyl-lactose moiety of gangliosides in lipid rafts because of the shift of β6α6 loop in the ganglioside molecules. This happens independent on FGF23 and FGFR [18]. Removal of sialic acids from the glycoprotein component of the ion channels affects their localization on the membrane and intensity of the ion transport [16-19]. Regulation of the activity of ion channels is essential for the healthy function of cardio-vascular system.
Similar to its paralog, β-klotho modifies physiological function of FGFR: endocrine FGF15/19 or FGF21 in complex with FGFR and β-klotho regulate the level of glucose, triglycerides, and cholesterol. FGF19 and β-klotho, bound to FGFR4, participate in homeostasis of bile acids. For example, mice deficient in FGF15, β-klotho or FGFR4 express higher level of Cyp7α1 and have increased synthesis of bile acids in liver [20]. Cardiomyocytes expressing β-klotho and FGFR1 are protected against ischemia through activation of the PI3K/Akt, AMPK, ERK1/2, and ROR-α pathways after binding of FGF21 by the receptor complex [21]. According to the structural studies, FGF21 binds to both extracellular subdomains of β-klotho, and this binary complex attaches to FGFR1c. The formed ternary complexes dimerize [22]. C-terminals of FGF21 and FGF19 bind to the same site on β-klotho [23].
Extracellular domain of klotho is cleaved by the membrane-bound proteinases ADAM10 and ADAM17. Their enzymatic activity is regulated by phosphorylation [18, 24, 25]. The linker between KL1 and KL2 contains four amino acids (Lys-Lys-Arg-Lys), which form a potential site for proteolysis [26]. The soluble klotho consists of three fragments cleaved by ADAM10 and ADAM17: complete extracellular domain consisting of both KL1 and KL2, and cleaved KL1 and KL2 domains. They act either as humoral pleiotropic factors or as local autocrine-paracrine factors and are involved in the signal transduction of growth factors, regulate ion homeostasis, and protect against oxidative stress [26]. Cells capable of binding the soluble klotho must express at least one of the receptors listed above or must have sialic acids on their surface. Cellular response to the binding depends on the type of receptors expressed by the cell. Presumably all mammalian tissues express one or another set of these receptors and, hence, can respond to hormonal signals of the soluble α-klotho.
There are two opposite opinions on functional differences between the soluble and membrane-bound forms of α-klotho. It has been demonstrated that only the membrane-bound but not the soluble form can serve as the FGF23 co-receptor [27, 28]. In this case its activity is limited only to the klotho-expressing cells. Contrary to this observation other authors claim that soluble klotho is also able to bind FGF receptors [29]. The recent structural study demonstrated that soluble klotho could form a ternary complex with FGFR [7] and, hence, it could act as a hormone in any tissues expressing FGFR. We believe that the question about the ability of soluble klotho to bind FGFR remains unresolved since the causes of the discrepancy remain unclear.
Subdomains KL1 and KL2 are functionally different despite their significant structural similarity. Thus, the full-length soluble klotho consisting of KL1 and KL2 inhibits IGF1 and bFGF signaling and binds FGF23. Unlike the full-length klotho, KL1 fragment does not interact with FGF23 [30]. On the other hand, KL2 does not interact with IGF1 and bFGF pathways, but is essential for FGF23 signaling [14]. These differences are confirmed by the structural studies [22]. It is important that the soluble forms of klotho participate in these interactions, hence the IGF1 signaling responds to the hormonal effect of klotho. It also interacts with Wnt, TGFβ1 receptors, lipid rafts, and ion channels. Data on the individual functions of KL1 and KL2 are very limited. A possibility of cleavage of γ-klotho can be considered only by analogy with the other two forms.
The secretable α-klotho is formed by alternative splicing. Molecular weight of this protein is 65 kDa, close to that of KL1 (70 kDa) and contains additional motif of ten amino acids. Similar to KL1 and KL2 it prevents cellular senescence and controls mineral homeostasis [26, 31]. However, some authors find the independent role of the secretable form and its presence in blood doubtful [32]. In their recent work Xu, Y. and Sun, Z. [33] proved that the HEK293 and IMCD cells transfected with a short fragment of α-klotho identical to the secretable klotho expressed it followed by its binding to s-formylglutathione hydrolase (FGH) thus regulating activity of this enzyme. The binary complex was found in cytoplasm and peri-nuclear area. This is a remarkable finding, but it still does not clarify physiological role of the secretable klotho, because the natural levels of this form are significantly lower than the one used in this study. It was also shown that the secretable α-klotho binds chaperones 60 and 70 or ribosomal proteins. Taking into account that they facilitate folding and post-translational modification of many proteins, we may presume that klotho participates in the regulation of their homeostasis, although this requires further examination [33].
Clearly, such functions require cytoplasmic localization of klotho, possibly due to its endocytosis, but this matter also requires further experimental clarification. We are unaware of such studies of the soluble α-klotho, and we cannot compare the properties of all its truncated forms under similar conditions.
The γ-klotho forms complexes with FGFR1b, FGFR1c, FGFR2c, and FGFR4 and can function as a co-receptor for FGF19 in cultured cells [34]. Structural studies of these complexes have not been performed yet. Nothing is known about the role of γ-klotho in cardiovascular system, but it plays an essential role in other organs. It has been found that FGF15/19 induces weight loss in obese mice by activating thermogenesis in the brown adipose tissue [35], regulates expression of ephrin A3 and ephrin 4b in the developing retina and determines its nasal-temporal differentiation, essential for correct information flow to visual cortex through the growing axons of ganglion cells. It controls growth of retina as well as lens differentiation and its growth in Danio rerio [36] and chicken [37]. The ability of FGF15/19 to transduce signals through the γ-klotho-FGFR complexes proves its role in the regulation of these processes, and expression of FGFR by cardio-vascular tissues makes this interaction possible.
ANTIOXIDANT AND ENDOTHELIAL-PROTECTIVE EFFECTS OF KLOTHO
Oxidative stress causes a number of pathological conditions occurring due to insufficiency of the cellular antioxidative defense. This is true for the inflammatory diseases, endothelial dysfunction, hypertension [38], and atherosclerosis [39]. The antioxidative properties of klotho protein have been reported in numerous studies. It increases viability of the human endothelial cells HUVECs subjected to H2O2-induced oxidative injury, activates antioxidative enzymes [superoxide dismutase (SOD), catalase, and heme oxygenase-1 (HO-1)], and facilitates inactivation of reactive oxygen species (ROS). In addition, it inhibits apoptosis of endothelial cells and improves their function. This activates secretion of nitric oxide (NO). Antioxidative defense depends on the activation of PI3K/Akt pathway and increased expression of Nrf2 (transcription factor – a key regulator of the cellular defense, activating expression of the genes encoding antioxidative defense proteins under oxidative conditions) [28].
The effect of α-klotho on the PI3K/Akt signaling pathway is complicated. In particular, by binding to the insulin/IGF1 receptors, klotho promotes resistance to oxidative stress by inhibiting the insulin (or IGF-1)/PI3K/Akt signaling cascade. This prevents phosphorylation of the transcription factor FOXO1 (forkhead box protein O1), which becomes active and translocates into the nucleus, where it binds to the promoter and activates expression of SOD2, encoding the mitochondrial Mn-dependent superoxide dismutase [40]. It is likely that klotho can activate FOXO1 by inhibiting not only Akt, but also serum- and glucocorticoid-inducible kinase (SGK), or by stimulation of c-Jun N-terminal kinase (JNK). SGK, as well as Akt, is a component of the insulin/PI3K pathway. The SGK-induced phosphorylation of FOXO1 and FOXO3 promotes its association with the regulatory 14-3-3 proteins, induces its nuclear export into cytoplasm and inactivates FOXO [41]. Thus, in this case too, the α-klotho-induced blockade of insulin receptors should limit oxidative stress. On the other hand, JNK directly interacts with FOXO. Induction of JNK promotes nuclear localization of these transcription factors and its inhibition decreases expression of the gene targets of FOXO1 [42, 43]. Moreover, stimulation of the PI3K/Akt signals can inhibit stress- and cytokine-induced activation of JNK [44]. This way α-klotho inhibits PI3K/Akt while interacting with insulin receptors and promotes activation of JNK and FOXO1. JNK can also phosphorylate 14-3-3 proteins, and this induces release and activation of the bound FOXO [45]. In addition, JNK phosphorylates FOXO4 at Thr447 and Thr451, which induces its nuclear translocation and SOD expression [46]. Interestingly, β-catenin, a component of the Wnt signaling pathway, can form complexes with FOXO1 and increase its transcriptional activity [47].
Antioxidant activity of α-klotho was also reported by Yao Y. et al. (2017) [48]. They found that preincubation with the recombinant klotho increased viability of the HUVECs treated with the oxidized low-density lipoproteins (ox-LDL) and under oxidative stress. The α-klotho-activated cytoplasmic Cu,Zn-dependent superoxide dismutase (SOD1) induced expression of PI3K, Akt, endothelial NO-synthase (eNOS), and accumulation of NO. These changes were accompanied by the decrease of accumulation of malondialdehyde (MDA) and ROS, expression of the inducible NO-synthase (iNOS), activity of Gp91(phox) (heme-binding subunit of NADPH-oxidase, which generates superoxide anion), and lectin-like receptors of ox-LDL. As the result, α-klotho suppressed oxidative stress, which was induced by ox-LDL in HUVEC, due to activation of SOD1 and NO synthesis, both of which neutralize ROS. Activation of the PI3K/Akt/eNOS pathway and suppression of the expression of ox-LDL receptors play a central role in these events [48]. We face here an obvious contradiction: in some cases, α-klotho activates and, in the others, inhibits the PI3K/Akt pathway. Taking this into consideration, it can be also mentioned that α-klotho suppresses activity of the IGF1/PI3K/Akt pathway and increases expression of SOD2. On the other hand, klotho does not inhibit PI3K/Akt during stimulation of other receptors like FGFR. This activates alternative protective mechanisms (Cu-Zn-SOD, eNOS, heme-oxygenase). Hence, it can be concluded that klotho flexibly regulates several alternative antioxidative mechanisms.
As we have mentioned above, the secretable α-klotho containing KL1 and additional peptide motif directly binds S-formylglutathione hydrolase (FGH) – a key enzyme in the pathway generating antioxidant glutathione (GSH). Klotho activates both the enzyme itself and expression of its gene by activating transcription factor Kid3 in the cultured HEK293 and IMCD cells in vitro and in mouse kidney. The functional interaction between α-klotho and FGH is possible due to the post-translational modification, N-glycosylation of asparagine N285. Inhibition of glycosylation or replacement of Asp by Ala at the position 285 leads to the loss of antioxidant activity of klotho. Probably, N-glycosylation of N285 is essential for the activity and N-glycan fragments are necessary for the interaction with FGH [33]. As it was mentioned, α-klotho was found in cytoplasm together with FGH, where its function is unknown. This warrants further investigation.
Further studies of the klotho signaling pathways would add clarity to our understanding of the mechanisms by which this protein promotes resistance to oxidative stress and finally suppresses aging and ROS-related pathological processes [40].
It is known, that klotho promotes endothelial integrity, increases generation of endothelial nitric oxide, and improves endothelium-dependent vasodilation. Incubation of HUVECs with uremic toxin indoxyl sulphate (IS) induces massive release of ROS, expression of monocyte chemoattractant protein-1 (Monocyte Chemoattractant Protein 1, MCP-1), reduces cell viability and nitric oxide synthesis. These are the signs of endothelial dysfunction. This is associated with elevation of the p38MAPK (mitogen-activated protein kinase) activity induced by its phosphorylation and nuclear translocation of the proinflammatory transcription factor NF-κB. Preincubation with α-klotho protein promotes cell viability and increases synthesis of NO, suppression of ROS accumulation, and expression of MCP-1. It also prevents activation of p38MAPK and NF-κB in HUVECs. Thus, α-klotho prevents endothelial dysfunction, probably partly by inhibition of ROS accumulation and p38MAPK activity, as well as by inhibition of the downstream member of the signaling cascade, NF-κB [49, 50]. Antagonism between α-klotho and NFκB is revealed on different levels of cellular processes, for example on the level of activation of NFκB and FOXO1 [51].
Circulating α-klotho promotes normalization of the endothelium-dependent vasodilation, prevents apoptosis, and enhances regeneration of endothelium. It supports integrity of the layer of endothelial cells and decreases vascular permeability by binding to the Ca2+ channels TRPC1 (Transient receptor potential channels) and receptors for the vascular endothelial growth factor, VEGFR-2. Receptor internalizes in response to the stimulation by VEGF and regulates Ca2+ influx [52, 53]. α-Klotho inhibits phosphorylation of the VEGF receptor [54] and prevents its endocytosis. Vascular endothelium in the klotho-deficient mice is more permeable due to the increased apoptosis, lower expression of cadherin, and high activity of the Ca2+-dependent calpain/caspase-3 [52, 53], and, likely, high signaling activity of VEGF.
The soluble klotho exhibits anti-inflammatory activity by suppressing activation of NF-κB and downregulating expression of the TNF-α-induced adhesion molecules ICAM-1 and VCAM-1 in the endothelium [53]. Transcription of α-klotho is inhibited by cytokine IL-1β during inflammation. Simultaneously ERK1/2 activity is inhibited [55]. Antagonism between α-klotho and pro-inflammatory transcription factor NF-κB is discussed in details in [13]. Apparently the systemic α-klotho therapy may have anti-inflammatory effect [56] modulating interaction between endothelium and immune cells. This research topic is of significant clinical and scientific interest. Summarizing the facts presented above, it can be stated that α-klotho exerts its protective effect on endothelium through its antioxidant and anti-inflammatory properties and its ability to regulate calcium homeostasis.
KLOTHO PROTEIN AND CORONARY HEART DISEASE
Molecular mechanisms of pathogenesis of cardio-vascular diseases including coronary heart disease are still being investigated. Klotho protein is one of the targets of these studies. Some authors emphasized correlation between its low plasma level and the risk of coronary heart disease [53, 57]. A study of 3555 patients with the stable coronary heart disease and left ventricular ejection fraction >40%, participating in the PEACE trial (trandolapril against placebo), revealed that low concentration of α-klotho was correlated with the increased frequency of deaths of cardio-vascular diseases or higher incidence of hospitalizations due to the heart failure. The correlation was not changed after multifactorial correction of clinical variables and biomarkers, such as glomerular filtration rate, cystatin-C, albumin to creatinin ratio in urea, concentration of FGF23, troponin T, pro-natriuretic N-end peptide B, C-reactive protein. Co-incidence of the low concentrations of α-klotho and high FGF23 elevated the risk of death or hospitalization caused by ischemia [58]. This ratio of both proteins is apparently beneficial for the klotho-independent activity of FGF23. There is no clear view on its independent effects. It is considered proven that FGF23 promotes left ventricle hypertrophy to the sane degree in the heterozygous klotho knockout mice and in the wild-type animals [59]. However, elevation of the FGF23 level and left ventricle hypertrophy in the heterozygous klotho-knockout mice was less pronounced than in the homozygous animals. While the authors explain the difference as a dose-dependent effect, protective effect of klotho may also be considered [59]. On the other hand, Xie, J. et al. (2015) consider that klotho deficiency, rather than the elevated level of FGF23, causes heart hypertrophy. In heterozygous mice with chronic kidney disease (CKD) the level of serum phosphate and FGF23 did not correlate with the development of hypertrophy of the left ventricle, but injection of exogenous α-klotho changed the situation [60]. Currently it is not possible to choose between the α-klotho deficiency and elevation of FGF23 as possible causes of the hypertrophy in human, in part due to the lack of reliable methods of detection of soluble klotho.
It was found that klotho deficiency may lead to the disruption of neovascularization induced by ischemia and cause overexpression of plasminogen activator inhibitor-1 (PAI-1), which stimulates fibrotic processes, sinoatrial node dysfunctions, and sudden cardiac death [53].
Ischemia/reperfusion activates ROS generation in mitochondria, oxidative damage, and death of cardiomyocytes [61]. Developing oxidative stress causes endothelial dysfunction. It is likely that antioxidative properties of α-klotho and its protection of endothelium are key factors in its anti-ischemia effect. It may be expected that systemic klotho therapy could produce beneficial effect in this case.
KLOTHO EFFECT ON ELECTRIC ACTIVITY OF THE HEART
Klotho is found in the pacemaker cells of the sinoatrial node. It has been demonstrated that the 20-h stress increases incidence of sudden death, which is related either to the loss of conductivity or blockade of the sino-atrial node. Detailed molecular and cellular mechanisms of this effect are still unknown. Presumably, expression of α-klotho in sinoatrial node is essential for the function of some ion channels regulating excitability of pacemaker cells [62]. It is known that α-klotho modulates ion channels in plasma membrane including Na+/phosphate co-transporters [63, 64], Na+/K+-ATPases [65], Ca2+-channels [66], heart-specific potassium channels (hERG, human Ether-a-go-go Related Gene). These channels contribute to electric activity of the heart and play an essential role in the coordination of heartbeat [67], heart repolarization, and participate in abnormal excitation of myocardium after hypertrophy. These facts emphasize possible beneficial role of α-klotho in the regulation of heart activity and the potentials of its therapeutic use.
POSSIBLE ROLE OF KLOTHO IN THE DEVELOPMENT OF ARTERIAL
HYPERTENSION
Arterial rigidity is one of the risk factors of arterial hypertension. Pulse wave velocity is an indicator of arterial rigidity [68]. The level of circulating α-klotho in patients with hypertension and elevated vascular rigidity is decreased by 45% [69]. Deficit of α-klotho could be an essential factor that promotes arterial rigidity. Rigidity, in turn, plays an important role in the pathogenesis of hypertension. This is further supported by the fact that the experimental haplodeficiency of this protein in the Kl+/– mice correlates with the significant increase in the pulse wave velocity and elevation of blood pressure, as well as increased level of aldosterone. Arterial rigidity is one of the earliest detected unfavorable structural and functional changes in the vascular wall, it significantly elevates the risk of coronary heart disease and stroke [69-71]. The Kl deficit in the cultured aortic smooth muscle cell activated autophagy leading to the decline of the level of elastin and increased expression of scleraxis, a key transcription factor of collagen synthesis, which plays an essential role in the development of arterial rigidity [72]. Furthermore, haplodeficiency of α-klotho is associated with activation of metalloproteinase MMP9, pro-fibrotic factors TGFβ-1 and TGFβ-3, RUNX2 (transcription factor regulating cell cycle in endothelial cells and osteoblasts). These effects could be neutralized by chloroquine or eplerenone, antagonists of the aldosterone receptor. Physiological effects in vivo, such as increase in the pulse wave velocity and elevated arterial blood pressure, correspond to the changes on the cellular level. Notably, α-klotho suppresses expression of CYP11B2, a key enzyme of aldosterone synthesis in adrenals, on the transcriptional level [73]. Thus, α-klotho is involved in various aspects of maintenance of normal arterial function and prevention of the development of arterial hypertension: regulation of aldosterone level and rigidity of the vascular wall. Finding the link between klotho deficiency and increased vascular rigidity is an essential discovery, since it defines a new direction of the studies in this area. Currently existing anti-hypertensive drugs mostly decrease peripheral resistance and do not affect pathological processes of remodeling and vascular rigidity. To this effect, α-klotho may be an important etiological factor and a potential target for intervention aimed to limit arterial remodeling and rigidity.
ROLE OF KLOTHO IN PATHOGENESIS OF CARDIOMYOPATHY
α-Klotho is synthesized mostly in kidney and secreted into the bloodstream. Klotho deficit caused by CKD induces uremic cardiomyopathy. Chronic injections of recombinant klotho to rats with CKD minimized heart remodeling. It has been concluded that recombinant klotho is a safe, efficient, and promising prophylactic and therapeutic tool for the prevention or slowing uremic cardiomyopathy [74].
According to the widely accepted views, excessive elevation of FGF23 and/or phosphate plays a central role in the pathogenesis of uremic cardiomyopathy. Thus, protective effect of α-klotho may be based on its interaction with FGFR and FGF23. However, it was found recently, that soluble klotho limits hypertrophy of myocardium via a FGF23- and phosphate-independent way, by suppressing abnormal activity of the calcium-dependent signaling in heart [60, 75]. It was found that various extracellular stimuli elevate intracellular calcium concentration by its release from the cellular organelles or by pumping the extracellular calcium through cationic TRPC channels. TRPC family includes 7 members (TRPC1, 2, 3, 4, 5, 6, 7; TRPC2 is not expressed in humans). Ca2+ influx through TRPC1, 3, 4, 5 and 6 in cardiomyocytes is essential for signal transduction including activation of the calmodulin-dependent serine threonine phosphatase (calcineurin). This phosphatase dephosphorylates and stimulates nuclear factor of the activated T cells (NFAT) and NFAT-dependent expression of a number of genes, such as heavy chain of β-myosin. This, in turn, promotes pathological hypertrophy and heart remodeling [76, 77]. Circulating α-klotho interacts with the IGF1 receptor; it prevents IGF1-dependent activation of PI3K and the downstream exocytosis of TRPC6 receptor in cardiac cells. This mechanism is essential for cardioprotection exerted by α-klotho [75]. Structural modeling and molecular docking revealed that the soluble α-klotho can inhibit TRPC6 channels also by binding to the 2-3-sialyl-lactose moiety in gangliosides of lipid rafts. Reposition of the loop β6α6 in the KL1 domain of this protein occurs during this interaction. The protein interacts with glycolipids and forms a strong complex. Binding of the soluble klotho to lipid rafts modulates their structural organization in the membrane and inhibits PI3K/Akt-dependent signal transduction and TRPC6 exocytosis. The authors believe that lipid rafts could be receptors for the soluble α-klotho [16, 18, 19], and that the protein exerts protection against the stress-induced heart hypertrophy in mice by this mechanism [18].
It was found that soluble α-klotho limits the CKD-induced vascular calcification via direct effect on the vascular smooth muscle cells and indirectly by regulation of the phosphate metabolism. In vitro klotho inhibits Na(+)-dependent absorption of phosphate and mineralization, caused by high phosphate content in the vascular smooth muscle cells [78]. Some results by other groups are not in good agreement with these observations [79]. Lindberg, K. et al. (2013) observed no effect of klotho on the mouse aortic ring calcification [80]. Lim, K. et al. (2012) demonstrated that FGF23 alleviates calcification of smooth muscle cells of human aorta and this effect depends on the klotho induction [81]. Jimbo, R. et al. (2014) found that FGF23 intensified Pi-stimulated calcification in the cultivated human vascular smooth muscle cells, overexpressing α-klotho [82]. Mice with klotho deficiency had high level of the circulating phosphate and calcitriol and medial arterial calcification, practically identical to that found in the FGF23-knockout mice [83]. Majority of the recent works, however, support the view that α-klotho protects blood vessels against calcification. Hu, M. C. et al. (2011) reported that its deficiency in the CKD mice promotes Pi-induced calcification and that soluble α-klotho may suppress sodium-dependent Pi absorption and calcification in rats [78]. Zhang, W. et al. (2015) demonstrated that soluble klotho inhibited Pi-induced calcification of the mesenchymal stem cells from human bone marrow by inactivating of the FGFR1/ERK pathway [29]. Zhao, Y. et al. (2015) and Chang, J. R. et al. (2016) found that α-klotho reduced calcification of blood vessels and pointed out that both, membrane-bound and soluble forms were protective. These discrepancies warrant further studies of this phenomenon [84, 85].
It is worth mentioning that the results of studies on the role of klotho in the development of heart hypertrophy vary. Some studies found no pathology in the α-klotho-deficient mice [86]. It developed only after the experimental stress, and this could indicate that this protein facilitates formation of stress tolerance in the heart [60].
Hu, M. C. et al. (2015) [87], however, observed hypertrophy of the myocardium with the subsequent fibrosis in the klotho-deficient mice younger than 12 weeks. Depletion of klotho was caused by a genetic defect, high phosphate consumption, aging, and CKD. The degrees of heart hypertrophy and fibrosis did not depend on the cause of deficiency but positively correlated with the phosphate concentration and were inversely proportional to the α-klotho level in plasma. In vitro the protein inhibited TGF-β1- and angiotensin II-induced hypertrophy and fibrosis of cardiomyocytes or fibrosis caused by high phosphate level [87].
The origins of the discrepancies in the results have to be clarified. Probably the differences are caused by genetic variances between the mouse strains including stress tolerance or some other reasons.
KLOTHO PARTICIPATION IN HEART AND VASCULAR REMODELING AND HEART
FAILURE
In the rat model of heart failure induced by intraperitoneal injection of isoproterenol, klotho transfection promoted increased ejection rate, speed of contraction and relaxation (± dP/dt max), as well as notable alleviation of fibrosis and remodeling of the myocardium compared to the non-transfected animals [88]. Probably the cardioprotective effect of α-klotho is related to its ability to regulate the level of intracellular calcium, one of the culprits of the changes in myocardium, mentioned above. Tang, G. et al. (2018) demonstrated that α-klotho modulates activity of the Na+/Ca2+-exchange and Na+/K+-ATPase in H9C2 cells, treated with isoproterenol [89].
In vitro experiments revealed that α-klotho inhibits angiotensin II-induced hypertrophy and proliferation of cardiomyocytes, and fibrosis in the cells of the cardiac connective tissue. These effects of angiotensin II are mediated by activation of the TGF-β1 signaling pathway. TGF-β1 is a dimeric polypeptide secreted by a number of cell types including activated monocytes and macrophages, fibroblasts, endotheliocytes, mast cells, and others. Of five known isoforms of this protein, TGF-β1 is expressed notably more than others and plays an essential role in remodeling and fibrosis of blood vessels and myocardium. At the early stage of myocardial infarction, it is engaged in inflammation and later it participates in remodeling of the left ventricular by activation of fibroblasts and hypertrophy of the intact myocardium [90]. Its serum level is related to the rate of myocardial fibrosis in the left atrium of the patients with paroxysmal atrial fibrillation [91]. Post-surgical relapse of the myocardial ischemia is associated with the elevated level of TGF-β1 [92]. Interaction of TGF-β1 with its receptor type II (TGFR-βII) induces phosphorylation of several Smad-proteins (Smad and Mad related proteins). The classic signal cascade includes heterodimerization of Smad2/3, its interaction with Smad4, and nuclear translocation. Smad proteins act as pro-fibrotic transcription factors activating COL1A1- (collagen I gene) and ACTA2 (Alpha-smooth muscle actin, α-SMA). They also activate expression of the prohypertrophic genes NPPA encoding for atrial natriuretic peptide, ANP, and MYH7 for the myosin heavy chain beta, MHC-β.
Soluble klotho can directly bind to TGFR-βII hindering its interaction with TGF-β1. This inhibits signal transduction through this pathway and suppresses expression of the angiotensin II receptor type 1 (AT1R) [11]. Klotho inhibits both expression of TGF-β1 and TGF-β1-dependent phosphorylation of Smad2/3 (canonical signaling pathway of TGF-β1) in the angiotensin II-infused cardiac tissues and angiotensin II-stimulated cultured cardiomyocytes and fibroblasts.
In addition, klotho inhibits expression of FGF23 in the heart with angiotensin II-induced hypertrophy and fibrosis in vivo and in vitro. FGF-23 is generated in bones. It plays an important role in the regulation of phosphate metabolism in blood, decreases its reabsorption in the proximal kidney tubules, and decreases its clearance. Multiple studies prove that high level of FGF-23 is a clinically-important risk factor for the development of severe complications, including cardiac morbidity and mortality. High level of FGF23 is linked to the increased mass and hypertrophy of the left ventricle; it correlates with lethality in the patients with terminal stage of renal failure and coronary disease [93]. Klotho involvement in the suppression of these pathological processes could be schematically described as follows. By decreasing the amount of TGF-β1, α-klotho indirectly inhibits expression of microRNA-132 (figure; [94]), which suppresses transcription factor FOXO3, essential for the expression of atrogin-1. This F-box protein activates ubiquitination and degradation of calcineurin-A in cardiomyocytes, while excessive calcineurin-A stimulates NFAT and activates expression of pro-fibrotic and pro-hypertrophic genes downstream of NFAT. Thus, by inhibiting microRNA-132 klotho limits the development of fibrosis and hypertrophy of the myocardium [95].
Role of klotho in the development of heart hypertrophy and fibrosis induced by angiotensin II. It was found that treatment with the exogenous klotho positively affected the angiotensin II-induced heart remodeling and its dysfunction, which partially could be explained by its inhibitory effect on the TGF-β1/miR-132 pathway. Downward arrows indicate negative regulation or inhibition by klotho.
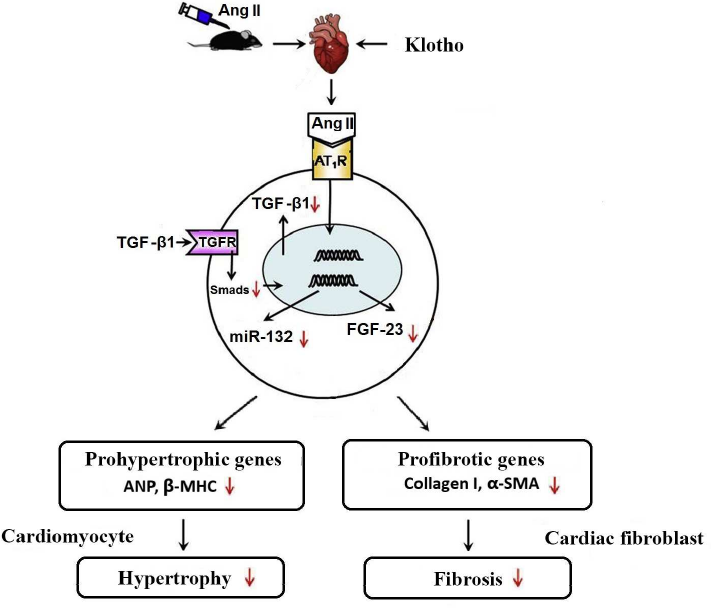
Wnt signaling pathway is also essential for the heart remodeling. It has been found that endothelial cells of endocardium can become progenitors of pericytes and myocytes, which are able to migrate into myocardium, differentiate, and integrate into the newly formed vascular wall. Wnt/Frizzled4, β-catenin, and Wnt ligands generated by the endothelial cells play a key role in these processes [96]. α-Klotho protein inhibits this signaling pathway, and this theoretically may slow down remodeling. On the other hand, pro-fibrotic activity of Wnt is neutralized by α-klotho and the net result of the klotho intervention could be positive, although this should be confirmed experimentally.
GENETIC PREDISPOSITION TO CARDIO-VASCULAR DISEASES LINKED TO THE
KLOTHO POLYMORPHISM
Cardio-vascular diseases have multifactorial etiology, and genetic predisposition to them is not uncommon. Cross-sectional studies revealed a link between the allele KL-VS and early coronary disease. Regression analysis demonstrated that KL-VS was an independent risk factor for coronary diseases, while the risk is modulated by a number of modifiable factors: hypertension masks, high density lipoproteins diminish, and smoking increases the risk [97]. There is a link between the KL-VS allele and ischemic stroke. Moreover, the homozygous patients in a younger group (< 40 years old) were at higher risk of the disease onset than the heterozygous patients. This association in the group older than 40 was insignificant [98].
Other types of klotho polymorphism were also found. They are associated with various deviations from the physiological norm. Thus, it has been found that a single-nucleotide polymorphism Kl rs650439 is significantly linked with the medium thickness of intima-media and atherosclerosis of the carotid artery in patients with hypertension [99].
Another polymorphism, G395A, in the promoter sequence of the human Kl gene may be a genetic risk factor for coronary heart disease and is independent on vasospastic angina [100].
Polymorphism C1818T in the exon 4 of Kl is associated with higher systolic blood pressure than the variant C1818C. It is likely related to the effect of C1818T on NO synthesis, which declines in people older than 40 years of age, bearers of this polymorphism. Low bioavailability of NO promotes endothelial dysfunction, vasoconstriction, thrombosis, and cardio-vascular diseases [101, 102]. Presumably, replacement therapy with klotho protein could be beneficial in the treatment of the pathologies mentioned above.
MODULATION OF KLOTHO ACTIVITY IS A NEW APPROACH TO THE THERAPY OF
CARDIO-VASCULAR DISEASES
While therapeutic agents designed purposely for modulation of klotho activity currently do not exist, some of the approved drugs either increase klotho expression or limit the decline in its synthesis in the pathologies, mentioned above. Thus, isoproterenol-caused intoxication decreased the heart rate, expression of klotho and HCN4 (hyperpolarization-activated cyclic nucleotide-gated (HCN) channels. These integral proteins are non-selective ligand-dependent cation channels in the plasma membrane of heart and brain. HCN are also called pacemaker channels, because they are involved in generation of the rhythmic activity of the cardiac and brain cells [103]. It has been found that the flavonoid astragaloside IV elevated expression of α-klotho, which in turn increased expression of HCN and f-channels and normalized heart rate. This finding may be just the first in the series of discoveries of the klotho-modulating drugs, which could be used to treat heart rhythm disorders [103].
Angioprotective effect of statins (atorvastatin and pitavastatin) is probably related to α-klotho through inactivation of RhoA [104]. Statins elevate expression of α-klotho mRNA in the cultured IMCD3 (Mouse internal medulla collecting duct epithelial cells) in a dose-dependent manner by inhibiting RhoA. The protein, in turn, regulates calcium metabolism and protects blood vessels against calcification, which increases their rigidity [104].
It has been demonstrated that inhibitors of the angiotensin-converting enzyme and blockers of the angiotensin II receptors induce expression of α-klotho due to the suppression of renin-angiotensin-aldosterone system, which inhibits synthesis of this protein [105].
Hypoglycemic drugs rosiglitazone and thiazolidinedione modulate expression of α-klotho in the vascular smooth muscle cells (VSMCs) because they stimulate PPARγ, a factor that activates its transcription. These drugs prevent tissue calcification probably by increasing the level of α-klotho [106].
Vascular calcification exacerbates ischemia in myocardium. It is the main risk factor of cardio-vascular mortality in patients. Considering that α-klotho plays a critical role in limiting calcification caused by the immunosuppressant rapamycin in vitro and in vivo through inhibition of mTOR [84], it could be a potential target in the search of therapeutic agents that limit pathological changes in blood vessels. A well-known inductor of α-klotho is vitamin D, whose excessive intake also causes calcification of blood vessels and internal organs. Many positive effects of this vitamin are probably linked to α-klotho, which, in turn, regulates the level of vitamin D and prevents its side effects [107].
Thus, we present the data indicating that drugs elevating expression of klotho may be valuable therapeutic agents, because this protein modulates a number of processes important for the development of cardio-vascular diseases. It suppresses lipid peroxidation and inflammation, prevents endothelial damage, atherosclerosis, calcification of blood vessels, inhibits heart and vascular fibrosis, and reduces vascular rigidity. While considering feasibility of the therapeutic applications of klotho we need to take into account possible side effects. They may be predicted from the analysis of the signaling mechanisms regulated by klotho. Thus, potential overdose may possibly lead to hypotension, hypophosphatemia, hypocalciemia, since the tandem of klotho and FGF23 promotes excretion of phosphate and calcium, although systemic administration of klotho in animal experiments caused neither of these effects. On the contrary, insufficient expression of klotho combined with the excessive level of FGF23 in fact leads to hypophosphatemia and hypocalciemia. As we have mentioned before, hypotensive effect of klotho is caused by increased elasticity of blood vessels and antagonism between the klotho and renin-angiotensin-aldosterone system. Another possible side effect may be caused by the interaction with insulin receptors, which may theoretically lead to insulin resistance. This possibility was tested in the experimental model and was rejected, since the protein did not participate directly in the induction of tolerance to insulin [108]. Pre-clinical studies cannot give final answers to the questions about the possible side effects in humans because of differences between the species. However, the clinically-tested inductors of klotho can be used for preliminary estimation. Thus, statins and vitamin D are used in clinical practice and their prolonged consumption did not cause the mentioned side effects. This indirectly indicates that elevation of the klotho concentration or its systemic intervention are probably safe. Unfortunately, rosiglitazone has a number of unwanted side effects, which are obviously not related to the increased synthesis of klotho protein [109, 110]. Hence, induction of klotho expression may have some limitations, but the systemic therapy with recombinant protein is devoid of them. Clinical testing may provide a final answer to this question, but this has not been performed yet.
CONCLUSION
For a long time, molecular pathogenetic mechanisms of cardio-vascular diseases continue to attract significant attention of scientific community. Studies of the role of protein-protein interaction in pathological processes may shed light on the origin of the heart and vascular pathologies and provide new opportunities for the discovery of therapeutic agents and new treatments for these diseases. Klotho protein plays an essential role in the mechanisms protecting cardio-vascular systems against functional disruption and development of cardiomyopathy, heart failure, arterial hypertension. Pleiotropic properties of klotho underlie its involvement in multiple cardioprotective mechanisms and their interaction. Regulation of its level in serum and its expression in cardiomyocytes by therapeutic agents may be of great importance in cellular metabolism and may become a perspective target for treatment of cardio-vascular diseases.
We already mentioned some unresolved issues (the differences between membrane-bound and soluble forms of klotho, enzymatic mechanisms, different functions of KL1 and KL2). Function of klotho in cytoplasm, where it may bind to the proteins other than chaperones or FGH has not been investigated yet, and this topic should be added to the list of the warranted studies. It is likely that it is precisely intracellular klotho that modulates gene expression by itself or with assistance of other proteins. This attracts attention to the origin of the cytoplasmic klotho and the routes of its possible internalization, for instance by co-endocytosis with the receptor proteins interacting with klotho. It is likely that klotho could participate in the fine tuning of the life-long homeostasis by interacting with canonical receptors and modulating their function. For example, high activity of canonical mechanisms of FGF and Wnt plays an important role in organogenesis at early stages of life. Adult organism maintains its homeostasis using various regulatory mechanisms, including gene expression and proteolytic degradation of proteins. Modulation of the activity of existing signaling pathways by klotho is yet another such mechanism. For example, the loss of such klotho-mediated control is accompanied by pathologies similar to the case when the excess of FGF23 and klotho deficiency are observed in the coronary heart disease. Insufficient control by klotho and high activity of Wnt, IGF1, TGFβ1 pathway promotes fibrosis and cancer development. It could be suggested that this tuning defines physiological meaning of klotho. Its role as a potential regulator and synchronizer of homeostasis has not been studied yet. Finally, majority of the published works are focused on α-klotho, and not on its paralogs β and γ, hence, this is another issue waiting to be clarified.
Ethics declarations. The authors declare no conflict of interest in financial or any other sphere. This article does not contain any studies with human participants or animals performed by any of the authors.
REFERENCES
1.Kuro-o, M., Matsumura, Y., Aizawa, H., Kawaguchi,
H., Suga, T., et al. (1997) Mutation of the mouse klotho gene leads to
a syndrome resembling ageing, Nature, 390, 45-51, doi:
10.1038/36285.
2.Shiraki-Iida, T., Aizawa, H., Matsumura, Y.,
Sekine, S., Iida, A., et al. (1998) Structure of the mouse klotho gene
and its two transcripts encoding membrane and secreted protein 1,
FEBS Lett., 424, 6-10, doi:
10.1016/S0014-5793(98)00127-6.
3.Li, S.-A., Watanabe, M., Yamada, H., Nagai, A.,
Kinuta, M., and Takei, K. (2004) Immunohistochemical localization of
Klotho protein in brain, kidney, and reproductive organs of mice,
Cell Structure Function, 29, 91-99, doi:
10.1247/csf.29.91.
4.Lim, K., Groen, A., Molostvov, G., Lu, T., Lilley,
K. S., et al. (2015) α-Klotho expression in human tissues, J.
Clin. Endocrinol. Metab., 100, 1308-1318, doi:
10.1210/jc.2015-1800.
5.Melnik, A. A. (2017) Protein Klotho and FGF23
fibroblasts growth, Pochki, 6, 132-138.
6.Ito, S., Fujimori, T., Hayashizaki, Y., and
Nabeshima, Y. (2002) Identification of a novel mouse membrane-bound
family 1 glycosidase-like protein, which carries an atypical active
site structure, Biochim. Biophys. Acta, 1576, 341-345,
doi: 10.1016/s0167-4781(02)00281-6.
7.Chen, G., Liu, Y., Goetz, R., Fu, L., Jayaraman,
S., et al. (2018) α-Klotho is a non-enzymatic molecular scaffold
for FGF23 hormone signalling, Nature, 553, 461-466, doi:
10.1038/nature25451.
8.Xie, T., and Leung, P. S. (2017) Fibroblast growth
factor 21: a regulator of metabolic disease and health span, Am. J.
Physiol. Endocrinol. Metab., 313, E292-E302, doi:
10.1152/ajpendo.00101.2017.
9.Katoh, M., and Nakagama, H. (2014) FGF receptors:
cancer biology and therapeutics, Med. Res. Rev., 34,
280-300, doi: 10.1002/med.21288.
10.Grabner, A., Amaral, A. P., Schramm, K., Singh,
S., Sloan, A., et al. (2015) Activation of cardiac Fibroblast Growth
Factor Receptor 4 causes left ventricular hypertrophy, Cell
Metab., 22, 1020-1032, doi: 10.1016/j.cmet.2015.09.002.
11.Doi, S., Zou, Y., Toga, O., Pastor, J. V., John,
G. B., et al. (2011) Klotho inhibits Transforming Growth Factor-β1
(TGF-β1) signaling and suppresses renal fibrosis and cancer
metastasis in mice, J. Biol. Chem., 286, 8655-8665, doi:
10.1074/jbc.M110.174037.
12.Lee, J., Jeong, D. J., Kim, J., Lee, S., Park, J.
H., et al. (2010) The anti-aging gene KLOTHO is a novel target for
epigenetic silencing in human cervical carcinoma, Mol. Cancer,
9, 109, doi: 10.1186/1476-4598-9-109.
13.Nesterova, A. A., Glinka, E. Yu., Tyurenkov, I.
N., and Perfilova, V. N. (2020) Protein klotho – universal
regulator of physiological processes in the organism, Usp. Fiziol.
Nauk, 51, 88-104.
14.Ligumsky, H., Rubinek, T., Merenbakh-Lamin, K.,
Yeheskel, A., Sertchook, R., et al. (2015) Tumor suppressor activity of
Klotho in breast cancer is revealed by structure–function
analysis, Mol. Cancer Res., 13, 1398-1407, doi:
10.1158/1541-7786.MCR-15-0141.
15.Sopjani, M., Rinnerthaler, M., Kruja, J.,
Dërmaku-Sopjani, M. (2015) Intracellular signaling of the aging
suppressor protein Klotho, Curr. Mol. Med., 15, 27-37,
doi: 10.2174/1566524015666150114111258.
16.Dalton, G., An, S.-W., Al-Juboori, S. I.,
Nischan, N., Yoon, J., Dobrinskikh, E., et al. (2017) Soluble klotho
binds monosialoganglioside to regulate membrane microdomains and growth
factor signaling, Proc. Natl. Acad. Sci. USA, 114,
752-757, doi: 10.1073/pnas.1620301114.
17.Wolf, M. T., An, S. W., Nie, M., Bal, M. S., and
Huang, C. L. (2014) Klotho up-regulates renal calcium channel transient
receptor potential vanilloid 5 (TRPV5) by intra- and extracellular
N-glycosylation-dependent mechanisms, J. Biol. Chem.,
289, 35849-35857, doi: 10.1074/jbc.M114.616649.
18.Wright, J. D., An, S. W., Xie, J., Lim, C., and
Huang, C. L. (2019) Soluble klotho regulates TRPC6 calcium signaling
via lipid rafts, independent of the FGFR-FGF23 pathway, FASEB
J., 33, 9182-9193, doi: 10.1096/fj.201900321R.
19.Wright, J. D., An, S.-W., Xie, J., Yoon, J.,
Nischan, N., et al. (2017) Modeled structural basis for the recognition
of α2-3-sialyllactose by soluble Klotho, FASEB J.,
31, 3574-3586, doi: 10.1096/fj.201700043R.
20.Zhang, J., Gupte, J., Gong, Y., Weiszmann, J.,
Zhang, Y., et al. (2017) Over-expression of fibroblast growth factor 21
increases bile acid biosynthesis by opposing FGF15/19 action,
EBioMedicine, 15, 173-183, doi:
10.1016/j.ebiom.2016.12.016.
21.Patel, V., Adya, R., Chen, J., Ramanjaneya, M.,
Bari, M. F., et al. (2014) Novel insights into the cardio-protective
effects of FGF21 in lean and obese rat hearts, PLoS One,
9, e87102, doi: 10.1371/journal.pone.0087102.
22.Lee, S., Choi, J., Mohanty, J., Sousa, L. P.,
Tome, F., et al. (2018) Structures of β-Klotho reveal a ‘zip
code’-like mechanism for endocrine FGF signaling, Nature,
553, 501-505, doi: 10.1038/nature25010.
23.Kuzina, E. S., Ung, P. M., Mohanty, J., Tome, F.,
Choi, J., et al. (2019) Structures of ligand-occupied β-Klotho
complexes reveal a molecular mechanism underlying endocrine FGF
specificity and activity, Proc. Natl. Acad. Sci. USA,
116, 7819-7824, doi: 10.1073/pnas.1822055116.
24.Rubinek, T., Shahmoon, S., Shabtay-Orbach, A.,
Ben Ami, M., Levy-Shraga, Y., et al. (2016) Klotho response to
treatment with growth hormone and the role of IGF-I as a mediator,
Metabolism, 65, 1597-1604, doi:
10.1016/j.metabol.2016.08.004.
25.Chen, C. D., Podvin, S., Gillespie, E., Leeman,
S. E., Abraham, C. R. (2007) Insulin stimulates the cleavage and
release of the extracellular domain of Klotho by ADAM10 and ADAM17,
Proc. Natl. Acad. Sci. USA, 104, 19796-19801, doi:
10.1073/pnas.0709805104.
26.Kim, J. H., Hwang, K. H., Park, K. S., Kong, I.
D., and Cha, S. K. (2015) Biological role of anti-aging protein klotho,
J. Lifestyle Med., 5, 1-6, doi:
10.15280/jlm.2015.5.1.1.
27.Kuro-o, M. (2009) Klotho and aging, Biochim.
Biophys. Acta, 1790, 1049-1058, doi:
10.1016/j.bbagen.2009.02.005.
28.Cui, W., Leng, B., and Wang, G. (2019) Klotho
protein inhibits H2O2-induced oxidative injury in
endothelial cells via regulation of PI3K/AKT/Nrf2/HO-1 pathways,
Can. J. Physiol. Pharmacol., 97, 370-376, doi:
10.1139/cjpp-2018-0277.
29.Zhang, W., Xue, D., Hu, D., Xie, T., Tao, Y.,
Zhu, T., Chen, E., and Pan, Z. (2015) Secreted klotho protein
attenuates osteogenic differentiation of human bone marrow mesenchymal
stem cells in vitro via inactivation of the FGFR1/ERK signaling
pathway, Growth Factors, 33, 356-365, doi:
10.3109/08977194.2015.1108313.
30.Abramovitz, L., Rubinek, T., Ligumsky, H., Bose,
S., Barshack, I., et al. (2011) KL1 internal repeat mediates Klotho
tumor suppressor activities and inhibits bFGF and IGF-I signaling in
pancreatic cancer, Clin. Cancer Res., 17, 4254-4266, doi:
10.1158/1078-0432.CCR-10-2749.
31.Neyra, J. A., and Hu, M. C. (2017) Potential
application of klotho in human chronic kidney disease, Bone,
100, 41-49, doi: 10.1016/j.bone.2017.01.017.
32.Mencke, R., Harms, G., Moser, J., van Meurs, M.,
Diepstra, A., Leuvenink, H. G., and Hillebrands, J. L. (2017) Human
alternative Klotho mRNA is a nonsense-mediated mRNA decay target
inefficiently spliced in renal disease, JCI Insight, 2,
94375, doi: 10.1172/jci.insight.94375.
33.Xu, Y., and Sun, Z. (2017) Regulation of
S-formylglutathione hydrolase by the anti-aging gene klotho,
Oncotarget, 8, 88259-88275, doi:
10.18632/oncotarget.19111.
34.Tacer, K. F., Bookout, A. L., Ding, X. S.,
Kurosu, H., John, G. B., et al. (2010) Research resource: comprehensive
expression atlas of the fibroblast growth factor system in adult mouse,
Mol. Endocrinol., 24, 2050-2064, doi:
10.1210/me.2010-0142.
35.Tomlinson, E., Fu, L., John, L., Hultgren, B.,
Huang, X., et al. (2002) Transgenic mice expressing human fibroblast
growth factor-19 display increased metabolic rate and decreased
adiposity, Endocrinology, 143, 1741-1747.
36.Nakayama, Y., Miyake, A., Nakagawa, Y., Mido, T.,
Yoshikawa, M., Konishi, M., and Itoh, N. (2008) Fgf19 is required for
zebrafish lens and retina development, Dev. Biol., 313,
752-766, doi: 10.1016/j.ydbio.2007.11.013.
37.Kurose, H., Okamoto, M., Shimizu, M., Bito, T.,
Marcelle, C., Noji, S., and Ohuchi, H. (2005) FGF19-FGFR4 signaling
elaborates lens induction with the FGF8-L-Maf cascade in the chick
embryo, Dev. Growth Differ., 47, 213-223a, doi:
10.1111/j.1440-169X.2005.00795.x.
38.Dinh, Q. N., Drummond, G. R., Sobey, C. G., and
Chrissobolis, S. (2014) Roles of inflammation, oxidative stress, and
vascular dysfunction in hypertension, Biomed. Res. Int.,
2014, 406960, doi: 10.1155/2014/406960.
39.Förstermann, U., Xia, N., and Li, H. (2017)
Roles of vascular oxidative stress and nitric oxide in the pathogenesis
of atherosclerosis, Circ. Res., 120, 713-735, doi:
10.1161/CIRCRESAHA.116.309326.
40.Yamamoto, M., Clark, J. D., Pastor, J. V.,
Gurnani, P., Nandi, A., Kurosu, H., et al. (2005) Regulation of
oxidative stress by the anti-aging hormone klotho, J. Biol.
Chem., 280, 38029-38034, doi: 10.1074/jbc.M509039200.
41.Wang, Y., Zhou, Y., and Graves, D. T. (2014) FOXO
transcription factors: their clinical significance and regulation,
Biomed. Res. Int., 2014, 925350, doi:
10.1155/2014/925350.
42.Kawamori, D., Kaneto, H., Nakatani, Y., Matsuoka,
T. A., Matsuhisa, M., Hori, M., and Yamasaki, Y. (2006) The forkhead
transcription factor Foxo1 bridges the JNK pathway and the
transcription factor PDX-1 through its intracellular translocation,
J. Biol. Chem., 281, 1091-1098, doi:
10.1074/jbc.M508510200.
43.Ju, Y., Xu, T., Zhang, H., and Yu, A. (2014)
FOXO1-dependent DNA damage repair is regulated by JNK in lung cancer
cells, Int. J. Oncol., 44, 1284-1292, doi:
10.3892/ijo.2014.2269.
44.Zhao, H. F., Wang, J., and Tony To, S. S. (2015)
The phosphatidylinositol 3-kinase/Akt and c-Jun N-terminal kinase
signaling in cancer: Alliance or contradiction? Int. J. Oncol.,
47, 429-436, doi: 10.3892/ijo.2015.3052.
45.Sunayama, J., Tsuruta, F., Masuyama, N., and
Gotoh, Y. (2005) JNK antagonizes Akt-mediated survival signals by
phosphorylating 14-3-3, J. Cell. Biol., 170, 295-304,
doi: 10.1083/jcb.200409117.
46.Essers, M. A., Weijzen, S., de Vries-Smits, A.
M., Saarloos, I., de Ruiter, N. D., Bos, J. L., and Burgering, B. M.
(2004) FOXO transcription factor activation by oxidative stress
mediated by the small GTPase Ral and JNK, EMBO J., 23,
4802-4812, doi: 10.1038/sj.emboj.7600476.
47.Essers, M. A., de Vries-Smits, L. M., Barker, N.,
Polderman, P. E., Burgering, B. M., and Korswagen, H. C. (2005)
Functional interaction between beta-catenin and FOXO in oxidative
stress signaling, Science, 308, 1181-1184.
48.Yao, Y., Wang, Y., Zhang, Y., and Liu, C. (2017)
Klotho ameliorates oxidized low density lipoprotein (ox-LDL)-induced
oxidative stress via regulating LOX-1 and PI3K/Akt/eNOS pathways,
Lipids Health Dis., 16, 77, doi:
10.1186/s12944-017-0447-0.
49.Yang, K., Nie, L., Huang, Y., Zhang, J., Xiao,
T., Guan, X., and Zhao, J. (2012) Amelioration of uremic toxin indoxyl
sulfate-induced endothelial cell dysfunction by Klotho protein,
Toxicol. Lett., 215, 77-83, doi:
10.1016/j.toxlet.2012.10.004.
50.Chung, C. P., Chang, Y. C., Ding, Y., Lim, K.,
Liu, Q., et al. (2017) α-Klotho expression determines nitric
oxide synthesis in response to FGF-23 in human aortic endothelial
cells, PLoS One, 12, e0176817, doi:
10.1371/journal.pone.0176817.
51.Prud’homme, G. J., Glinka, Y., Kurt, M.,
Liu, W., and Wang, Q. (2017) The anti-aging protein Klotho is induced
by GABA therapy and exerts protective and stimulatory effects on
pancreatic beta cells, Biochem. Biophys. Res. Commun.,
493, 1542-1547, doi: 10.1016/j.bbrc.2017.10.029.
52.Kusaba, T., Okigaki, M., Matui, A., Murakami, M.,
Ishikawa, K., et al. (2010) Klotho is associated with VEGF receptor-2
and the transient receptor potential canonical-1 Ca2+
channel to maintain endothelial integrity, Proc. Natl. Acad. Sci.
USA, 107, 19308-19313, doi: 10.1073/pnas.1008544107.
53.Semba, R. D., Cappola, A. R., Sun, K.,
Bandinelli, S., Dalal, M., et al. (2011) Plasma klotho and
cardiovascular disease in adults, J. Am. Geriatr. Soc.,
59, 1596-1601, doi: 10.1111/j.1532-5415.2011.03558.x.
54.Kokkinaki, M., Abu-Asab, M., Gunawardena, N.,
Ahern, G., Javidnia, M., et al. (2013) Klotho regulates retinal pigment
epithelial functions and protects against oxidative stress, J.
Neurosci., 33, 16346-16359, doi:
10.1523/JNEUROSCI.0402-13.2013.
55.Zhao, Y., Meng, C., Wang, Y., Huang, H., Liu, W.,
et al. (2016) IL-1 inhibits – Klotho expression and FGF1
signaling in hepatocytes, Physiol. Endocrinol. Metab.,
310, E289-300, doi: 10.1152/ajpendo.00356.2015.
56.Prud’homme, G. J., Glinka, Y., Kurt, M.,
Liu, W., and Wang, Q. (2020) Systemic Klotho therapy protects against
insulitis and enhances beta-cell mass in NOD mice, Biochem. Biophys.
Res. Commun., 525, 693-698, doi:
10.1016/j.bbrc.2020.02.123.
57.Navarro-González, J. F., Donate-Correa,
J., Muros de Fuentes, M., Pérez-Hernández, H.,
Martínez-Sanz, R., and Mora-Fernández, C. (2014) Reduced
Klotho is associated with the presence and severity of coronary artery
disease, Heart, 100, 34-40, doi:
10.1136/heartjnl-2013-304746.
58.Bergmark, B. A., Udell, J. A., Morrow, D. A.,
Jarolim, P., Kuder, J. F., et al. (2019) Klotho, fibroblast growth
factor-23, and the renin-angiotensin system – an analysis from
the PEACE trial, Eur. J. Heart Fail., 21, 462-470, doi:
10.1002/ejhf.1424.
59.Faul, C., Amaral, A. P., Oskouei, B., Hu, M. C.,
Sloan, A., and Isakova, T. (2011) FGF23 induces left ventricular
hypertrophy, J. Clin. Invest., 121, 4393-4408, doi:
10.1172/JCI46122.
60.Xie, J., Yoon, J., An, S-W., Kuro-o, M., and
Huang, C.-L. (2015) Soluble klotho protects against uremic
cardiomyopathy independently of fibroblast growth factor 23 and
phosphate, J. Am. Soc. Nephrol., 26, 1150-1160, doi:
10.1681/ASN.2014040325.
61.Zhou, X., Li, S., Wang, Z., Yu, L., and Jiang, H.
(2015) Klotho protein: a potential therapeutic agent during myocardial
ischemia and reperfusion, Int. J. Cardiol., 191, 227-228,
doi: 10.1016/j.ijcard.2015.05.029.
62.Takeshita, K., Fujimori, T., Kurotaki, Y., Honjo,
H., Tsujikawa, H., et al. (2004) Sinoatrial node dysfunction and early
unexpected death of mice with a defect of klotho gene expression,
Circulation, 109, 1776-1782, doi:
10.1161/01.CIR.0000124224.48962.32.
63.Dermaku-Sopjani, M., Sopjani, M., Saxena, A.,
Shojaiefard, M., Bogatikov, E., et al. (2011) Downregulation of
NaPi-IIa and NaPi-IIb Na-coupled phosphate transporters by coexpression
of Klotho, Cell Physiol. Biochem., 28, 251-258, doi:
10.1159/000331737.
64.Hu, M. C., Shi, M., Zhang, J., Pastor, J.,
Nakatani, T., and Lanske, B. (2010) Klotho: a novel phosphaturic
substance acting as an autocrine enzyme in the renal proximal tubule,
FASEB J., 24, 3438-3450, doi: 10.1096/fj.10-154765.
65.Sopjani, M., Alesutan, I., Dermaku-Sopjani, M.,
Gu, S., Zelenak, C., et al. (2011) Regulation of the
Na+/K+-ATPase by Klotho, FEBS Lett.,
585, 1759-1764, doi: 10.1016/j.febslet.2011.05.021.
66.Boros, S., Bindels, R. J., and Hoenderop, J. G.
(2009) Active Ca(2+) reabsorption in the connecting tubule, Pflugers
Arch., 458, 99-109, doi: 10.1007/s00424-008-0602-6.
67.Munoz, C., Pakladok, T., Almilaji, A., Elvira,
B., Seebohm, G., et al. (2013) Klotho sensitivity of the hERG channel,
FEBS Lett., 587, 1663-1668, doi:
10.1016/j.febslet.2013.04.011.
68.Kaess, B. M., Rong, J., Larson, M. G., Hamburg,
N. M., Vita, J. A., et al. (2012) Aortic stiffness, blood pressure
progression, and incident hypertension, JAMA, 308,
875-881, doi: 10.1001/2012.jama.10503.
69.Cavalcante, J. L., Lima, J. A., Redheuil, A., and
Al-Mallah, M. H. (2011) Aortic stiffness: current understanding and
future directions, J. Am. Coll. Cardiol., 57, 1511-1522,
doi: 10.1016/j.jacc.2010.12.017.
70.Gao, D., Zuo, Z., Tian, J., Ali, Q., Lin, Y.,
Lei, H., and Sun, Z. (2016) Activation of SIRT1 attenuates klotho
deficiency-induced arterial stiffness and hypertension by enhancing
AMP-activated protein kinase activity, Hypertension, 68,
1191-1199, doi: 10.1161/HYPERTENSIONAHA.116.07709.
71.Mencke, R., and Hillebrands, J.-L. (2016) The
Role of the anti-ageing protein klotho in vascular physiology and
pathophysiology, Nature, 553, 461-466, doi:
10.1016/j.arr.2016.09.001.
72.Chen, K., and Sun, Z. (2019) Autophagy plays a
critical role in Klotho gene deficiency-induced arterial stiffening and
hypertension, J. Mol. Med. (Berl.), 97, 1615-1625, doi:
10.1007/s00109-019-01841-6.
73.Zhou, X., Chen, K., Wang, Y., Schuman, M., Lei,
H., and Sun, Z. (2016) Antiaging gene Klotho regulates adrenal CYP11B2
expression and aldosterone synthesis, JASN, 27,
1765-1776, doi: 10.1681/ASN.2015010093.
74.Hu, M. C., Shi, M., Gillings, N., Flores, B.,
Takahashi, M., and Kuro-o, M., and Moe, O. W. (2017) Recombinant
α-Klotho may be prophylactic and therapeutic for acute to chronic
kidney disease progression and uremic cardiomyopathy, Kidney
Int., 91, 1104-1114, doi: 10.1016/j.kint.2016.10.034.
75.Xie, J., Cha, S. K., An, S. W., Kuro-o, M.,
Birnbaumer, L., and Huang, C. L. (2012) Cardioprotection by Klotho
through downregulation of TRPC6 channels in the mouse heart, Nat.
Commun., 3, 123, doi: 10.1038/ncomms2240.
76.Wu, X., Eder, P., Chang, B., and Molkentin, J. D.
(2010) TRPC channels are necessary mediators of pathologic cardiac
hypertrophy, Proc. Natl. Acad. Sci. USA, 107, 7000-7005,
doi: 10.1073/pnas.1001825107.
77.Eder, P., and Molkentin, J. D. (2011) TRPC
channels as effectors of cardiac hypertrophy, Circ. Res.,
108, 265-272, doi: 10.1161/CIRCRESAHA.110.225888.
78.Hu, M. C., Shi, M., Zhang, J., Quiñones,
H., Griffith, C., Kuro-o, M., and Moe, O. W. (2011) Klotho deficiency
causes vascular calcification in chronic kidney disease, J. Am. Soc.
Nephrol., 22, 124-136, doi: 10.1681/ASN.2009121311.
79.Yamada, S., and Giachelli, C. M. (2017) Vascular
calcification in CKD-MBD: Roles for phosphate, FGF23, and Klotho,
Bone, 100, 87-93, doi: 10.1016/j.bone.2016.11.012.
80.Lindberg, K., Olauson, H., Amin, R., Ponnusamy,
A., Goetz, R., et al. (2013) Arterial klotho expression and FGF23
effects on vascular calcification and function, PLoS One,
8, e60658.
81.Lim, K., Lu, T. S., Molostvov, G., Lee, C., Lam,
F. T., Zehnder, D., and Hsiao, L. L. (2012) Vascular Klotho deficiency
potentiates the development of human artery calcification and mediates
resistance to fibroblast growth factor 23, Circulation,
125, 2243-2255.
82.Jimbo, R., Kawakami-Mori, F., Mu, S., Hirohama,
D., Majtan, B., et al. (2014) Fibroblast growth factor 23 accelerates
phosphate-induced vascular calcification in the absence of Klotho
deficiency, Kidney Int., 85, 1103-1111.
83.Shimada, T., Kakitani, M., Yamazaki, Y.,
Hasegawa, H., Takeuchi, Y., et al. (2004) Targeted ablation of Fgf23
demonstrates an essential physiological role of FGF23 in phosphate and
vitamin D metabolism, J. Clin. Invest., 113, 561-568.
84.Zhao, Y., Zhao, M. M., Cai, Y., Zheng, M. F.,
Sun, W. L., et al. (2015) Mammalian target of rapamycin signaling
inhibition ameliorates vascular calcification via Klotho upregulation,
Kidney Int., 88, 711-721, doi: 10.1038/ki.2015.160.
85.Chang, J. R., Guo, J., Wang, Y., Hou, Y. L., Lu,
W. W., et al. (2016) Intermedin1-53 attenuates vascular calcification
in rats with chronic kidney disease by upregulation of α-Klotho,
Kidney Int., 89, 586-600, doi:
10.1016/j.kint.2015.12.029.
86.Agarwal, I., Ide, N., Ix, J. H., Kestenbaum, B.,
Lanske, B., et al. (2014) Fibroblast growth factor-23 and cardiac
structure and function, J. Am. Heart Assoc., 3, e000584,
doi: 10.1161/JAHA.113.000584.
87.Hu, M. C., Shi, M., Cho, H. J., Adams-Huet, B.,
Paek, J., et al. (2015) Klotho and phosphate are modulators of
pathologic uremic cardiac remodeling, J. Am. Soc. Nephrol.,
26, 1290-1302, doi: 10.1681/ASN.2014050465.
88.Jia, Z., Wei, L., Liu, Q., Zhu, Z., Yang, J.,
Yang, X., Gan, S., Chen, W., and Zhang, L. (2015) Impact of
transfection with recombinant adenovirus vector-mediated Klotho gene on
myocardial remodeling in a rat model of heart failure, Zhonghua Xin
Xue Guan Bing Za Zhi, 43, 219-226.
89.Tang, G., Shen, Y., Gao, P., Song, S. S., and Si,
L. Y. (2018) Klotho attenuates isoproterenol-induced hypertrophic
response in H9C2 cells by activating
Na+/K+-ATPase and inhibiting the reverse mode of
Na+/Ca2+-exchanger, In vitro Cell Dev. Biol.
Animal., 54, 250-256, doi: 10.1007/s11626-017-0215-5.
90.Moskalev, A. V., Rudoi, A. S., Apchel, A. V.,
Zueva, V. O., and Kazymova, O. E. (2016) Features of the biology of
transforming growth factor β and immunopathology, Bull. Russ.
Military Med. Acad., 54, 206-216.
91.Zaslavskaya, E. L., Morozov, A. N., Ionin, V. A.,
Ma, I., Nifontov, S. E., et al. (2018) The role of transforming growth
factor beta1 and galectin-3 in the formation of left atrial fibrosis in
patients with paroxysmal atrial fibrillation and metabolic syndrome,
Russ. J. Cardiol., 154, 60-66.
92.Korzhenevskaya, K. V., Gavrisheva, N. A., Panov,
A. V., Ses, T. P., Alugishvili, M. S., Kozlov, V. V. (2010)
Transforming growth factor-β1 in different clinical course of
coronary heart disease after coronary artery bypass grafting, Med.
Immunol., 12, 521-528.
93.Inoue, N., Maeda, R., Kavakami, H., Shokawa, T.,
Yamamoto, H., Ito, C., and Sasaki, H. (2009) Aortic pulse wave velocity
predicts cardiovascular mortality in middle-aged and elderly Japanese
men, Circ. J., 73, 549-553.
94.Ding, J., Tang, Q., Luo, B., Zhang, L., Lin, L.,
et al. (2019) Klotho inhibits angiotensin II-induced cardiac
hypertrophy, fibrosis, and dysfunction in mice through suppression of
transforming growth factor-β1 signaling pathway, Eur. J.
Pharmacol., 859, 172549, doi:
10.1016/j.ejphar.2019.172549.
95.Ucar, A., Gupta, S. K., Fiedler, J., Erikci, E.,
Kardasinski, M., et al. (2012) The miRNA-212/132 family regulates both
cardiac hypertrophy and cardiomyocyte autophagy, Nat. Commun.,
3, 1078.
96.Chen, Q., Zhang, H., Liu, Y., Adams, S., Eilken,
H., et al. (2016) Endothelial cells are progenitors of cardiac
pericytes and vascular smooth muscle cells, Nat. Commun.,
7, 12422, doi: 10.1038/ncomms12422.
97.Arking, D. E., Becker, D. M., Yanek, L. R.,
Fallin, D., Judge, D. P., et al. (2003) KLOTHO allele status and the
risk of early-onset occult coronary artery disease, Am. J. Hum.
Genet., 72, 1154-1161, doi: 10.1086/375035.
98.Majumdar, V., Nagaraja, D., and Christopher, R.
(2010) Association of the functional KL-VS variant of Klotho gene with
early-onset ischemic stroke, Biochem. Biophys. Res. Commun.,
403, 412-416, doi: 10.1016/j.bbrc.2010.11.045.
99.Oguro, R., Kamide, K., Kokubo, Y., Shimaoka, I.,
Congrains, A., et al. (2010) Association of carotid atherosclerosis
with genetic polymorphisms of the klotho gene in patients with
hypertension, Geriatr. Gerontol. Int., 10, 311-318, doi:
10.1111/j.1447-0594.2010.00612.x.
100.Imamura, A., Okumura, K., Ogawa, Y., Murakami,
R., Torigoe, M., Numaguchi, Y., and Murohara, T. (2006) Klotho gene
polymorphism may be a genetic risk factor for atherosclerotic coronary
artery disease but not for vasospastic angina in Japanese, Clin.
Chim. Acta, 371, 66-70, doi: 10.1016/j.cca.2006.02.021.
101.Majumdar, V., Jose, D., and Christopher, R.
(2011) Influence of Klotho genotypes on plasma NO(x) levels in South
Indian population, Thromb. Res., 128, 251-255, doi:
10.1016/j.thromres.2011.04.002.
102.Pavlatou, M. G., Remaley, A. T., and Gold, P.
W. (2016) Klotho: a humeral mediator in CSF and plasma that influences
longevity and susceptibility to multiple complex disorders, including
depression, Transl. Psychiatry, 6, e876, doi:
10.1038/tp.2016.135.
103.Qiu, X., Guo, Q., Xiong, W., Yang, X., and
Tang, Y. Q. (2016) Therapeutic effect of astragaloside-IV on
bradycardia is involved in up-regulating klotho expression, Life
Sci., 144, 94-102, doi: 10.1016/j.lfs.2015.11.021.
104.Narumiya, H., Sasaki, S., Kuwahara, N., Irie,
H., Kusaba, T., et al. (2004) HMG-CoA reductase inhibitors up-regulate
anti-aging klotho mRNA via RhoA inactivation in IMCD3 cells,
Cardiovasc. Res., 64, 331-336, doi:
10.1016/j.cardiores.2004.07.011.
105.Yoon, H. E., Ghee, J. Y., Piao, S., Song, J.
H., Han, D. H., et al. (2011) Angiotensin II blockade upregulates the
expression of Klotho, the anti-ageing gene, in an experimental model of
chronic cyclosporine nephropathy, Nephrol. Dial. Transplant.,
26, 800-813, doi: 10.1093/ndt/gfq537.
106.Cheng, L., Zhang, L., Yang, J., and Hao, L.
(2017) Activation of peroxisome proliferator-activated receptor γ
inhibits vascular calcification by upregulating Klotho, Exp. Ther.
Med., 13, 467-474, doi: 10.3892/etm.2016.3996.
107.Haussler, M. R., Whitfield, G. K., Haussler, C.
A., Sabir, M. S., Khan, Z., Sandoval, R., and Jurutka, P. W. (2016)
1,25-Dihydroxyvitamin D and Klotho: a tale of two renal hormones coming
of age, Vitam. Horm., 100, 165-230, doi:
10.1016/bs.vh.2015.11.005.
108.Lorenzi, O., Veyrat-Durebex, C., Wollheim, C.
B., Villemin, P., Rohner-Jeanrenaud, F., Zanchi, A., and Vischer, U. M.
(2010) Evidence against a direct role of klotho in insulin resistance,
Pflugers Arch., 459, 465-473, doi:
10.1007/s00424-009-0735-2.
109.Bilan, V. P., Salah, E. M., Bastacky, S.,
Jones, H. B., Mayers, R. M., et al. (2011) Diabetic nephropathy and
long-term treatment effects of rosiglitazone and enalapril in obese
ZSF1 rats, J. Endocrinol., 210, 293-308, doi:
10.1530/JOE-11-0122.
110.Dagenais, G. R., Gerstein, H. C., Holman, R.,
Budaj, A., Escalante, A., et al. (2008) Effects of ramipril and
rosiglitazone on cardiovascular and renal outcomes in people with
impaired glucose tolerance or impaired fasting glucose: results of the
diabetes reduction assessment with ramipril and rosiglitazone
medication (DREAM) trial, Diabetes Care, 31, 1007-1014,
doi: 10.2337/dc07-1868.