REVIEW: At the Crossroads of Bioenergetics and Antibiotic Discovery
K. Lewisa
Antimicrobial Discovery Center, Department of Biology, Northeastern University, Boston, MA 02115 USA
Received July 10, 2020; Revised August 19, 2020; Accepted August 19, 2020
Dr. Vladimir Skulachev was my mentor, and his pioneering work in the field of bioenergetics inspired the discoveries described in this review, written in the form of a personal account of events. Examining basic mechanisms of chemiosmotic coupling unexpectedly led us to transenvelope multidrug resistance pumps (MDR pumps) that severely limit development of novel antibiotics. One of the major advances of Skulachev and his group was the discovery of the mitochondrial membrane potential with the use of permeant cations such as TPP+, which served as electric probes. We describe our finding of their natural counterparts in plants, where they act as antimicrobials. The most challenging problems in antimicrobial drug discovery are antibiotic tolerance of chronic infections caused by dormant persister cells; antibiotic resistance, responsible for the current antimicrobial resistance crisis (AMR); and finding novel compounds acting against Gram-negative bacteria, protected by their powerful multidrug resistance pumps. Our study of persisters shows that these are rare cells formed by stochastic fluctuation in expression of Krebs cycle enzymes, leading to a drop in ATP, target shutdown, and antibiotic tolerance. Searching for compounds that can corrupt targets in the absence of ATP, we identified acyldepsipeptide (ADEP) that activates the ClpP protease, forcing cells to self-digest. Growing previously uncultured bacteria led us to teixobactin, a novel cell wall acting antibiotic. Teixobactin avoids efflux by targeting lipid II and lipid III, precursors of peptidoglycan and wall teichoic acid, located on the surface. The targets are immutable, and teixobactin is the first antibiotic with no detectable resistance. Our search for compounds acting against Gram-negative bacteria led to the discovery of darobactins, which also hit a surface target, the essential chaperone BamA.
KEY WORDS: bioenergetics, persisters, antibiotics, acyldepsipeptide, teixobactin, darobactinDOI: 10.1134/S0006297920120019
Abbreviations: ADEP, acyldepsipeptide; AMR, antimicrobial resistance crisis; MDR pumps, multidrug resistance pumps; MFS, Major Facilitator superfamily of transporters; pmf, proton motive force; RND, resistance-nodulation-cell division family of transporters; WHO, World Health Organization.
Dedicated to Vladimir P. Skulachev, my friend and mentor
INTRODUCTION
I had the good fortune to learn science in the Skulachev lab. As an undergrad, I was fascinated by the principle puzzle Skulachev and his group were working on – the basic mechanism of oxidative phosphorylation. The mechanism of ATP synthesis in glycolysis, “substrate level phosphorylation”, was well understood – breakdown of glucose creates high-energy intermediates such as phosphoenol pyruvate, and the phosphate is transferred to ADP to make ATP. The chemistry is elegant, but not surprising. It was assumed that oxidative phosphorylation works the same way – oxidation of pyruvate in the Krebs cycle donates hydrogen to the electron transport chain in mitochondria, and oxidation drives formation of high-energy phosphorylated intermediates, such as NADH dehydrogenase~Pi. However, attempts to isolate such intermediates failed. Moreover, while it was possible to measure increase in ATP in response to oxidation of a substrate in isolated mitochondria or even in submitochondrial membrane particles, this was not observed with the isolated protein complexes. The moment the membrane was gone, so was oxidative phosphorylation. This riddle led Peter Mitchell to propose a radical theory – mitochondria are batteries, and their membrane is charged by the electron transport chain that pumps protons across it. The protons flow back through the ATP synthase, discharging the battery, and ATP is made [1-3]. The central tenet of the hypothesis was that the mitochondrial membrane is charged, which seemed impossible to measure, since the organelle is smaller than a microelectrode. An important breakthrough came from what are now known as “Skulachev cations” – compounds such as tetraphenyl phosphonium that can penetrate into mitochondria and act as electrical probes. They will accumulate, if the membrane is charged. Measuring a change in the concentration of these cations outside of mitochondria allowed Skulachev and his team to detect a membrane potential, and calculate it using the Nernst equation [4]. This important discovery offered proof for the chemiosmotic theory. Another important observation published in Nature as well described the action of uncouplers of oxidative phosphorylation [5]. These compounds from different chemical classes uncouple respiration from ATP synthesis. Skulachev and his team found that the uncoupling activity of compounds such as CCCP correlated with their activity as protonophores, shuttling protons across the membrane and collapsing the proton motive force (pmf). This provided additional validation to the chemiosmotic theory. By the time I joined Skulachev’s lab, these discoveries had already been published, but the spirit of going after big questions and finding solutions to seemingly impossible problems was very much alive.
FROM LOCALIZED COUPLING TO MULTIDRUG RESISTANCE PUMPS (MDR
PUMPS)
In 1987, I moved with my family to the US, and had a short postdoctoral stint in the laboratory of Julius Adler, who discovered the principal mechanism of bacterial chemotaxis. Upon listening patiently to my various ideas, Julius had this to say: “you need a generalized approach to discover new molecules” – meaning specific hypotheses will not lead to new chemical matter. Armed with that wisdom, several months later I got my first faculty job as an Assistant Professor at the Applied Biology Department at MIT. Three days after I joined, the Department was disbanded, and like other untenured Faculty I was reassigned, in my case to Biology and had a couple of years to serve out my contract. Those events had little effect on me – I had my own lab, startup funding, and freedom to choose what to do. But there was a problem.
Bioenergetics was becoming a mature science, shifting towards protein structural studies. While I enjoy reading about protein structure/function, my interests lie in early discovery. Thinking about starting something new, I wondered about a lingering controversy at the heart of bioenergetics – delocalized vs. localized coupling. While Mitchell proposed that protons cross the membrane, Williams suggested that they could also move laterally, directly from a respiratory chain component to the ATP synthase [6]. There was a minor body of unconvincing literature in support of that proposition. In discussing the problem, Skulachev famously posed this question: “do you build a bridge across the river or along it?”. Localized coupling would have one major advantage though, it would function in organelles or cells with a damaged membrane, and could operate as a safety option. If localized coupling existed, I figured that the right place to look for it would be in bacteria, with their unmatched ability to adapt to changing conditions. I thought of a simple experiment – add CCCP, and look for an adaptive response. Adding CCCP at a subinhibitory concentration to E. coli was of course tolerated, and then a stepwise increase produced cells that continued to grow after the uncoupler surpassed what was supposed to be its inhibitory concentration. Since cells were adapting to a synthetic compound they never encountered in nature, we thought they must be recognizing not CCCP, but the consequences of its action – a drop in pmf, and turning on some protective mechanism, such as localized coupling. Selecting cells with a genomic library cloned in an expression vector for survival in CCCP would then lead us to the mechanism. I told my talented postdoc, Olga Lomovskaya (currently VP of Biology at Qpex), that whatever we find, it must be interesting, but it will not be a CCCP transporter. Indeed, attempts to extrude CCCP across the membrane would be futile, this would only speed up its shuttling of protons (Fig. 1). Olga selected cells with increased resistance to CCCP, sequenced the plasmid insert, and found that it coded for a transporter of the Major Facilitator superfamily [7].
Fig. 1. Discovery of a transenvelope pump. A protonophor, CCCP, dissipates pmf by shuttling protons across the cytoplasmic membrane. In Gram-negative bacteria, MDR pumps extrude chemically unrelated amphipathic compounds such as CCCP across the outer membrane permeability barrier.
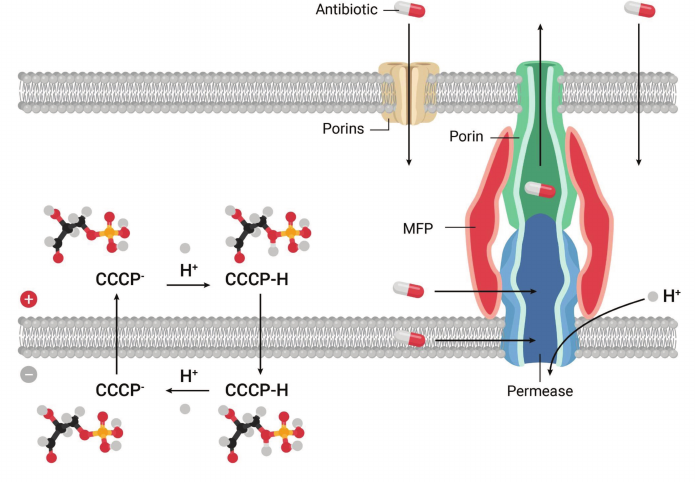
This was a reminder that making accurate predictions in biology is highly problematic. There was another gene in this operon, which showed homology to a “membrane fusion” protein participating in the transenvelope transporter of hemolysin from E. coli. This provided a clue, and we proposed that EmrAB, as we named it, is a bacterial multidrug pump that is a proton antiporter, and effluxes compounds all the way across the outer membrane of the cell, which serves as a barrier for hydrophobic molecules (Fig. 1). Another unexpected finding was that of the mechanism that upregulated the pump in response to CCCP. This turned out to be an EmrAB repressor, acting as a CCCP-binding multidrug sensor [8].
This line of experiments suggested to me that local coupling does not exist, but in the process, we did discover something fairly interesting. Gobind Khorana, now my colleague at MIT, presented our EmrAB paper to the PNAS. We proposed that bacteria protect themselves from toxic compounds by a combination of the outer membrane barrier and MDR pumps that extrude toxins across it. EmrAB was the first example of a transenvelope MDR, but it has a rather narrow repertoire. After our paper came out, Hiroshi Nikaido published another transenvelope pump, AcrAB [9]. This one has a very broad spectrum, and plays an important role in the efflux of antibiotics. MDRs were setting us on a path of drug discovery.
Are there preferred substrates of the MDR pumps that could point to their natural function? For bacteria in general, and especially for Gram-positive species that lack an outer membrane barrier, the best substrates are hydrophobic cations [10], the Skulachev cations we discussed. This makes sense, since the membrane potential can drive accumulation of such a compound 1,000-fold, making them very dangerous to the cell. However, all of the published MDR substrates for Gram positive species such as B. subtilis [11] or S. aureus were synthetic – ethidium bromide, benzalkonium chloride, chlorhexidine etc. This suggested that Nature produced these compounds as well, but how would one find them? The fact that bacteria protect themselves well from such compounds with MDRs meant that these are not effective antibiotics. With that in mind, I armed myself with the thick volume of Merck catalog and leafed through it, looking for compounds that would be hydrophobic cations, naturally produced, without known antimicrobial activity. This led me, quite rapidly, to Berberine alkaloids, made by a variety of plants. Berberine looks very much like ethidium bromide – it is a quaternary ammonium alkaloid that intercalates into DNA. We bought it from Sigma and showed that berberine has an excellent activity against Gram-positive bacteria if you knock out their MDRs [10]. Berberine accumulates in the cell driven by the membrane potential [12], damages the membrane and intercalates into DNA. If not for the MDRs, the plant came up with a perfect antimicrobial – the membrane and DNA are immutable targets. But why would the plant continue making antimicrobials bacteria are so well protected from? More importantly, what should the plant ideally do in order to empower berberine? Let me pose here for a moment to describe where this type of a question comes from. A Russian engineer Genrich Altshuller decided to formalize the process of invention by producing a set of rules, and described this in a thin book that I came across when I was a student. As I see it, Altshuller’s main goal was to disable common sense that prevents us from asking the right question (“do not go in that direction since it does not make sense”). His most useful rule was to formulate what result you would like to have ideally without worrying about technical feasibility. Once you answered that question, then you can figure out how to make it work. As Altshuller put it: “if your ideal result seems absurd, you are on to something”. Altshuller has an international following – his TRIZ method is widely used (there is a good entry in Wikipedia), but not in Biology. I reformulate his ideal result to ask the question from the perspective of a living organism, and in the particular case we are considering, what should the plant ideally do in order to empower berberine? Well, it should disable the MDR pump, thus make an MDR inhibitor. We set out to find this missing inhibitor by fractionating the barberry plant and testing for activity in the presence of subinhibitory berberine. This led to the discovery of methoxyhydnocarpin (MHC), a potent MDR inhibitor [13]. Berberine and MHC formed a highly effective synergistic couple. Interestingly, berberine has been used in both Chinese and Native American traditional medicine, it is sold over the counter, and taken orally it is likely to modulate the microbiome, hopefully to our advantage. Limited penetration across epithelial cells of the GI tract makes it safe, but intercalation into DNA is not a great property for a systemic therapeutic to have. A different hydrophobic cation did become a drug – Dr. Skulachev had a neat idea of using TPP+ to address nature’s best antioxidant, plastoquinone, to mitochondra, the main source of ROS in humans. Plastoquinone functions in the electron transport chain of chloroplasts, where O2 is produced, and can reduce ROS. Since plastoquinone can be reduced by the electron transport chain of both chloroplasts and mitochondria, it is a renewable antioxidant. The plastoquinone-TPP+ chimera, named SkQ1, accumulates in mitochondria, driven by the membrane potential, and decreases ROS by acting as an antioxidant, and by mildly reducing the pmf [14]. It is used as an antioxidant to treat dry eye disease, and may have anti-aging properties as well.
While it was fairly easy to find inhibitors of MF MDRs of Gram-positive bacteria, the main clinical need is to target the RND family AcrAB-like MDRs of Gram-negative species. This turned out to be challenging, and after years of efforts from a number of groups, we do not have an MDR inhibitor that would cover pumps from a number of different species and is non-toxic. If we wanted to make a difference, we would have to discover antibiotics that bypass the MDRs.
THE QUEST FOR NEW ANTIBIOTICS
The study of MDRs brought me into contact with scientists from The Big Pharma – Pfizer, Merck, Wyeth, Novartis, and Astra Zeneca. Most of their effort went into making analogs of existing compounds to act against drug-resistant bacteria or to improve pharmacological properties. There was also an emerging consensus that novel compounds would be needed to properly address the problem of resistance. I wondered, when was the last time a professor working in academia discovered a useful antibiotic? That happened to be in 1944, when Selman Waksman discovered streptomycin [15]. The last novel antibiotic introduced into the clinic was the narrow-spectrum daptomycin (acting only against Gram-positive bacteria), discovered in 1978 at Eli Lilly (Fig. 2). All of this was not particularly encouraging, but also seemed like an exciting challenge (see [16] for a detailed review on antibiotic discovery).
Fig. 2. The timeline of antibiotic discovery. The year of discovery of the major classes of antibiotics (lower panel), and the year they were introduced into the clinic (upper panel) are shown. (*) Denotes synthetic compounds. Blue, narrow-spectrum; red, broad-spectrum.
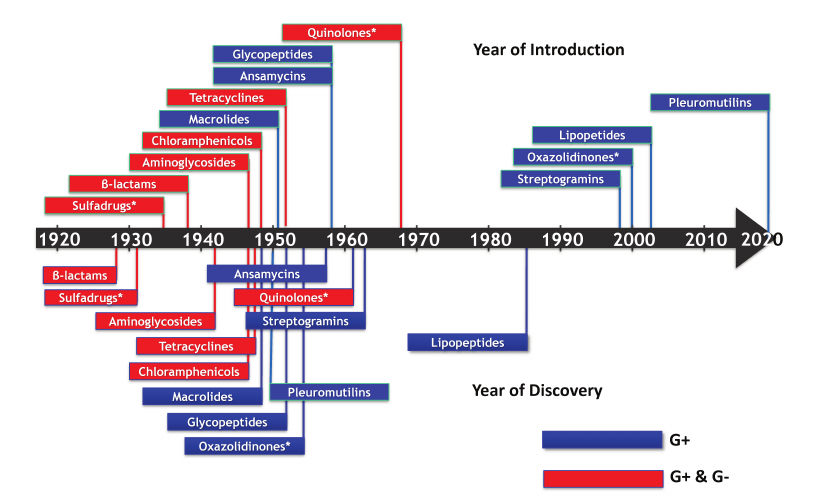
Selman Waksman discovered streptomycin by introducing a simple systematic screen of soil actinomycetes on a Petri dish overlaid with a target pathogen, looking for zones of inhibition. After the discovery of streptomycin that became the first drug to treat tuberculosis, additional large-scale screens by the industry led to the discovery of other aminoglycosides, as well as tetracyclines, chloramphenicoles, macrolides, and other compounds. The golden era of antibiotic discovery was launched, but was short-lived. Discovery of novel compounds largely ceased by 1960. Apart from the accidentally discovered synthetic fluoroquinolones, no broad-spectrum antibiotic capable of acting against Gram-negative species has been discovered since the 1960s. Not surprisingly, pathogens kept on acquiring resistance, and we find ourselves in the midst of the antimicrobial resistance crisis (AMR). Bacteria seem to have used all logical possibilities to develop resistance – destruction of the antibiotic; decreased penetration; efflux; target modification; target switching; antibiotic sequestration [17]. Of “critical concern” according to the WHO are Gram-negative bacteria such as E. coli, K. pneumoniae, P. aeruginosa and A. baumannii [18]. Mortality from the carbapenemase-producing K. pneumoniae (KPC), for example, is 40% [19, 20]. We have a pretty good idea why the golden era ended – this was a result of overmining. Broad-spectrum antibiotics are highly valuable to Actinomycetes, they spread readily among different species by horizontal transmission, are abundant, and easy to find. And, all the major classes were discovered by 1950.
UNCULTURED BACTERIA AND TEIXOBACTIN
The microbial world is bigger than actinomycetes and probably harbors novel useful antibiotics. One obvious source to look at is uncultured bacteria that make up 99% of all microbial diversity. Placing a droplet of bacteria from an environmental sample on a slide to count them, and plating a similar droplet on a Petri dish shows that only 1% of cells produce colonies. The result of this simple experiment is known as “The Great Plate Count Anomaly”, and has been the oldest unsolved problem in microbiology, dating back to the 19th century [21]. There have been numerous attempts over the last 100 years to improve recovery by tinkering with growth conditions, but this did not work. In Altshuller’s terms, how do you ideally grow uncultured bacteria? Ideally, you grow them where you know they do grow, in their natural environment. The challenge is then to come up with a gadget that would allow you to grow bacteria in pure culture in an environment that has as much as 109 cells/ml.
We reasoned that the design can be borrowed from a conventional dialysis bag used to desalt proteins. In collaboration with my colleague Slava Epstein we designed a “diffusion chamber”, where bacteria are taken from the environment such as marine sediment or soil, diluted, mixed with agar, and sandwiched between 2 semi-permeable membranes of the device [22]. The chamber is then returned to the environmental sample the cells came from. Nutrients and growth factors diffuse through the chamber, tricking bacteria into perceiving this as their natural environment, and they form colonies. Uncultured bacteria started to grow in the lab.
Recovery by this approach is around 50%. Very conveniently, once colonies formed in the chamber, with high probability they would grow on regular media, we call this “domestication”. Apparently, the limiting step in having most species grow in vitro is the formation of a sizable cell population. In search of a mechanism of “uncultivability” we considered that growth factors might be donated to bacteria by their neighbors. To test this idea, we plated a heavy inoculum from the marine sediment onto a Petri dish, and considered that some of the colonies might belong to uncultured species that happened to be in the vicinity of a cultivable microorganism. Reinoculating neighboring colonies together and separately showed that this is indeed the case. Fractionating the supernatant of the cultivable helper and testing for growth induction of the uncultured bacteria led us to the growth factors. These turned out to be siderophores [23].
In aerobic environments, iron is present in the form of insoluble FeIII. Bacteria release siderophores that chelate FeIII and bring it into the cell to be reduced to FeII that is used to build the Fe-S clusters and the hemes of the respiratory chain components. Bioenergetics was again reminding us of its presence. Uncultured bacteria from taxonomically unrelated groups have lost their ability to synthesize siderophores, and steal them from their neighbors. This may save resources, but comes at a loss of liberty – uncultured bacteria can not settle new territory. Dependence on siderophores accounts for about 10% of uncultured bacteria in the environment. It seemed that finding additional growth factors will allow us to close the gap in the great plate count anomaly and solve the problem of uncultured bacteria. This however did not happen. The next growth factor we discovered was hemin, accounting for <1% of uncultured species, and that was it.
Using a similar approach, we investigated uncultured bacteria of the human microbiome, they make up about 30% of the species in that environment. We found helper-dependent pairs of colonies, but were unable to isolate the growth factor. A common helper turned out to be E. coli, so we took advantage of an ordered knockout library of E. coli strains, screened it, and found mutants that did not act as helpers. These happened to carry deletions in the menaquinone biosynthetic pathway [24]. This was completely unexpected – menaquinone is a highly hydrophobic compound, and an integral membrane component of the anaerobic respiratory chain, shuttling hydrogen between dehydrogenases and cytochromes. Adding menaquinone to the plate recapitulated the action of helper bacteria, allowing uncultured microorganisms to grow. This explained why we failed to isolate the growth factor – the menaquinone concentration in solution was very low. As in the case of siderophores, bacteria that stringently depend on the presence of a respiratory chain borrow its essential component from their neighbors. Similarly to the external environment, about 10% of gut uncultured bacteria are dependent on quinones, and we found only 1 additional, minor growth factor. This turned out to be gamma-amino butyric acid, GABA, a major neurotransmitter [25]. The uncultured bacterium which we named Evtepia gabavorous only consumes GABA and no other nutrient, quite unusual if not unprecedented case in microbiology. GABA producers that we identified are of practical interest. My talented graduate student Phil Strandwitz who made this discovery founded Holobiome, a Biotech company, with the goal of putting GABA producers in a pill to treat an assortment of maladies such as anxiety and depression (there is a recent Science feature on Holobiome https://www.sciencemag.org/news/2020/05/meet-psychobiome-gut-bacteria-may-alter-how-you-think-feel-and-act). Sometimes science takes us on unanticipated detours.
While it has been interesting to find growth factors, why the remaining 90% of species are uncultured is an intriguing puzzle waiting to be solved.
Apart from the basic science puzzle, we appreciated the potential of uncultured bacteria for producing secondary metabolites, and Dr. Epstein and I started NovoBiotic to properly exploit them. A large-scale screen produced a number of new antimicrobials, but most of these failed in development due to toxicity, poor stability, and other issues – a common attrition problem with drug discovery. The 25th compound, Novo25, showed excellent activity against Gram-positive bacteria, but failed an important test – there was no resistance development. No resistant mutants usually means a non-specific compound, such as detergents that bacteria like to make. However, Novo25 showed no cytotoxicity against mammalian cells [26]. This was clearly not the behavior of a detergent. We then performed detailed evolutionary experiments with S. aureus, starting with subinhibitory doses of Novo25 and increasing the dose daily, but again, got no mutants. All currently approved antibiotics produce resistant mutants in this experiment. The compound was clearly unusual. Since we could not obtain resistant mutants, this suggested that Novo25 does not target proteins. Indeed, a protein will always mutate to avoid antibiotic binding. We bet on lipid II, precursor of peptidoglycan, and showed that adding a purified compound to the medium protects from Novo25. Named teixobactin (teixos-wall), this compound is a member of a novel class of cell-wall acting antibiotics (Fig. 3). It binds to the PiPi-sugar of lipid II, while the nature of the sugar is not important. This allows teixobactin to bind to the similar lipid III, precursor of wall teichoic acid. The target of teixobactin, PiPi-sugar seems unsuitable – indeed, how would it provide selectivity of action, why would the compound not bind to nucleoside phosphates such as ADP? The answer lies in the unexpected complexity of the mechanism of binding recently described by Markus Weingarth and colleagues in a study with a synthetic teixobactin analog [27]. Teixobactin first lands on the surface of the cell, with two hydrophobic isoleucine residues anchoring it to the membrane (Fig. 3). The ring headgroup weakly binds to PiPi-sugar of lipid II, but then teixobactin molecules bound to their target interact with each other to form a vast β-sheet.
Fig. 3. The mechanism of action of teixobactin. a) Teixobactin binds to lipid II, precursor of peptidoglycan, and lipid III, precursor of wall teichoic acid. b) Teixobactin binds to PiPi-sugar of lipid II, and a vast β-sheet of anti-parallel molecules forms, producing a supramolecular structure that explains high selectivity and potency of the antibiotic. c) Eleftheria terrae, a Gram-negative bacterium, produces teixobactin and exports it across the outer membrane with a transenvelope pump. This large teixobactin molecule cannot diffuse back through the outer membrane, which provides protection against this compound. The target Gram-positive bacteria do not have an outer membrane and are exposed to teixobactin.
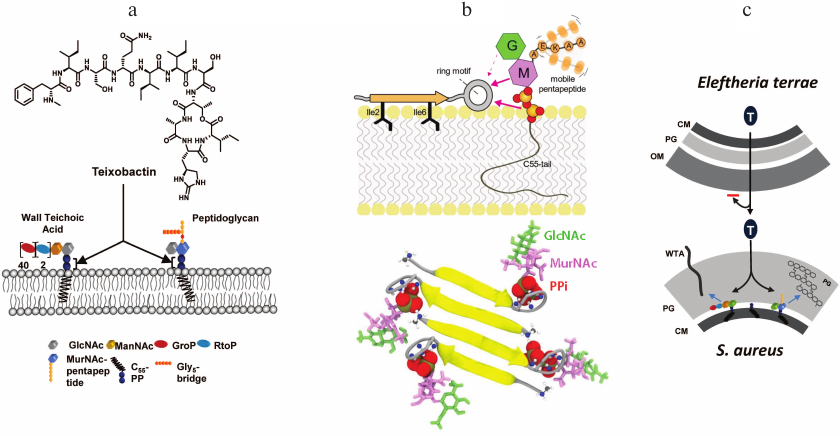
Selectivity of action and potency come from the creation of this supramolecular structure. The L- and D-amino acids are strategically positioned in teixobactin to create anti-parallel strands of a β-sheet in which two adjacent molecules are shifted, keeping the headgroup free to attack the target.
Importantly, the binding sites are located on the surface of the membrane, so teixobactin is not subject to efflux. Binding to two immutable targets on the cell surface largely explains the remarkable lack of resistance development to teixobactin. But there is always a possibility of a resistant mechanism traveling on a plasmid, often originating from the producer organism. However, the producer of teixobactin is a Gram-negative bacterium, Eleftheria terrae, and it protects itself from teixobactin by exporting it across its outer membrane. The pump is a proton antiporter, homologous to the AcrAB of E. coli. The target organisms are Gram-positive bacteria and cannot possibly borrow an outer membrane from the producer. Antibiotics can of course be enzymatically destroyed, i.e., β-lactamases cleaving penicillin, but these enzymes are only known for commonly found antibiotics. Teixobactin is rare, and is the first example of a compound evolved to be protected from resistance development.
When we published the paper, it became the most discussed study of the year (Feature, Science http://news.sciencemag.org/scientific-community/2015/12/which-studies-got-most-media-buzz-2015). While we were not prepared for this level of attention, in retrospect, it is understandable – the ability of bacteria to develop resistance to antibiotics has been a given, and realizing that the old paradigm may be wrong creates considerable hope for finding effective ways to resolve the AMR. Teixobactin is now in late-stage IND-enabling studies, slated to enter human clinical trials in a year.
BIOFILMS, DORMANT CELLS, AND ATP
The field of bacterial MDR pumps we helped to start was maturing, and like the chemiosmotic mechanism of coupling before it, was entering the stage of protein crystallography and detailed structure/function studies. It was time to move on.
Looking for a good problem to work on, I decided to investigate the puzzling recalcitrance of chronic infections associated with biofilms to antibiotic therapy. The basic observation is straightforward – bacteria form a biofilm, a mass of cells covered by exopolymers, and once they do this, the infection is very difficult to treat with antibiotics. Cells isolated from biofilms however do not grow in the presence of these antibiotics, meaning that they are susceptible. This makes no sense, a beautiful paradox.
At that time, a talented postdoc Alexei Brooun (presently at Merck) joined my lab, and was willing to take on this problem. He started with a simple descriptive experiment, monitoring time-dependent killing of a P. aeruginosa biofilm by ofloxacin [28]. What he discovered was striking – the majority of cells in a biofilm died quite rapidly, but there was a small surviving subpopulation that seemed impervious to the antibiotic (Fig. 4) [28, 29]. Once regrown, these cells showed no resistance, so they were not resistant mutants. I found this hard to believe – the principle mechanism of biofilm recalcitrance was in clear view, the result of a simple experiment that must have been performed before. We took a thick stack of biofilm papers and leafed through it, looking for a time-killing experiment. Sure enough, we found 4 such papers, with a subpopulation of surviving cells. For reasons I do not understand, this surviving population was overlooked.
Fig. 4. Persister cells. A bacterial population resistant to an antibiotic will continue to grow in its presence. If a strain is not resistant, the bulk of the population dies, leaving a small fraction of antibiotic-tolerant persister cells.
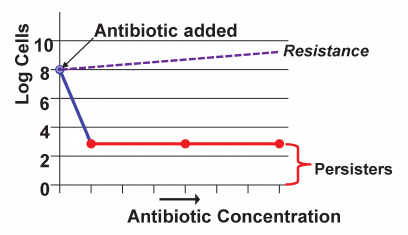
I realized that we rediscovered persisters, a little-known phenomenon described by the Irish microbiologist Joseph Bigger in 1944 [30]. Bigger was testing the recently introduced penicillin against S. aureus, and found that it did not sterilize the population. The remaining cells were not resistant mutants, and could be regrown to form a new population that produced a new fraction of rare survivors which he aptly named “persisters”. After Bigger published his discovery in Lancet, the paper was promptly forgotten. Forty years later, Harris Moyed resurrected persisters, and working with E. coli set up a simple selection to find the underlying mechanism. Applying ampicillin to a growing culture, he collected surviving cells, and repeated the procedure, selecting for mutants that would make more persisters. He was indeed able to isolate such “high persister mutants” – hip, and mapped them to a hipBA locus [31]. A gain of function mutation in HipA (hipA7) produced cells that made 1,000 times more persisters. However, knocking out the hipBA locus had no effect on persister formation. This line of inquiry was hitting a dead-end, and once again, persisters were forgotten.
The presence of persisters explained the biofilm phenotype, and with it, the most important property of chronic infections in general. A common feature of chronic infections is the ability of a pathogen to hide from the immune system. Biofilm exopolymers protect cells from the large components of the immune system, M. tuberculosis hide in macrophages, and H. pylori causes infection in the stomach, largely devoid of an immune response. Persisters survive antibiotic treatment, and once its concentration drops, restore the population, fueling relapsing chronic infections. That was our theory, but testing it proved to be very challenging. Persisters form a small and temporary subpopulation, not clear how to perform an experiment in vivo linking them to drug tolerance. A clue to solving this problem came from the old Moyed experiment described above, where a population is treated with a high concentration of antibiotic, then allowed to regrow, and after several cycles, there is an enrichment in hip mutants. But this is exactly what happens when people are treated with antibiotics. In a way, millions of people participated in this experiment, and all we needed to do was look at its result. We set out to find hip mutants among clinical isolates of patients with chronic infections. These we could readily detect – for example, almost half of the isolates of P. aeruginosa from patients with cystic fibrosis, where the infection lasts for decades, were hip mutants [32]. Importantly, many of the hip mutants carried no resistance mutations. This means that survival in the presence of antibiotics favors persisters, linking them causally to recalcitrance of a chronic infection. We went on to show that hip mutants are selected for in the course of treating tuberculosis patients [33] and in a relapsing urinary tract infection with E. coli [34]. In the case of E. coli, a good number of hip isolates carried the same gain of function mutations in the HipA toxin that Moyed discovered many years ago. While we still do not know what the natural function of the wild type HipA may be, its gain-of-function alleles certainly play a role in clinical tolerance of antibiotics. HipBA is a toxin/antitoxin module; HipA is a protein kinase [35] that phosphorylates glu-tRNA synthase, inhibiting translation [36]. We also determined the mechanism of the hipA7 gain-of-function mutation – HipA forms an inactive dimer, and the mutation loosens the interaction among subunits, allowing ATP to reach the active site of the kinase. With this mechanistic understanding, we gave the phenomenon a name – “heritable multidrug tolerance”, by analogy with heritable resistance [34].
How persisters that are not formed through HipA7 survive antibiotics remained a mystery that we set out to solve. There are many TA modules of different classes scattered throughout the chromosomes of bacteria, but their function is unclear. We thought that if we could identify a TA module in E. coli that was induced under particular conditions, then it will become the major component responsible for persister formation. Looking at the upstream sequences of known TAs, the TisAB module caught our attention, since the promoter region contained a Lex-box. The Lex-box is an operator region for binding the LexA repressor, which is the global regulator of the SOS response. When DNA is damaged, LexA is cleaved, activating expression of DNA repair enzymes. Toby Dorr, my talented grad student (now on the faculty of Cornell) took on this project and found that DNA damage by fluoroquinolone antibiotics turns on expression of the TisB toxin, and with it, production of persister cells [37]. TisB is an unusual toxin, it is an endogenous antimicrobial peptide. This seems like an oxymoron; the function of antimicrobial peptides is to be exported and kill other bacteria. These compounds come in a variety of classes, but the common theme is a short hydrophobic cationic peptide that forms an ion channel in the membrane, collapsing the pmf and ultimately killing the cell. In collaboration with Sergei Bezrukov, a leading expert on ion channels who works at the NIH, we showed that incorporating TisB into an artificial “black membrane” produces typical voltage-gated ion channels [38]. Apparently, E. coli uses two very different defenses to avoid killing by DNA damaging agents. The majority of cells express repair enzymes and try to survive. A small fraction of them express TisB, which decreases the membrane potential, leading to a drop in ATP and shutdown of the major biosynthetic pathways, putting cells into dormancy. The major biosynthetic processes are also the main targets of bactericidal antibiotics – peptidoglycan synthesis/penicillin binding proteins (β-lactams), protein synthesis/ribosome (aminoglycosides), and DNA synthesis/DNA gyrase. Bactericidal antibiotics kill not by inhibiting these targets, but by corrupting them (Fig. 5).
Fig. 5. The low energy hypothesis of persister formation. Bactericidal antibiotics corrupt active targets, killing the cell. If ATP is low, this produces drug tolerant persister cells that survive.
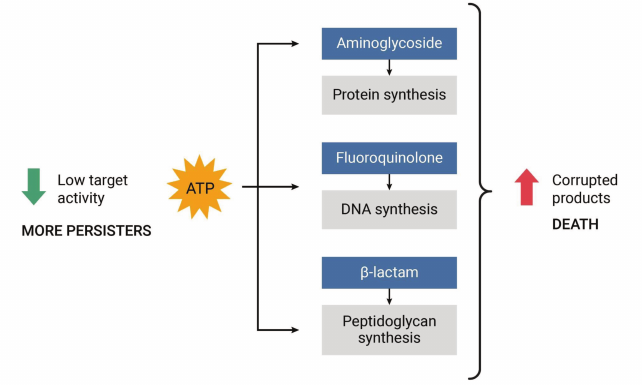
For example, aminoglycosides kill by causing mistranslation, and toxic misfolded peptides kill the cell. In a dormant cell, ATP is low, functions shut down, and there is nothing to corrupt.
Discovery of the TisB function provided a clue for persister formation in a regular, unstressed population. We knew that persisters are most abundant in a stationary population, where ATP is low. It seemed that persisters may be rare cells in which ATP drops due to stochastic variation in expression of energy producing components [39, 40]. This turned out to be the case, we found that sorted cells with low levels of expression of Krebs cycle enzymes are tolerant to killing by antibiotics [41].
The “low energy” mechanism of persister formation (Fig. 5) is simple and rather obvious. Indeed, we have known for quite a while that antibiotics require active targets to kill the cell, and that a decrease in ATP will of course produce dormancy and drug tolerance. I actually thought about this simple possibility a decade or so ago, and discounted it as improbable. At the time, studies started to come out describing stochastic variability in cellular phenotypes, and the classical example was that of the Lac repressor. There are only ~10 molecules of this repressor in an E. coli cell, and there will be significant variation in its content among cells in a population due to inevitable noise in expression. Abundant proteins, on the other hand, were thought to be free of such noise in their levels of expression. Now we know that this is not the case, expression of abundant enzymes generating energy is noisy, and leads to persisters.
TO KILL A DORMANT CELL
How do you kill a persister? The obvious answer is you do not. All known antibiotics fail to kill persisters, and we understand why – these are dormant cells with inactive targets. Seems like a perfect dead-end for drug discovery.
Ignoring reality, it is useful to ask a question: how would an anti-persister compound ideally work? Such a compound would have to corrupt an important target and kill a cell without the requirement for ATP. Based on this, an old abandoned antibiotic comes to mind – acyldepsipeptide (ADEP). Discovered at Eli Lilly in 1985, ADEP is produced by Streptomyces hawaiensis and has good activity against Gram-positive pathogens, but the company was looking for broad-spectrum antibiotics, and the compound was dropped. The mechanism of its action was reported [42], and that is what caught our attention. ADEP targets ClpP, a bacterial protease. ClpP recognizes misfolded proteins with the aid of an ATP-dependent chaperone, and digests them. ADEP keeps the pore of the protease open, which can now digest incoming peptides without the need for ATP (Fig. 6). This seemed to perfectly match what we were looking for, but there was a problem.
Fig. 6. Anti-persister compounds. a) Acyldepsipeptide keeps the pore of the ClpP protease of S. aureus and other Gram-positive bacteria open, forcing the cell to self-digest. b) Lassomycin activates ATP hydrolysis by the C1 chaperone of the mycobacterial ClpP1P2C1 protease, depleting ATP to the point of no return. c) Ciprofloxacin and rifampicin kill a population of S. aureus, leaving intact persisters; ADEP sterilizes the culture. d) ADEP4, a more active analog of the natural ADEP; and lassomycin.
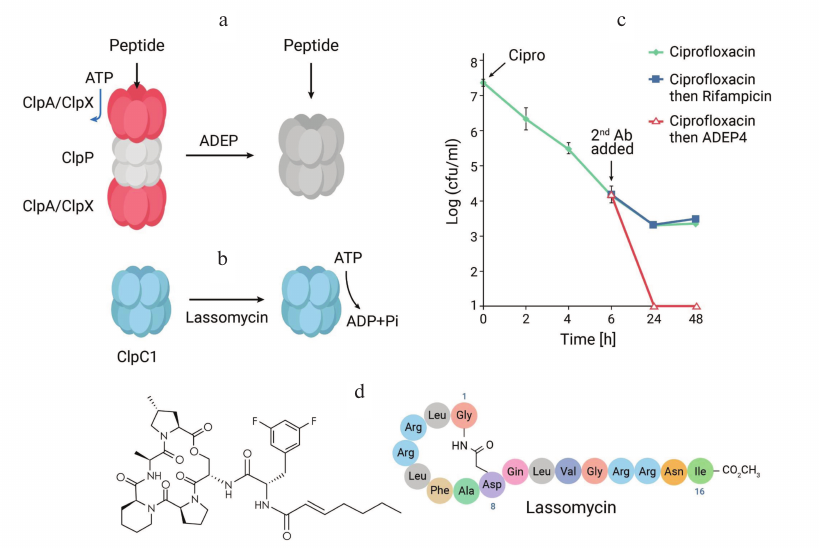
There were two papers suggesting that ADEP would only act against actively growing cells. One study reported that ADEP-ClpP acted only against nascent peptides exiting the ribosome [43], and the other claimed that the primary target is EnvZ, the protein that forms the septation ring in the process of division [44]. Both processes take place in actively growing cells. It appeared that my guess about ADEP was wrong.
There was however one issue with these studies – they examined proteolysis on a timescale of minutes, typical for biochemistry experiments. But antibiotics act on a timescale of hours and days. Brian Conlon, a talented postdoc who is now on the faculty of the University of North Carolina, Chapel Hill, reexamined the action of ADEP (which we had to custom-synthesize). He exposed it to a stationary culture of S. aureus for 24 h. The result was massive proteolysis of over 400 mature proteins. ADEP was forcing the cell to self-digest [45].
Further experiments showed that ADEP sterilizes a population of S. aureus in vitro and in a mouse model of infection. The remarkable properties of this compound point the way to control chronic infections for which we currently do not have adequate treatments (this is precisely why they are chronic). ADEP however does not have great pharmacological properties, it is rather unstable in vivo and has toxicity issues. A biotech company Arietis (USA) produced hundreds of ADEP analogs, and is getting close to a clinical candidate [46].
Another compound to kill persisters came from our screen of uncultured bacteria against M. tuberculosis [47]. The biggest problem in natural products antibiotic discovery is the enormous background of toxic, and to a lesser extent, known compounds. Ideally, would be good to be able to know if an extract contains a promising new compound before doing any chemistry. One solution to this seemingly improbable proposition is to screen for selective compounds. Several infectious diseases are caused by single pathogens, and a narrowly selective compound would be highly desirable. Apart from sparing the microbiome, the target of such a compound selective against a particular group of bacteria would be absent in humans, thus non-toxic. We screened extracts of uncultured species against M. tuberculosis, and counterscreened against S. aureus. This screen was based on a bet that nature actually makes selective compounds. Since natural antibiotics selective against M. tuberculosis were virtually unknown, any active extract would contain a novel and interesting compound, and we would know this before doing any chemistry. This screen uncovered several novel compounds, including lassomycin.
Lassomycin is a lasso-fold peptide, and resistant mutations were located in the gene coding for the C1 chaperone of the ClpP1P2C1 protease of M. tuberculosis. This protease is essential, and only distantly related to the ClpP proteases of other bacteria, explaining selectivity of the compound against Mycobacteria. The C1 chaperone recognizes misfolded peptides, and with the aid of ATP feeds them to the protease. We found that lassomycin dramatically activates the ATPase of the chaperone. Lassomycin was also able to kill persisters of M. tuberculosis. Apparently, ATP depletion to the point of no return kills regular cells and persisters.
Why the ClpP protease is under attack from two differently acting anti-persister compounds is unclear. But what we can learn from these first examples is a general principle that nature uses to kill persisters – corrupting hydrolases (Fig. 6). Hydrolysis per se does not require energy. There are numerous tightly controlled hydrolases in the cell – proteases, lipases, phosphatases and nucleases. These are all potential targets for anti-persister compounds.
SCREENING PLANET EARTH
Given their formidable penetration barrier, no wonder that it is difficult to find antibiotics acting against Gram-negative bacteria. With the lack of success in developing MDR pump inhibitors, it became clear that the problem could be approached from the opposite direction – testing unrelated compounds to understand the properties that favor penetration [48]. In two studies, Paul Hergenrother and his team did exactly that: measured penetration of a large number of unrelated compounds into E. coli, ranked them by their ability to penetrate, and came up with “rules of permeation” with the aid of cheminformatics [49, 50]. Some of the parameters have been previously established – a molecular weight cut-off of 600 Da, and low hydrophobicity, cLogP of -0.1 [51], but the additional ones are interesting and not at all obvious: low number of rotatable bonds (rigid structure), low three-dimensionality (flat is good), and positive charge in the form of an amino group, -NH3+, are all good for permeation. The molecular weight is dictated by the diameter of outer membrane porins; low hydrophobicity, rigid structure, flat/elongated shape and positive charge all apparently favor passage through the negatively charged β-barrels of porins. These are emerging rules that are likely to be further refined and expanded.
Using the rules of permeation holds the promise of rationally designing antibiotics. When this will become a reality is unclear. For now, natural product antibiotic discovery seems like a more realistic bet. Compounds acting against Gram-negative bacteria have been overmined, but only from Actinomycetes, and other bacterial groups must have developed their own compounds to act against their Gram-negative competitors. But where does one look for these producers? Screening projects start with cells typically obtained from 10 g of soil; whether this sample contains a producer of a drug lead is of course unknown. Repeating this process numerous times still does not tell us anything about the possibility of finding something interesting. Ideally, one would want to screen not grams of soil, but the entire biosphere. This sounds like a fantasy, but there a solution – the screen has already been performed.
Let us assume that there is a group of bacteria that share the requirements for antibiotics with us: activity against Gram-negative pathogens, low toxicity, and good pharmacokinetics (PK) – an ability to move through tissues without being rapidly sequestered or destroyed. This group of bacteria would have collected the antibiotics we are interested in from the biosphere by horizontal transmission of DNA. These considerations lead us to nematophilic bacteria. Nematode symbionts, Photorhabdus and Xenorhabdus, are members of the gut microbiome and are closely related to other Enterobacteriaceae, such as E. coli. Nematodes invade insect larvae and release their symbionts. Nematophiles first produce neurotoxins to immobilize their prey, and then release various antimicrobials to fend off invading environmental microorganisms [52, 53]. However, the most immediate competitors probably come not from the environment, but from other members of the nematode gut. Interestingly, Gram-negative bacteria that are common opportunistic pathogens of humans are abundant in the microbiome of entomopathogenic nematodes [54]. The antimicrobial compounds of nematophilic bacteria must be non-toxic to the nematode, and be able to spread well through the tissues of the larvae. This suggests antimicrobials with low toxicity and good pharmacokinetics active against Gram-negative pathogens.
We screened a small collection of Photorhabdus, around 20 species, looking for zones of inhibition on agar overlaid with E. coli. Most of the tested bacteria did not produce zones of inhibition, which could be due to poor expression of “silent” biosynthetic gene clusters (BGCs) in vitro. A concentrated extract from P. khanii produced a small zone of E. coli growth inhibition on a Petri dish, while spotting a colony had no effect. We isolated the antibiotic, and determined its mass by MS, which is 966 Da [55]. This is quite a bit larger than the 600 Da cutoff for penetration of compounds across the outer membrane, which was puzzling. Structural analysis showed that the compound is a heavily modified 7-mer peptide (Fig. 7).
Fig. 7. The mechanism of action of darobactin. Upper panel, darobactin A produced by a number of Photorhabdus species is the first member of the class, and darobactin C is provided as an example of an analog. The dar operon codes for a propeptide that is modified by the radical SAM enzyme DarE, and the DarCDE transporter exports darobactin. Lower panel, darobactin targets the BamA chaperone and insertase of outer membrane proteins.
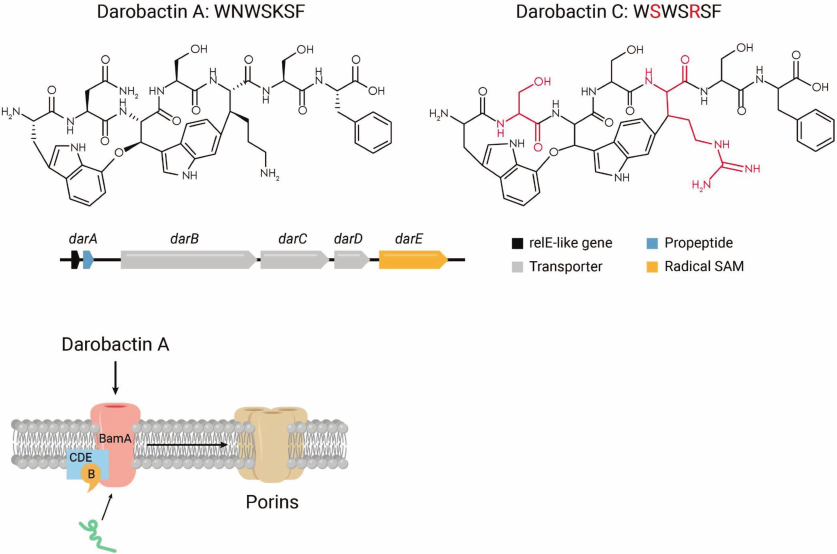
We named the compound darobactin, from the Greek/Russian Dar, gift. It contains two fused rings, one of them formed by linking unactivated carbons between tryptophan and lysine. This would require a free radical reaction, and the BGC indeed includes a “radical SAM” enzyme, DarE. Mutants resistant to darobactin were mapped to BamA, an essential chaperone that folds and inserts porins into the outer membrane. This resolves the puzzle of the size – the target is on the surface, obviating the need for darobactin to penetrate. BamA is a β-barrel protein which is not an enzyme and does not have a well-ordered catalytic site that can be targeted by an inhibitor. Indeed, considerable efforts to target BamA by screening synthetic compound libraries have not been successful, this is a typical “undruggable” target. Sebastian Hiller, a top expert in structure/function of chaperones and our collaborator, obtained a co-crystal structure of darobactin with BamA (Hundeep et al., in review).
Darobactin A turned out to be the first member of a large, and growing class of compounds. Searching the database for homologs of the dar operon uncovered 8 analogs, 5 of which we published. This reminds me of the discovery of aminoglycosides, a class of antibiotics with numerous members. The first to be discovered, streptothrycin, did not become a drug; the second, streptomycin, did, but is not widely used, while the later-discovered gentamicin and tobramycin are commonly used therapeutics. It took 23 years to get from streptothrycin to tobramycin [56]. It took us only several days to identify the 8 analogs of darobactin A computationally. Based on its properties, darobactin A is a promising lead, but the probability of it being the best lead is about 10%. We do not need to obtain and culture the producers of darobactins B-I, the sequence is sufficient to synthesize the operons and plug them into an expression vector in E. coli for production.
We set out to tap into the screen of the biosphere that Photorhabdus had performed, and discovered darobactin. Where did it come from? The dar operon has an unusually low GC content, so this sequence clearly comes from a horizontal transmission event. Where it came from, we do not know. Photorhabdus originated 370 million years ago, probably long enough to screen the entire biosphere. What else have they captured?
GAZING INTO THE FUTURE BY CONTEMPLATING THE PAST
Looking ahead – not a bad idea to consider lessons from bacteria that had several billion years to produce antibiotics. Natural-product antibiotics can corrupt, rather than inhibit targets, as we see from the examples of ADEP or aminoglycosides, and appear to be indifferent to whether a target is druggable (from our perspective) or not. Their simpler synthetic cousins are good at inhibiting well-structured active sites of enzymes. Compare this to teixobactin that binds pyrophosphate-sugar of lipid II, and does so strongly and selectively, but only because it assembles into a β-lattice supramolecular structure; or darobactin that manages to target a β-barrel chaperone by mimicking a β-strand. Current synthetic chemistry is not (yet) capable of predicting compounds with such sophisticated modes of action. We will therefore continue to mine the natural antimicrobiome. Of course, we search for a perfect antibiotic. But does it exist?
For an answer to this question, let us consider the antibiotic arsenal assembled by three different types of eukaryotes. Fungi borrowed their broad-spectrum antibiotics from bacteria, and they are penicillin and cephalosporin, both β-lactams. A seemingly unremarkable small animal, springtail captured antibiotic genes by horizontal transmission, and they code for penicillin and cephalosporin. On a considerably shorter prospecting timescale, H. sapiens assembled an arsenal of antibiotics, and the best ones are penicillin and cephalosporin. These compounds are small, water-soluble, hit multiple penicillin binding proteins – peptidoglycan transpeptidases absent in humans – and are non-toxic. The remaining major classes of the clinically useful antibiotics – aminoglycosides, tetracyclines, chloramphenicols and macrolides – all target the bacterial ribosome, and act against mitochondria as well, with toxic consequences. This is probably why other eukaryotes passed on these compounds. Moving forward, we would be well advised to emulate other eukaryotes and focus on compounds that act selectively against bacterial targets, such as teixobactin and darobactin.
β-Lactams are excellent but not perfect. There are numerous β-lactamases, and some of the resistance comes from target modification. Besides, these compounds only kill growing cells. A major problem in antibiotic discovery is resistance development, that is why the main classes of clinically used antibiotics engage in multitargeting. β-Lactams act against multiple penicillin-binding proteins, fluoroquinolones target homologous DNA gyrase/topoisomerase, and the remaining classes act against the ribosome, binding to rRNA coded by multiple gene copies. These 3 targets are a small part of the ~500 essential proteins present in bacteria [57, 58], a crippling restriction for discovery. Bacteria apparently solve the problem of resistance by attacking their neighbors with combinations of compounds – β-lactam + β-lactamase, or quinupristin + dalfopristin. We should be doing the same, introducing novel compounds not as singles, but as combinations. This will liberate all targets for discovery. Two imperfect antibiotics will make a perfect combination. An antipersister compound combined with another novel antibiotic will cure acute and chronic infections, and will have a lasting impact.
Compounds we discovered so far are changing the way we think of antibiotics and their targets. Teixobactin challenges the old dogma of unavoidable resistance; both teixobactin and darobactin hit undruggable targets, suggesting that all targets are fair game for antibiotics; lassomycin and ADEP show us how to kill “invincible” persister cells by dysregulating hydrolases. We do not know the size of the global antimicrobiome, but we may be assured that it is vast, produced by some 1012 bacterial species inhabiting the planet [59]. No doubt, many more uniquely interesting antibiotics are waiting to be discovered. Finding them is an exciting intellectual challenge, and the lessons I learned many years ago in the Skulachev lab and now pass on to my students will serve us well.
Acknowledgments. I thank Boris Chernyak for helpful comments. Recent work in the author’s lab described in this Review was supported by NIH grants P01 AI118687 and R01 AI141966.
Ethics declarations. The author declares participation in the following Companies: Novobiotic Pharmaceuticals; Arietis Pharma; Holobiome; and Daros. This article contains description of studies with the involvement of animal subjects, but not humans performed by the author.
Open access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
REFERENCES
1.Mitchell, P. (1961) Coupling of phosphorylation to
electron and hydrogen transfer by a chemi-osmotic type of mechanism,
Nature, 191, 144-148, doi: 10.1038/191144a0.
2.Mitchell, P. (1966) Chemiosmotic Coupling in
Oxidative and Photosynthetic Phosphorylation, Glynn Research
Laboratories, Bodmin.
3.Mitchell, P., and Moyle, J. (1967) Chemiosmotic
hypothesis of oxidative phosphorylation, Nature, 213,
137-139, doi: 10.1038/213137a0.
4.Liberman, E. A., Topaly, V. P., Tsofina, L. M.,
Jasaitis, A. A., and Skulachev, V. P. (1969) Mechanism of coupling of
oxidative phosphorylation and the membrane potential of mitochondria,
Nature, 222, 1076-1078, doi: 10.1038/2221076a0.
5.Skulachev, V. P., Sharaf, A. A., and Liberman, E.
A. (1967) Proton conductors in the respiratory chain and artificial
membranes, Nature, 216, 718-719, doi:
10.1038/216718a0.
6.Williams, R. J. (1961) Possible functions of chains
of catalysts, J. Theor. Biol., 1, 1-17, doi:
10.1016/0022-5193(61)90023-6.
7.Lomovskaya, O., and Lewis, K. (1992) Emr, an
Escherichia coli locus for multidrug resistance, Proc. Natl.
Acad. Sci. USA, 89, 8938-8942.
8.Lomovskaya, O., Lewis, K., and Matin, A. (1995)
EmrR is a negative regulator of the Escherichia coli multidrug
resistance pump EmrAB, J. Bacteriol., 177, 2328-2334.
9.Li, X. Z., Ma, D., Livermore, D. M., and Nikaido,
H. (1994) Role of efflux pump(s) in intrinsic resistance of
Pseudomonas aeruginosa: active efflux as a contributing factor
to beta-lactam resistance, Antimicrob. Agents Chemother.,
38, 1742-1752.
10.Hsieh, P. C., Siegel, S. A., Rogers, B., Davis,
D., and Lewis, K. (1998) Bacteria lacking a multidrug pump: a sensitive
tool for drug discovery, Proc. Natl. Acad. Sci. USA, 95,
6602-6606.
11.Neyfakh, A. A., Bidnenko, V. E., and Chen, L. B.
(1991) Efflux-mediated multidrug resistance in Bacillus
subtilis: similarities and dissimilarities with the mammalian
system, Proc. Natl. Acad. Sci. USA, 88, 4781-4785.
12.Severina, I. I., Muntyan, M. S., Lewis, K., and
Skulachev, V. P. (2001) Transfer of cationic antibacterial agents
berberine, palmatine and benzalkonium through bimolecular planar
phospholipid film and Staphylococcus aureus membrane, IUBMB
Life Sci., 52, 321-324.
13.Stermitz, F. R., Lorenz, P., Tawara, J. N.,
Zenewicz, L. A., and Lewis, K. (2000) Synergy in a medicinal plant:
antimicrobial action of berberine potentiated by
5′-methoxyhydnocarpin, a multidrug pump inhibitor, Proc. Natl.
Acad. Sci. USA, 97, 1433-1437, doi:
10.1073/pnas.030540597.
14.Vyssokikh, M. Y., Holtze, S., Averina, O. A.,
Lyamzaev, K. G., Panteleeva, A. A., et al. (2020) Mild depolarization
of the inner mitochondrial membrane is a crucial component of an
anti-aging program, Proc. Natl. Acad. Sci. USA, 117,
6491-6501, doi: 10.1073/pnas.1916414117.
15.Schatz, A., Bugie, E., and Waksman, S. A. (1944)
Streptomycin, a substance exhibiting antibiotic activity against
gram-positive and gram-negative bacteria., Proc. Soc. Exp. Biol.
Med., 55, 66-69.
16.Lewis, K. (2020) The science of antibiotic
discovery, Cell, 181, 29-45, doi:
10.1016/j.cell.2020.02.056.
17.Alekshun, M. N., and Levy, S. B. (2007) Molecular
mechanisms of antibacterial multidrug resistance, Cell,
128, 1037-1050, doi: 10.1016/j.cell.2007.03.004.
18.Tacconelli, E., Carrara, E., Savoldi, A.,
Harbarth, S., Mendelson, M., et al. (2018) Discovery, research, and
development of new antibiotics: the WHO priority list of
antibiotic-resistant bacteria and tuberculosis, Lancet Infect.
Dis., 18, 318-327, doi: 10.1016/s1473-3099(17)30753-3.
19.Ramos-Castaneda, J. A., Ruano-Ravina, A.,
Barbosa-Lorenzo, R., Paillier-Gonzalez, J. E., Saldana-Campos, J. C.,
Salinas, D. F., and Lemos-Luengas, E. V. (2018) Mortality due to KPC
carbapenemase-producing Klebsiella pneumoniae infections: systematic
review and meta-analysis: mortality due to KPC Klebsiella pneumoniae
infections, J. Infect., 76, 438-448, doi:
10.1016/j.jinf.2018.02.007.
20.Xu, L., Sun, X., and Ma, X. (2017) Systematic
review and meta-analysis of mortality of patients infected with
carbapenem-resistant Klebsiella pneumoniae, Ann. Clin.
Microbiol. Antimicrob., 16, 18, doi:
10.1186/s12941-017-0191-3.
21.Winterberg, H. (1898) Zur Methodik der
Bakterienzahlung, Zeitschr. Hyg., 29, 75-93.
22.Kaeberlein, T., Lewis, K., and Epstein, S. S.
(2002) Isolating “uncultivable” microorganisms in pure
culture in a simulated natural environment, Science, 296,
1127-1129.
23.D’Onofrio, A., Crawford, J. M., Stewart, E.
J., Witt, K., Gavrish, E., Epstein, S., Clardy, J., and Lewis, K.
(2010) Siderophores from neighboring organisms promote the growth of
uncultured bacteria, Chem. Biol., 17, 254-264, doi:
10.1016/j.chembiol.2010.02.010.
24.Fenn, K., Strandwitz, P., Stewart, E. J., Dimise,
E., Rubin, S., Gurubacharya, S., Clardy, J., and Lewis, K. (2017)
Quinones are growth factors for the human gut microbiota,
Microbiome, 5, 161, doi: 10.1186/s40168-017-0380-5.
25.Strandwitz, P., Kim, K. H., Terekhova, D., Liu,
J. K., Sharma, A., et al. (2019) GABA-modulating bacteria of the human
gut microbiota, Nat. Microbiol., 4, 396-403, doi:
10.1038/s41564-018-0307-3.
26.Ling, L. L., Schneider, T., Peoples, A. J.,
Spoering, A. L., Engels, I., et al. (2015) A new antibiotic kills
pathogens without detectable resistance, Nature, 517,
455-459, doi: 10.1038/nature14098.
27.Shukla, R., Medeiros-Silva, J., Parmar, A.,
Vermeulen, B. J. A., Das, S., et al. (2020) Mode of action of
teixobactins in cellular membranes, Nat. Commun., 11,
2848, doi: 10.1038/s41467-020-16600-2.
28.Brooun, A., Liu, S., and Lewis, K. (2000) A
dose-response study of antibiotic resistance in Pseudomonas
aeruginosa biofilms, Antimicrob. Agents Chemother.,
44, 640-646.
29.Spoering, A. L., and Lewis, K. (2001) Biofilms
and planktonic cells of Pseudomonas aeruginosa have similar
resistance to killing by antimicrobials, J. Bacteriol.,
183, 6746-6751, doi: 10.1128/JB.183.23.6746-6751.2001.
30.Bigger, J. W. (1944) Treatment of staphylococcal
infections with penicillin, Lancet, ii, 497-500.
31.Moyed, H. S., and Bertrand, K. P. (1983) hipA, a
newly recognized gene of Escherichia coli K-12 that affects frequency
of persistence after inhibition of murein synthesis, J.
Bacteriol., 155, 768-775.
32.Mulcahy, L. R., Burns, J. L., Lory, S., and
Lewis, K. (2010) Emergence of Pseudomonas aeruginosa strains
producing high levels of persister cells in patients with cystic
fibrosis, J. Bacteriol., 192, 6191-6199, doi:
10.1128/JB.01651-09.
33.Torrey, H. L., Keren, I., Via, L. E., Lee, J. S.,
and Lewis, K. (2016) High persister mutants in Mycobacterium
tuberculosis, PLoS One, 11, e0155127, doi:
10.1371/journal.pone.0155127.
34.Schumacher, M. A., Balani, P., Min, J., Chinnam,
N. B., Hansen, S., Vulic, M., Lewis, K., and Brennan, R. G. (2015)
HipBA-promoter structures reveal the basis of heritable multidrug
tolerance, Nature, 524, 59-64, doi:
10.1038/nature14662.
35.Correia, F. F., D’Onofrio, A., Rejtar, T.,
Li, L., Karger, B. L., Makarova, K., Koonin, E. V., and Lewis, K.
(2006) Kinase activity of overexpressed HipA is required for
growth arrest and multidrug tolerance in Escherichia coli, J.
Bacteriol., 188, 8360-8367.
36.Kaspy, I., Rotem, E., Weiss, N., Ronin, I.,
Balaban, N. Q., and Glaser, G. (2013) HipA-mediated antibiotic
persistence via phosphorylation of the glutamyl-tRNA-synthetase,
Nat. Commun., 4, 3001, doi: 10.1038/ncomms4001.
37.Dorr, T., Vulic, M., and Lewis, K. (2010)
Ciprofloxacin causes persister formation by inducing the TisB toxin in
Escherichia coli, PLoS Biol., 8, e1000317, doi:
10.1371/journal.pbio.1000317.
38.Gurnev, P. A., Ortenberg, R., Dorr, T., Lewis,
K., and Bezrukov, S. M. (2012) Persister-promoting bacterial toxin TisB
produces anion-selective pores in planar lipid bilayers, FEBS
Lett., 586, 2529-2534, doi:
10.1016/j.febslet.2012.06.021.
39.Conlon, B. P., Rowe, S. E., Gandt, A. B., Nuxoll,
A. S., Donegan, N. P., et al. (2016) Persister formation in
Staphylococcus aureus is associated with ATP depletion,
Nat. Microbiol., 1, 16051, doi:
10.1038/nmicrobiol.2016.51.
40.Shan, Y., Brown Gandt, A., Rowe, S. E.,
Deisinger, J. P., Conlon, B. P., and Lewis, K. (2017) ATP-dependent
persister formation in Escherichia coli, MBio, 8,
doi: 10.1128/mBio.02267-16.
41.Zalis, E. A., Nuxoll, A. S., Manuse, S., Clair,
G., Radlinski, L. C., Conlon, B. P., Adkins, J., and Lewis, K. (2019)
Stochastic variation in expression of the tricarboxylic acid cycle
produces persister cells, MBio, 10, doi:
10.1128/mBio.01930-19.
42.Brotz-Oesterhelt, H., Beyer, D., Kroll, H. P.,
Endermann, R., Ladel, C., et al. (2005) Dysregulation of bacterial
proteolytic machinery by a new class of antibiotics, Nat. Med.,
11, 1082-1087, doi: 10.1038/nm1306.
43.Kirstein, J., Hoffmann, A., Lilie, H., Schmidt,
R., Rubsamen-Waigmann, H., Brotz-Oesterhelt, H., Mogk, A., and Turgay,
K. (2009) The antibiotic ADEP reprogrammes ClpP, switching it from a
regulated to an uncontrolled protease, EMBO Mol. Med., 1,
37-49, doi: 10.1002/emmm.200900002.
44.Sass, P., Josten, M., Famulla, K., Schiffer, G.,
Sahl, H. G., Hamoen, L., and Brotz-Oesterhelt, H. (2011) Antibiotic
acyldepsipeptides activate ClpP peptidase to degrade the cell division
protein FtsZ, Proc. Natl. Acad. Sci. USA, 108,
17474-17479, doi: 10.1073/pnas.1110385108.
45.Conlon, B. P., Nakayasu, E. S., Fleck, L. E.,
LaFleur, M. D., Isabella, V. M., et al. (2013) Activated ClpP kills
persisters and eradicates a chronic biofilm infection, Nature,
503, 365-370.
46.Griffith, E. C., Zhao, Y., Singh, A. P., Conlon,
B. P., Tangallapally, R., et al. (2019) Ureadepsipeptides as ClpP
Activators, ACS Infect. Dis., 5, 1915-1925, doi:
10.1021/acsinfecdis.9b00245.
47.Gavrish, E., Sit, C. S., Cao, S., Kandror, O.,
Spoering, A., et al. (2014) Lassomycin, a ribosomally synthesized
cyclic peptide, kills Mycobacterium tuberculosis by targeting
the ATP-dependent protease ClpC1P1P2, Chem. Biol., 21,
509-518, doi: 10.1016/j.chembiol.2014.01.014.
48.Lewis, K. (2012) Antibiotics: recover the lost
art of drug discovery, Nature, 485, 439-440, doi:
10.1038/485439a.
49.Richter, M. F., Drown, B. S., Riley, A. P.,
Garcia, A., Shirai, T., Svec, R. L., and Hergenrother, P. J. (2017)
Predictive compound accumulation rules yield a broad-spectrum
antibiotic, Nature, 545, 299-304, doi:
10.1038/nature22308.
50.Parker, E. N., Drown, B. S., Geddes, E. J., Lee,
H. Y., Ismail, N., Lau, G. W., and Hergenrother, P. J. (2020)
Implementation of permeation rules leads to a FabI inhibitor with
activity against Gram-negative pathogens, Nat. Microbiol.,
5, 67-75, doi: 10.1038/s41564-019-0604-5.
51.O’Shea, R., and Moser, H. E. (2008)
Physicochemical properties of antibacterial compounds: Implications for
drug discovery, J. Med. Chem., 51, 2871-2878.
52.Crawford, J. M., and Clardy, J. (2011) Bacterial
symbionts and natural products, Chem. Commun. (Camb.),
47, 7559-7566, doi: 10.1039/c1cc11574j.
53.Tobias, N. J., Shi, Y. M., and Bode, H. B. (2018)
Refining the natural product repertoire in entomopathogenic bacteria,
Trends Microbiol., 26, 833-840, doi:
10.1016/j.tim.2018.04.007.
54.Tambong, J. T. (2013) Phylogeny of bacteria
isolated from Rhabditis sp. (Nematoda) and identification of
novel entomopathogenic Serratia marcescens strains, Curr.
Microbiol., 66, 138-144, doi: 10.1007/s00284-012-0250-0.
55.Imai, Y., Meyer, K. J., Iinishi, A., Favre-Godal,
Q., Green, R., et al. (2019) A new antibiotic selectively kills
Gram-negative pathogens, Nature, 576, 459-464, doi:
10.1038/s41586-019-1791-1.
56.Krause, K. M., Serio, A. W., Kane, T. R., and
Connolly, L. E. (2016) Aminoglycosides: an overview, Cold Spring
Harb. Perspect. Med., 6, a027029, doi:
10.1101/cshperspect.a027029.
57.Juhas, M., Eberl, L., and Glass, J. I. (2011)
Essence of life: essential genes of minimal genomes, Trends Cell.
Biol., 21, 562-568, doi: 10.1016/j.tcb.2011.07.005.
58.Grazziotin, A. L., Vidal, N. M., and Venancio, T.
M. (2015) Uncovering major genomic features of essential genes in
Bacteria and a methanogenic Archaea, FEBS J., 282,
3395-3411, doi: 10.1111/febs.13350.
59.Locey, K. J., and Lennon, J. T. (2016) Scaling
laws predict global microbial diversity, Proc. Natl. Acad. Sci.
USA, 113, 5970-5975, doi: 10.1073/pnas.1521291113.