REVIEW: Inhibitors of β-Lactamases. New Life of β-Lactam Antibiotics
A. M. Egorov1, M. M. Ulyashova1, and M. Yu. Rubtsova1,a*
1Faculty of Chemistry, Lomonosov Moscow State University, 119991 Moscow, Russia* To whom correspondence should be addressed.
Received July 9, 2020; Revised August 14, 2020; Accepted August 15, 2020
β-Lactam antibiotics account for about 60% of all produced antibiotics. Due to a high activity and minimal side effects, they are the most commonly used class of antibacterial drugs for the treatment of various infectious diseases of humans and animals, including severe hospital infections. However, the emergence of bacteria resistant to β-lactams has led to the clinical inefficiency of these antibiotics, and as a result, their use in medicine has been limited. The search for new effective ways for overcoming the resistance to β-lactam antibiotics is an essential task. The major mechanism of bacterial resistance is the synthesis of β-lactamases (BLs) that break the antibiotic β-lactam ring. Here, we review specific inhibitors of serine β-lactamases and metallo-β-lactamases and discuss approaches for creating new inhibitors that would prolong the “life” of β-lactams.
KEY WORDS: antibiotic resistance of bacteria, β-lactamases, inhibitors, β-lactam antibioticsDOI: 10.1134/S0006297920110024
Abbreviations: AMA, aspergillomarasmin A; BL, β-lactamase; ESBL, extended-spectrum β-lactamase; DBO, diazabilcyclooctane; MBL, metallo-β-lactamase; PBP, penicillin-binding protein.
INTRODUCTION. THE ERA OF ANTIBIOTICS AND THE ROLE OF
β-LACTAMS
For many centuries, infectious diseases had been a constant threat for public health. Until the early 1900s, they accounted for up to 25% of deaths [1]. The discovery in 1929 by the British microbiologist Alexander Fleming of the antimicrobial properties of the Penicillium notatum mold resulted in the identification of the first β-lactam antibiotic, penicillin, and marked the emergence of a novel class of pharmaceuticals.
β-Lactam antibiotics produced by various bacteria and fungi have existed in nature for over 2 billion years. Their targets are penicillin-binding proteins (PBPs) that catalyze various reactions in the synthesis of peptidoglycan, the major element of the bacterial cell wall [2]. The antibacterial activity of these antibiotics is based on the structural likeness of the β-lactam ring to the terminal D-Ala-D-Ala-fragment of the peptidoglycan pentapeptides. β-Lactams form a covalent complex with serine in the PBP transpeptidase domain and irreversibly inhibit peptidoglycan synthesis, resulting in the lysis of bacterial cells and the loss of their viability.
A high efficacy of penicillin and its natural analogs, together with their relatively low toxicity and low cost of industrial synthesis, have facilitated the development of novel β-lactam antibiotics [3]. The goal of chemical modification of β-lactams was to extend the action spectrum, improve the pharmacokinetic properties, and to counteract the resistance mechanisms of bacteria. Currently, this class of antibiotics includes four groups with the four-membered β-lactam ring: penicillins, cephalosporins, carbapenems, and monobactams (Fig. 1).
Fig. 1. Structures of the main groups of β-lactam antibiotics.
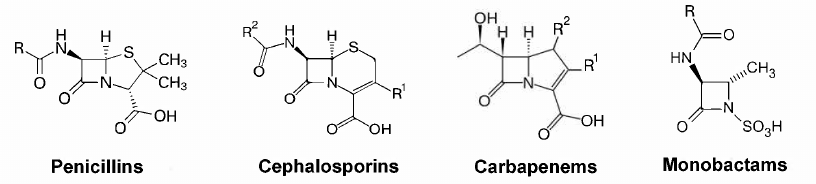
The discovery of penicillin was one of the most important discoveries of the XX century. Successful chemical modifications of penicillins, cephalosporins, and carbapenems have resulted in the development of several dozens of drugs for suppressing multiple bacterial infections, including severe hospital-acquired forms. Human organism has no targets for the action of β-lactam antibiotics, which explains their low toxicity. Due to their properties, β-lactam antibiotics have remained the most commonly used antibacterial preparations for more than 70 years. They make up about 60% of all antibiotics used in medicine and agriculture for the therapy and prevention of infectious diseases of human and animals [4]. Some antibiotics of this class belong to the so-called reserve group.
The clinical efficacy of β-lactam antibiotics is limited mainly due to the evolution of microorganisms, resulting in the development of microbial antibiotic resistance. The World Health Organization (WHO) reported that in 2019, the resistance to antimicrobial preparations was one of the ten main threats to the public health worldwide [5]. As a result, the “life” of many β-lactams in medical practice is limited and the development of new antibacterial preparations of this class has diminished.
THE “TRAGEDY” of β-LACTAM ANTIBIOTICS,
β-LACTAMASES, AND RESISTANCE MECHANISMS
The first reports on the isolation of microbial strains with a natural resistance to penicillin appeared almost at the same time as the discovery of this antibiotic. Such resistance is a protective mechanism that has developed during bacterial evolution as a result of competitive struggle with other microorganisms. The selective pressure on microorganisms provided by the wide and frequently uncontrolled use of antibiotics in clinical medicine and especially, in veterinary and agriculture, has led to the emergence of various resistance mechanisms, such as efflux (antibiotic transport out of the cell) and modification in the structure of porins (membrane transfer proteins), antibiotics themselves, or their targets [6].
All mechanisms of microbial resistance to antibiotics involve bacterial enzymes [7]. The most common mechanism of the resistance to β-lactams is expression of specific hydrolases, β-lactamases (BLs, EC 3.5.2.6), that catalyze hydrolysis of the antibiotic β-lactam ring. One of the first described cases was hydrolysis of penicillins in Gram-positive bacteria. Later, it was found that the main resistance mechanism in Gram-positive bacteria is associated with the acquisition of new genes that encode additional PBPs with a reduced affinity for β-lactams, e.g., mecA gene in the methicillin-resistant Staphylococcus aureus (MRSA) [8]. The most common antibiotic resistance mechanism in Gram-negative bacteria is the synthesis of BLs. BLs form a superfamily consisting of ~2800 enzymes [9]. The information on BLs is constantly updated on the National Center for Biotechnology Information (NCBI) site (https://www.ncbi.nlm.nih.gov/pathogens/beta-lactamase-data-resources).
A common property of all BLs is the ability to catalyze hydrolysis of the amide bond in the β-lactam ring, which is the major structural element responsible for the antibacterial activity. BL-encoding genes are characterized by a high rate of mutations. These genes are usually found in mobile genetic elements (plasmids, transposons, and integrons), which facilitates their rapid spread among infectious bacteria, as well as bacteria inhabiting natural environment (water, soil) [10]. A combination of BL-encoding genes and other resistance genes leads to the emergence of the multidrug resistance (MDR), extensive drug resistance (XDR), and pandrug resistance (PDR) [11]. As a result, many infectious agents have become deadly dangerous and now represent a global threat for human health.
BLs and PBPs have a common precursor protein and possess common structural elements. In both PBPs and serine BLs, the active site serine forms a covalent complex with the antibiotic molecule (acyl-enzyme), leading to the break of the amide bond in the β-lactam ring. The formation of the antibiotic complex with the PBP is irreversible, which results in the enzyme inhibition.
Based on the amino acid sequences homology, there are four molecular classes of BLs: A, B, C, and D. Enzymes of the A, C, and D classes are serine hydrolases; members of the B class are metalloenzymes that contain one or two cofactor Zn2+ ions in the active site. Each BL class is subdivided into enzymes types with different substrate specificity and sensitivity to inhibitors. β-Lactam-resistant bacteria that cause human infections most frequently produce class A BLs of the TEM, CTX-M, and SHV types; during the last years, an exponential growth in the number of strains producing carbapenemases of the B, D, and A classes has been recorded [9].
All serine BLs are compact protein globules with the secondary structure including 11 α-helices, 5 β-sheets, and irregularly structured loops (Fig. 2a). Serine BLs have the sandwich-type structure and consist of three α/β/α domains connected with a network of ionic and hydrogen bonds [12]. Serine BLs have a compact nucleus consisting of spatially close α-helix and β-sheet that contain 3 to 4 amino acid-long conserved motifs (including the catalytic serine). Although this globular structure is rigid in general, its flexibility is provided by the movements of the loops, which often contain mutations [13, 14]. An important conserved structural element is the Ω-loop located in the lower part of the entrance to the active site pocket that contains catalytically important conserved Glu residue (Fig. 2b). This residue plays a crucial role in the deacylation of the acyl-enzyme complex, resulting in the release of the cleaved antibiotic molecule from the BL active site [15].
Fig. 2. a) Structure of the class A β-lactamase TEM-1 (catalytic Ser70 is presented as a space-filling model). b) Structure of the Ω-loop in the TEM-type BLs.
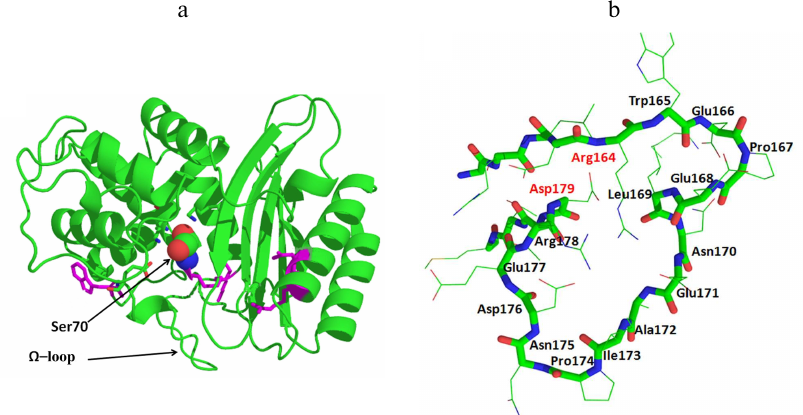
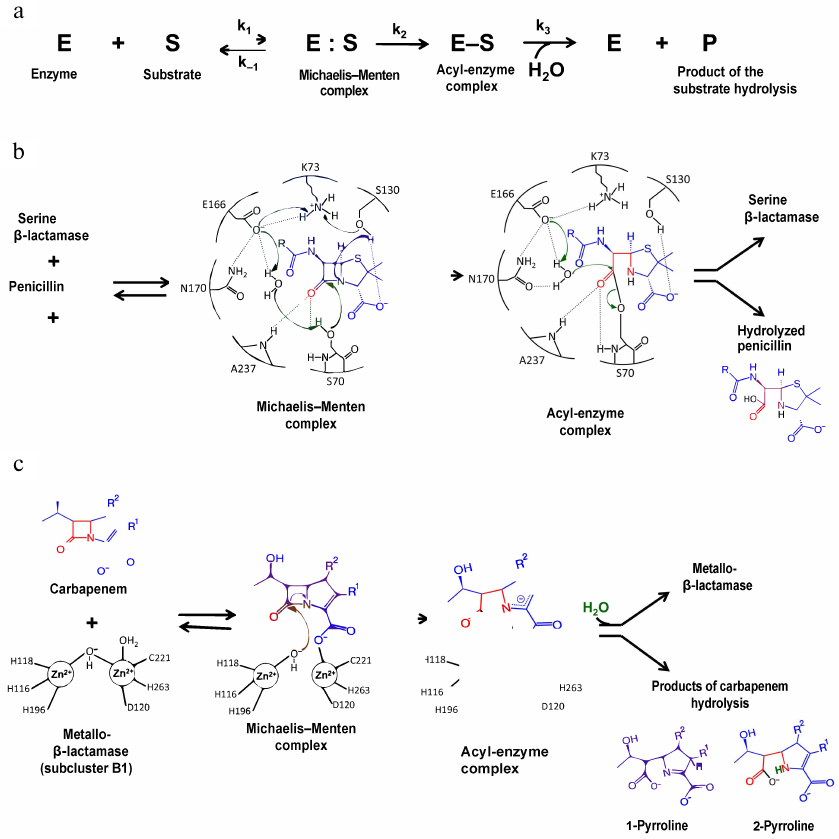
The mechanism of the β-lactam hydrolysis by serine BLs from different classes is similar and includes three main stages (Fig. 3a). All these enzymes form a covalent acyl-enzyme complex with the serine residue, which is stabilized by other amino acids (depending on the active site structure). At the first stage, the antibiotic molecule is oriented and bound in the enzyme active site, and the hydroxyl group of the catalytic serine is activated. In the class A BLs of the TEM type, activation of the catalytic serine requires deprotonation of Lys73. The deprotonation mechanism includes formation of a chain of hydrogen bonds with the involvement of the water molecule and Glu166 and Asn170 residues of the Ω-loop (Fig. 3b) [16]. An alternative mechanism of Lys73 deprotonation occurs with the participation of Ser130 residue of the SDN (Ser130-Asn132) loop. Then, the serine hydroxyl group acts as a nucleophile and attacks the carbonyl group of the β-lactam ring with the formation of a highly reactive tetrahedral acylated intermediate that is converted to the low-energy acyl-enzyme complex in which serine is covalently bound with the antibiotic molecule. At the third stage, the water molecule coordinated in the enzyme active site by Glu166 and other charged residues of the Ω-loop attacks the covalent acyl-enzyme complex. The β-lactam ring breaks as a result of the amide bond diacylation and antibiotic molecule leaves the active site, while the enzyme molecule is released for the next catalytic act [17].
Fig. 3. a) Hydrolysis of β-lactam antibiotics by BLs; b) mechanism of penicillin hydrolysis by class A BLs; c) mechanism of carbapenem hydrolysis by metallo-BLs.
The structure of BLs can be modified by mutations in the enzyme active site and peripheral regions of the protein globule. Some of them (key mutations) lead to the changes in the structural elements of the active site, thus altering the catalytic activity and the substrate specificity of the mutant enzyme. For example, mutations of Arg164 in the N-terminus of the Ω-loop in the TEM-type BLs and Asp179 in the C-terminus of the Ω-loop in the SHV-type BLs change the loop conformation and cause an increase in the active site volume, allowing formation of stable enzyme complexes with oxyimino-cephalosporins with bulky side groups [18]. Such modifications have resulted in the origin of the extended-spectrum BLs (ESBLs) that can efficiently hydrolyze cephalosporins of the II-IV generations [9]. Another direction in the BL evolution is the emergence of new types of enzymes with an extended substrate specificity, e.g., class A KPC-type and class D OXA-type carbapenemases.
At present, class A serine BLs of the TEM, SHV, CTX-M, and KPC types are the most common BLs. Each type includes several dozens to several hundred enzymes originating from the same ancestor protein (e.g., TEM-1 or SHV-1) and representing mutant forms that contain from one to several amino acid substitutions. The TEM- and SHV-type enzymes are subdivided into the broad-spectrum BLs that hydrolyze only penicillins and generation I cephalosporins, and ESBLs with mutated key residues that hydrolyze penicillins and cephalosporins of the I-IV generations. The SHV-type BLs are characterized by the presence of insertions in the amino acid sequence; in particular, SHV-16 BL contains a repeat of the Asp163- Thr168 fragment of the Ω-loop that includes catalytically important Glu166 [19]. This enzyme is characterized by an increased conformational mobility of the catalytic region, which promotes the availability of the active site to generation III cephalosporins but decreases the thermostability of the enzyme. It might be related to the redistribution of the internal contacts between the amino acids of the loop and their contacts with other amino acids of the protein globule [20].
BLs of the CTX-M type hydrolyze generation III-IV cephalosporins. All these enzymes are ESBLs, and their prevalence in the clinically important infectious agents has now become global [21]. Based on the homology of amino acid sequences, these BLs are subdivided into five subclusters that display lower homology than the TEM- and SHV-type enzymes [22]. The CTX-M enzymes differ in the catalytic activity toward different generation III cephalosporins (cefotaxime, ceftazidime, and cefepime). This BL group is also characterized by the presence of hybrid enzymes composed of fragments of BLs from different subclusters. Thus, CTX-M-116, -123, and -132 are hybrids of CTX-M-15 and CTX-M-14 [23-25]. The key substitutions in these enzymes enhance the efficiency of ceftazidime hydrolysis; the most common substitutions are Asp240Gly and Pro167Ser/Thr in the Ω-loop.
The catalytic residue in class C BLs is Ser64, while Tyr150 acts as a common base and Asn152 (analog of Asn132 in class A BLs) is involved in the correct orientation of the substrate molecule. The catalytic residue in class D BLs is Ser67, but the mechanism of its activation for the subsequent nucleophilic attack on the β-lactam ring remains unclear. The role of the common base in these enzymes presumably belongs to Lys70, the position of which in the enzyme structure is optimal for activating the water molecule and similar to the position of Lys73 in class A BLs.
Class B contains multiple metallo-BLs (MBLs) that have one or two Zn2+ ions as cofactors and hydrolyze three groups of β-lactam antibiotics (penicillins, cephalosporins, and carbapenems) (Fig. 3c) [16, 17]. These enzymes had diverged from a PBP ancestor earlier in the evolution than other BLs and now belong to a large superfamily of metallohydrolases. Based on the primary structure homology, substrate specificity, and number of Zn2+ ions, MBLs are divided into three subclusters: B1, B2, and B3. Enzymes of the B1 and B3 subclusters contain two Zn2+ ions; enzymes of the B2 subcluster contain one Zn2+ ion. MBLs are characterized by a low degree of homology (less than 20% in the subclusters); even the residues participating in the coordination of metal ions are not conserved in the enzymes from different subclusters. VIM, IMP, and NDM enzymes of the B1 subcluster belong to the most clinically important MBLs.
The active site of MBL is located at the bottom of a wide shallow groove between two β-sheets. In the subcluster B1 enzymes, the Zn2+ ion in the first Zn-binding center has a tetrahedral environment formed by His116, His118, and His196; in the second Zn-binding site, Zn2 is penta-coordinated by His263, Cys221, and Asp120 (Fig. 3c) [16, 26]. Coordination of Zn2+ ions involves two water molecules, one of which (bridging water molecule) connects two metal ions, whereas the other (apical water molecule) forms contacts with the second Zn2+ ion. Metal ions in the subcluster B3 enzymes are coordinated by the same residues in the first binding site, whereas in the second binding site, cysteine is substituted by histidine. MBLs of the B1 and B3 subclusters are characterized by the mobile L3 loop located near the entrance to the active site between the α3-helix and β7-sheet. Substrate binding alters the loop conformation, leading to the changes in the effective values of the dielectric permeability and stabilization of the intermediate forms of the enzyme-substrate complex [26, 27].
Antibiotic binding by the two-zinc MBLs occurs with the coordination of the second Zn2+ by the substrate carboxyl group and release of the hydroxide ion (Fig. 3c) [16]. The metal ion pulls the electron density from the carbonyl oxygen atom, thus increasing and stabilizing its positive charge. The hydroxide ion coordinated by the Zn2+ ion, which is a stronger nucleophile than the water molecule, attacks the carbonyl carbon atom of the antibiotic molecule. An intermediate product is formed with a negative charge delocalized in the pyrrolidine ring, followed by the protonation of the C2 atom with the involvement of the water molecule and formation of 1-pyrroline or protonation of the N4 atom and formation of 2-pyrroline.
Mono-zinc MBLs of the B2 subcluster are represented only by carbapenemases. These enzymes efficiently hydrolyze carbapenems but exhibit low activity toward penicillins and cephalosporins, which might be related to the presence of asparagine residue instead of conserved His116 typical of B1 and B3 subclusters of MBL.
The evolution of BLs has favored the development of protective mechanisms in bacteria and promoted the loss of efficiency of the β-lactam antibiotics. According to the predictions by the WHO, by the middle of the XXI century, the number of deaths from the infections caused by microorganisms resistant to antimicrobial preparations could exceed the number of cancer-related deaths, and the humankind will return to the pre-antibiotic era [28]. Taking into account that the latest new class of antibiotics was discovered more than 30 years ago, developing the approaches for the recovery of the activity of β-lactam antibiotics is an urgent task of the global healthcare.
Here, we discuss the approaches for overcoming the resistance to β-lactam antibiotics based on the inhibition of BLs.
INHIBITORS OF β-LACTAMASES
Despite the differences in the primary structure of serine BLs, the action mechanism of these enzymes is rather conserved. Therefore, the original search for the BL inhibitors had been based on the competitive inhibition with β-lactam ring-containing low-molecular-weight compounds that were screened for the ability to form a more stable acyl-enzyme complex resistant to deacetylation.
Generation I inhibitors of serine BLs. Generation I inhibitors of BLs include sulfones and oxapenems with the β-lactam ring in their structure (Fig. 4a). The main representatives of this group are clavulanic acid, sulbactam, and tazobactam.
Fig. 4. a) Structural formulas of the generation I inhibitors of BLs; b) scheme of inhibition of class A serine BLs by β-lactam inhibitors; c) inhibition of class A BLs by clavulanic acid and mechanisms of the acyl-enzyme complex transformation.
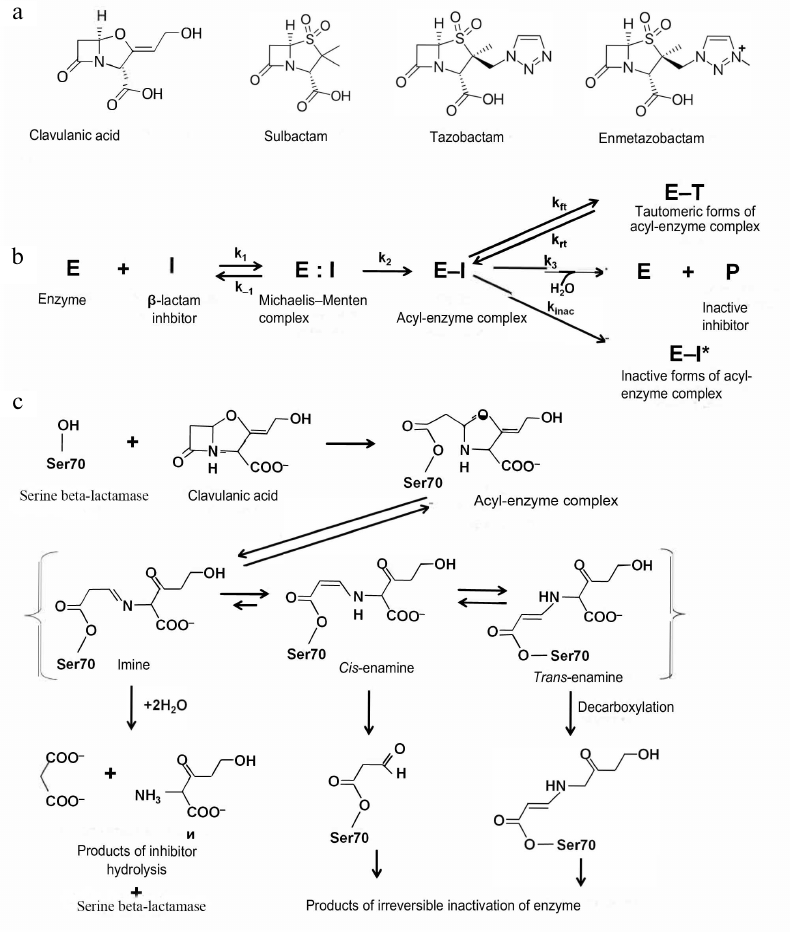
Clavulanic acid was the first inhibitor of serine BLs that was found in 1980s via screening of natural compounds isolated from Streptomyces clavuligerus [29]. It has the β-lactam ring condensed with a 5-membered ring containing oxygen atom and carboxyl group as a substituent (Fig. 4a). Clavulanic acid exhibits low antimicrobial activity, however, when used in a combination with amoxycillin, minimum inhibitory concentration (MIC) of antibiotic against Gram-negative bacteria producing class A BLs significantly decreases [30]. Later, other compounds of this group, sulbactam and tazobactam, were synthesized [31, 32]. These two inhibitors are sulfones of penicillanic acid, in which the β-lactam ring is condensed with a carboxyl group-containing 5-membered ring (Fig. 4a). They differ by substituents in the C2 position (methyl group in sulbactam and triazolyl-containing fragment in tazobactam).
Although these three compounds are inhibitors of class A serine BLs, they display different enzyme specificity: clavulanic acid and tazobactam exhibit comparable inhibitory activity against enzymes of the TEM, SHV, and CTX-M types and are more efficient than sulbactam [33]. However, sulbactam has a higher antibacterial activity and interacts with PBPs. The inhibitory activities of sulbactam and clavulanic acid against the KPC-type enzymes and other class A serine carbapenemases are low.
A new β-lactam inhibitor is the tazobactam derivative enmetazobactam, which is a methylated sulfone of penicillanic acid (Fig. 4a). It easily penetrates into bacterial cells and is active against many class A serine BLs, especially ESBLs [34].
All generation I inhibitors of class A serine BLs have a common inhibition mechanism (Fig. 4b) [17]. The binding of these inhibitors in the enzyme active site is similar to the binding of β-lactam antibiotics. Initially, a non-covalent complex (E:I) is produced, in which the carboxyl fragment of the inhibitor molecule is situated in the conserved region of the active site within the limits of electrostatic interaction with Arg244 and Lys234 and forms hydrogen bonds with Thr235 and Ser130 [35]. The carbonyl oxygen is located in the oxyanion pocket formed by the nitrogen atoms of the peptide backbone of Ser70 and Ala237, providing correct positioning of the carbonyl carbon for the nucleophilic attack by the catalytic Ser70 and formation of the E-I [36]. There are three possible pathways for the acyl-enzyme complex transformations: irreversible inactivation (E-I*); reversible inactivation with the production of the E-T tautomer, and slow deacylation with the regeneration of the active enzyme E. Molecules ensuring irreversible inactivation would be preferable inhibitors of BLs.
The mechanism of the class A serine BL inhibition by clavulanic acid is shown in Fig. 4c. The acyl-enzyme complex is formed by the mechanism similar to the enzyme interaction with the antibiotics: the serine hydroxyl group is activated with participation of Lys73, Ser130, and Glu166 [22]. After acylation, the inhibitor molecule is β-eliminated, leading to the opening of the 5-membered oxazole ring with the formation of the transitory intermediate imine of the acyl-enzyme. This complex can be tautomerized with the generation of intermediate enamines in the trans- or cis-conformation. Trans- and cis-enamines have a conjugated system of double bonds with the carbonyl bond that hinders the nucleophilic attack of the carbonyl carbon by the water molecule and prevents rapid deacylation [37]. Depending on the structure of the BL active site and inhibitor properties, the intermediate imine/enamine complex can regroup several times with the generation of inactive forms of the enzyme or can be deacylated slowly with the regeneration of active BL. There are several possible pathways for the irreversible inactivation, such as formation of enzyme complexes with the 70- or 88-Da inhibitor fragments bound to Ser70, decarboxylation of the enamine form and subsequent slow hydrolysis of the produced esters, and nucleophilic attack of the linear imine-acyl complex by Ser130 resulting in the inhibitor fragmentation and formation of the enzyme complex with the 52-Da inhibitor fragment, which further degrades, leading to the generation of several forms of inactivated enzyme.
Generation I inhibitors are effective against class A serine BLs, but inactive against class C and D serine BLs. Only tazobactam displays a low activity against class C BLs [38], while its derivative enmetazobactam inhibits some BLs of the OXA type [34]. Since the structure of the MBL active site is fundamentally different from that of serine BLs, inhibitors of this group are inefficient against MBLs and, moreover, can be hydrolyzed by them (i.e., serve as substrates) [39].
Inhibitory activity of pharmaceutical combinations of generation I inhibitors with β-lactams. All the above-described β-lactam inhibitors are used in clinical practice as combinations with antibiotics. To be used as combination, the antibiotic and the inhibitor should have similar pharmacokinetic properties, i.e., they should act simultaneously or the inhibitor binding with the enzyme should precede the substrate binding. The most commonly used pharmaceutical combinations are amoxicillin/clavulanic acid, ticarcillin/clavulanic acid, ampicillin/sulbactam, and piperacillin/tazobactam [40, 41]. Recently, a new combination of ceftolozane, a new antibiotic from the cephalosporin group, with tazobactam was approved, which is efficient in the treatment of infections with complications [42]. This combination was shown to be highly efficient against the ESBL-expressing bacteria of the Enterobacteriaceae family and the multidrug resistant Pseudomonas aeruginosa [43, 44]. A combination of cefepime with the new β-lactam inhibitor enmezobactam is now in the phase III of clinical trials; this combination was shown to suppress in vitro the resistance of the Enterobacteriaceae family microorganisms producing the KPC-type carbapenemases [45].
Despite successful clinical use of combinations of penicillin and cephalosporin with the generation I β-lactam inhibitors against class A serine BLs (which still remain the most common BLs), it has become obvious that this approach has serious limitations. Since the action mechanism of all β-lactam inhibitors includes slow phases of acyl-enzyme complex deacylation, none of these inhibitors is irreversible. The evolution of the resistance mechanisms of serine BLs has led to the emergence of mutations of some peripheral residues (e.g., residues 69, 130, 244, 275, and 276 in the TEM-type BLs), leading to the resistance to the β-lactam inhibitors [13]. Mutations of some BL residues disturb the network of hydrogen bonds involved in Ser70 deprotonation, thus hindering formation of the acyl-enzyme complex. Other mutations in BLs affect the distribution of charged groups, resulting in the water molecule shift and decreased affinity for the inhibitors.
The search for competitive inhibitors based on substrate analogs with the same action mechanism was principally limited, as the acyl-enzyme complexes with these inhibitors can be broken through other mechanisms, and the wide use of inhibitors in the pharmaceutical combinations has stimulated the development of resistance to them. Moreover, the search for suitable antibiotic/inhibitor combinations is difficult, since the choice of pairs is limited because of the differences in the pharmacokinetic properties.
The emergence of infectious microorganisms simultaneously resistant to different classes of antibiotics has become a very serious challenge for clinical medicine. As a rule, these bacteria produce several BLs of different classes; therefore, the use of β-lactam inhibitors for their suppression is limited because of narrow specificity, the absence of activity against class B, C, and D BLs, and ability of MBLs to degrade such inhibitors. A new group of β-lactam preparations, carbapenems, has been developed for suppression of the multidrug-resistant microorganisms (Fig. 1) and it has been proposed to use combinations of these antibiotics for the treatment of severe infections. The first preparation developed was a combination of ertapenem and high concentrations of doripenem or meropenem, that was used for the suppression of multidrug-resistant microorganisms of the Enterobacteriaceae family [46]. The following mechanism was suggested: ertapenem characterized by a high affinity for the KPC-type carbapenemases forms stable complexes with these enzymes and inhibits them, whereas meropenem or doripenem act as antibacterial drugs on PBP and suppress bacterial growth. Such combinations have been shown to suppress bacterial growth thousand and ten times in vitro and in vivo, respectively. Clinical trials have confirmed the efficacy of such combined therapy in comparison with other antibacterial preparations, but only against pathogens producing the KPC-type BLs [47].
The new generation of inhibitors. The appearance of a new group of β-lactam antibiotics, carbapenems, for the therapy of severe hospital-acquired infections has led to emergence of carbapenemases, BLs capable of hydrolyzing these new antibiotics. Carbapenemases vary significantly in the structure and belong to different molecular classes. Because of the growth of infections caused by carbapenemase-producing bacteria, it was necessary to create new inhibitors with a broader inhibitory spectrum in comparison with the generation I inhibitors.
Inhibitors of the diazabicyclooctane group. The first non-β-lactam inhibitors of serine BLs that belong to the bridged diazabicyclooctane (DBOs) were proposed in mid-1990s [48]. At present, this group includes avibactam, relebactam, nacubactam, zidebactam, and durlobactam (Fig. 5a) that have a 5-membered bicyclical DBO scaffold as a common structural element. The differences between the inhibitors are determined by the substituents at the C2 atom; durlobactam has a double bond in the DBO scaffold. Instead of the carboxyl group typical for β-lactam inhibitors, these inhibitors have a negatively charged sulfate group.
Fig. 5. a) Structural formulas of the DBO inhibitors; b) scheme of reversible inhibition of serine BLs by avibactam; c) mechanism of inhibition of serine BLs by avibactam.
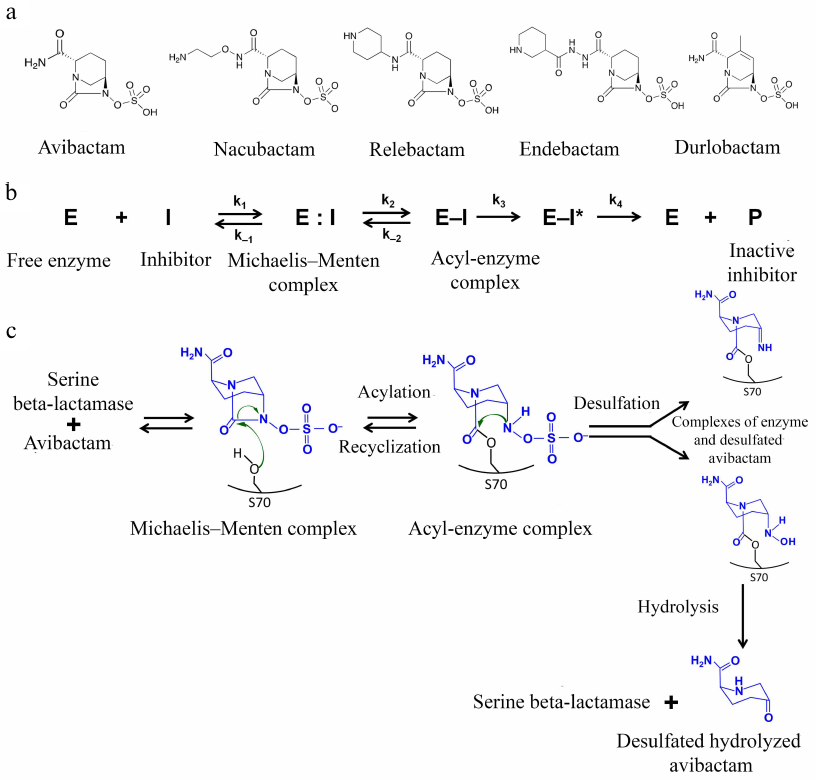
Inhibition of BLs is provided by the interaction of the active site serine with the amide group of the DBO ring (Fig. 5b). The correct orientation of the inhibitor molecule in the active site is ensured by retaining the distance between the carbonyl oxygen and negatively charged oxygen atom of the sulfate group, which is similar to the distance between the equivalent atoms (carbonyl oxygen and carboxylic group oxygen) in β-lactam inhibitors. The carboxamide side chain interacts with the charged Asn/Gln residues of the conserved triad, with the involvement of the catalytic serine (Ser-X-Asn) and Ω-loop. As a result of the acyl-enzyme complex formation with the catalytic serine, the C7-N6 bond is broken and the 5-membered DBO ring opens [49].
The structure of the acyl-enzyme complexes with DBO inhibitors is different from the structure of the enzyme complexes with β-lactam inhibitors: in the former, the sulfate group interacts with the conserved Lys-Thr-Gly triad and the N6 atom interacts with the conserved Ser130 in class A serine BLs or Tyr150 in class C serine BLs. Formation of additional bonds with the inhibitor molecule in the enzyme active site determines the broad specificity of these inhibitors against different BL classes.
Inhibitors of this type are characterized by different mechanisms of the acyl-enzyme complex modification: in the class A BLs, the inhibitor forms a hydrogen bond with the water molecule, and as a result, Glu166 of the Ω-loop is protonated, which makes further deacylation with the participation of the loop residues impossible [50]. Hydrolysis of the inhibitor molecule is also difficult because of the presence of the aromatic system nitrogen atom that is bound to the carbon atom of the carbonyl group. Instead, these inhibitors ensure two other mechanisms for the break of the acyl-enzyme complex (Fig. 5c): recyclization of the DBO ring with the release of the native inhibitor molecule, which can interact again with the enzyme, and slow desulfation resulting in the inhibitor hydrolysis, which prevents its further participation in the suppression of the BL activity. The recyclization is observed in class C and A BLs (except the KPC-type enzymes); it also occurs in the class D enzymes, but at a much lower rate [50]. In the KPC-type enzymes desulfation occurs mainly.
Avibactam. The advantages of avibactam in comparison with the generation I β-lactam inhibitors include a high activity against clinically significant class A CTX-M-15 BL and class C AmpC BL and prolonged half-elimination period [51]. The use of avibactam in a combination with ceftazidime was found to be effective against multidrug-resistant bacteria of the Enterobacteriaceae family and P. aeruginosa [52]. However, avibactam does not inactivate MBLs and most class D enzymes (except OXA-48); therefore, this combination cannot be used against carbapenem-resistant strains of Acinetobacter spp. and P. aeruginosa and MBL-producing strains of Enterobacteriaceae [53].
Currently, the aztreonam/avibactam combination is tested against the multidrug-resistant strains of Enterobacteriaceae producing serine BLs together with MBLs [54]. This choice is determined by the MBL inability to hydrolyze monobactams, and avibactam is added for the inhibition of ESBLs and serine carbapenemases.
Relebactam. Relebactam is different from avibactam in the presence of the piperidine ring. It is used in a combination with imipenem in the treatment of urinary tract and abdominal infections caused by the carbapenem-resistant strains of Enterobacteriaceae and P. aeruginosa producing KPC-type BLs of the class A and BLs of the class C [55]. The imipenem/relabactam combination is active against Klebsiella pneumoniae and Enterobacter spp. resistant to imipenem due to the expression of ESBLs or class C AmpC BLs, as well as due to impaired cell wall permeability. However, this combination was ineffective against Enterobacteriaceae strains expressing OXA-48 BLs or MBLs [56].
Nacubactam, zidebactam, and durlobactam are new inhibitors of the DBO group. Their advantages include inherent antibacterial activity due to the interaction with some PBPs. The nacubactam/meropenem and zidebactam/cefepime combinations are active against class A and C serine BLs as well as MBLs [57, 58]. Zidebactam inhibits class D BL OXA-48. Durlobactam was developed to expand the inhibition spectrum against various class D BLs (OXA-48 bacteria of the Enterobacteriaceae family and OXA-23/24/58 Acinetobacter baumannii) [59]. This inhibitor is used in a combination with sulbactam, which displays an antibacterial activity and interacts with some PBPs of Acinetobacter spp., Neisseria gonorrhoeae, and Haemophilus influenza in addition to the ability to inhibit class A serine BLs [60]. It has been shown that the combination of these two inhibitors efficiently suppresses the multidrug-resistant A. baumannii.
Boronic acids derivatives as inhibitors of BLs. A novel group of BL inhibitors are boronic acid compounds capable of inhibiting microbial serine proteases [61]. However, the first inhibitors of this group produced various side effects because of the inhibition of mammalian serine proteases. Therefore, cyclic boronates have been studied to improve the selectivity of theses inhibitors toward BLs (Fig. 6a). Vaborbactam was the first compound that inhibited class A serine BLs (CTX-M, SHV, TEM, KPC types) and C class BLs, but not mammalian serine proteases [62].
Fig. 6. a) Structures of BL inhibitors based on cyclic derivatives of boronic acid; b) formation of the tetrahedral acyl-enzyme complex of the boron atom of the boronic acid derivatives with the BL active site serine and Zn2+ ion of metallo-BL.
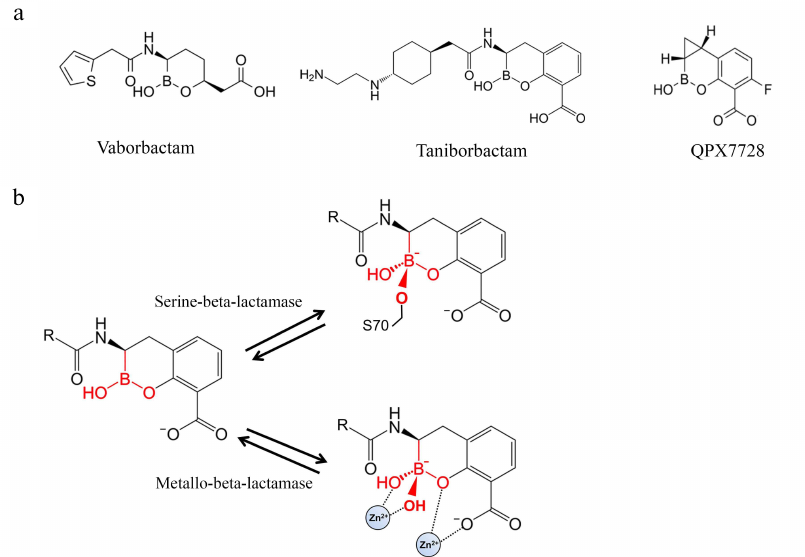
When a boronic acid derivative inhibitor binds to the enzyme active site, its carboxyl fragment is situated in the carboxyl-binding pocket. The aromatic system atoms form hydrophobic contacts with the side groups of Tyr/Trp, and the amide group interacts with Ser237, Asn132, and Asn102 (similarly to the amide group of penicillins) [61]. Boronic acid can form covalent bonds with alcohols; therefore, the boron atom of the inhibitor imitates the carbonyl carbon in β-lactams and acts as a target for the nucleophilic attack of the catalytic serine hydroxyl group. As a result, a tetrahedral complex is produced with the formation of the covalent bond between the serine and the boron atom – an analog of the transitory state of the acyl-enzyme complex of BLs with β-lactams (Fig. 6b). Boronic acid derivatives act as the transitory state inhibitors, because their complex with the enzyme has a tetrahedral conformation imitating the transitory state of the tetrahedral oxyanion in the acylation and deacylation reactions [63].
Screening of cyclic boronates identified inhibitors of all four BL classes, including MBLs [64, 65]. The advantage of these inhibitors is their ability to inhibit PBP5 [61]. Thus, taniborbactam (VNRX-5133) is active against class A and C serine BLs, some class D carbapenemases (OXA-48), and MBLs of the VIM and NDM types produced by carbapenem-resistant bacteria of the Enterobacteriaceae family and P. aeruginosa [66]. This inhibitor is 50-50,000 times more efficient against clinically significant MBLs than vaborbactam.
X-ray crystallographic analysis of complexes of two clinically significant BL (serine CTX-M-15 and MBL VIM-2) with taniborbactam revealed formation of the tetrahedral intermediates with the involvement of boron atom and catalytic serine of BL CTX-M-15 or Zn2+ ion of MBL VIM-2 resulting in the emergence of additional stabilizing interactions, e.g., interactions of the inhibitor N-(2-aminoethyl)cyclohexylamine moiety with negatively charged amino acids (Glu149 in MBLs VIM-2 and NDM-1) [67]. The cefepime/taniborbactam combination was active both in vitro and in vivo against ESBLs and carbapenemase-producing strains of Enterobacteriaceae and P. aeruginosa [66].
QPX7728 is a new cyclic boronate inhibitor that demonstrates a broad inhibitory activity against ESBLs, class A carbapenemases, and class C BLs. Its efficiency is comparable to or even exceeds the efficiency of the DBO group inhibitors (avibactam, relebactam, and vaborbactam) [68]. Unlike other inhibitors, QPX7728 is also active against MBLs (NDM, VIM, IMP, etc.) and class D carbapenemases, in particular, enzymes of the OXA-48 bacteria from the Enterobacteriaceae family and OXA-23/24/58 A. baumannii. A broader inhibitory spectrum of QPX7728 can be due to the more compact structure of this inhibitor because of the absence of bulky substituents, which allows its molecule to enter completely into the active site of the enzyme.
The information on the specificity of serine BL inhibitors is presented in the table. Generation I inhibitors are active mainly against A class BLs (except carbapenemases). A novel inhibitor from this group, enmetazobactam, is active against class A KPC-type carbapenemases and some D class BLs of the OXA-type. Next-generation inhibitors from the DBO group and boronic acid derivatives are characterized by a broader spectrum of action: they inhibit serine BLs, class A carbapenemases, class C BLs, some BLs and carbapenemases of the D class, and MBLs.
Activity of inhibitors of serine BLs and MBLs
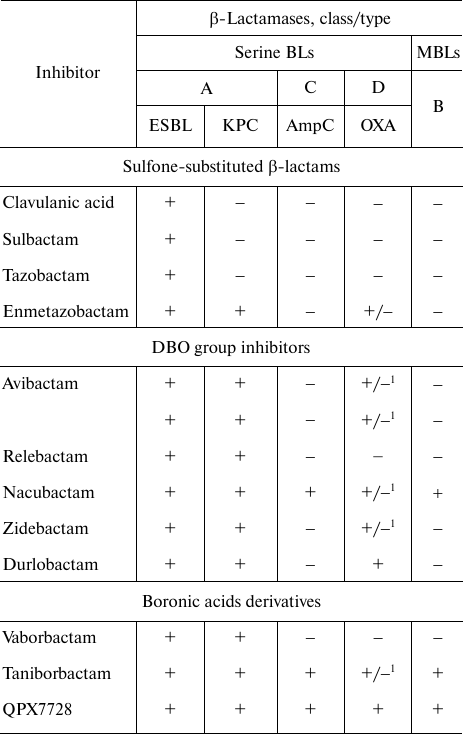
Note. 1) BL OXA-48 (BL OXA-23, OXA-24/40, and OXA-58 are not
inhibited).
Inhibitors of metallo-BLs. The last decades are characterized by the growing expansion of multi- and pan-resistant Gram-negative pathogenic bacteria that synthesize several BLs. Among these enzymes, a special role belongs to highly active MBLs with a broad substrate specificity toward virtually all groups of β-lactams. Generation I inhibitors of serine BLs are almost inactive against class B MBLs; therefore, the search for the inhibitors of these enzymes is especially urgent [69, 70]. By present, several hundred compounds capable of inhibiting MBLs have been described; however, none of them has been approved for the clinical application [16]. The development of MBL inhibitors is difficult because of the structural diversity of these enzymes and differences in the hydrolysis mechanisms. There are two main strategies for searching for potential MBL inhibitors: Zn2+-dependent and Zn2+-independent approaches. Each strategy has limitations. The Zn-dependent inhibitors display non-specific effects more frequently, because many biochemical processes in humans involve metal-dependent enzymes. The search for the Zn-independent inhibitors with a broad specificity is hindered by a limited number of conserved amino acids in MBLs and the presence of a large pocket-shaped active site in these enzymes. Besides, bivalent cations (Ca2+, Mg2+), which are present in high (mM) concentrations in the blood plasma and tissues, can compete for the binding with the Zn-dependent inhibitors.
Zn-dependent inhibitors. Zn-dependent inhibitors of MBLs are subdivided into groups according to their action mechanism: (i) chelating agents extracting metal ions from the active site and (ii) inhibitors competing with antibiotics for the binding with metal ions and amino acids of the active site.
Chelating agents. A common mechanism of MBL inhibition is removal of Zn2+ ions from the active site by chelating agents. The most known chelate, EDTA, cannot be applied in clinics because of its high toxicity. It is used only for the identification of MBLs in the diagnostics of resistant bacteria [71]. Another type of these inhibitors is zinc-selective analogs of spiro-dihydroindole thiadiazole, the efficiency of which has been shown in mouse models [72].
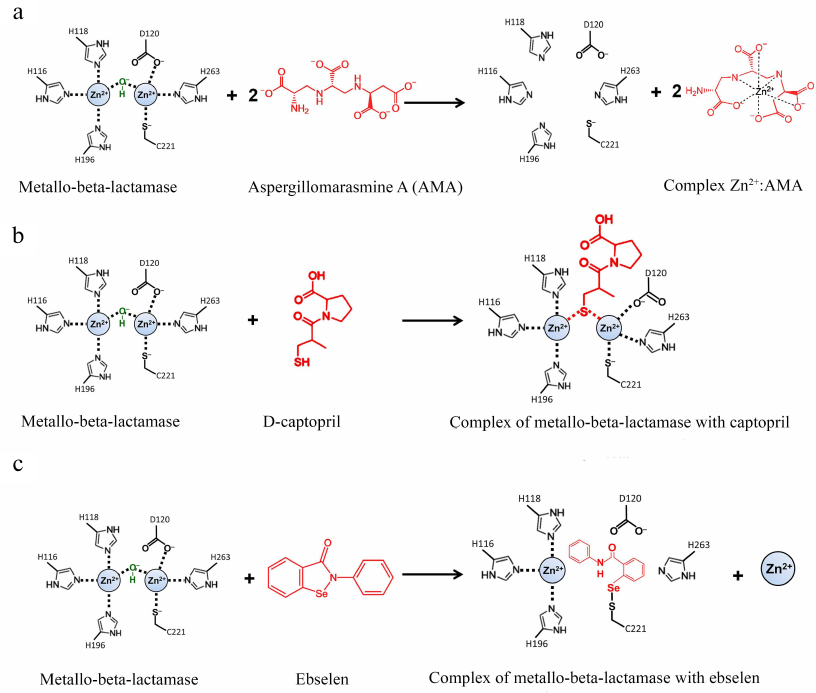
The natural compound aspergillomarasmine A (AMA) is a very promising chelating agent that can efficiently inhibit MBLs of the NDM, VIM, and IMP types [73, 74]. AMA extracts Zn2+ from the enzyme active site; two AMA equivalents can efficiently remove one equivalent of Zn2+ (Fig. 7a).
Fig. 7. Inhibition of metallo-BLs by aspergillomarasmine A (a), D-captopril (b), and ebselen (c).
Other known chelating agents are 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA), 1,4,7-triazacyclononane-1,4,7-triacetic acid (NOTA) [75], and their derivatives [76]. When combined with meropenem, these compounds are active against various pathogenic MBL-producing bacteria. All these compounds demonstrate a low toxicity for mammalian cells.
Zn-binding compounds. Boronic acid cyclic derivatives were described in detail in the previous section. They inhibit both serine BLs and MBLs and are the most promising inhibitors of MBL [64]. Their analogs cyclobutanes are also inhibitors with a broad specificity [77].
Compounds containing thiol groups are Zn-binding inhibitors with a high complex formation constant [78, 79]. L-captopril, which is a known inhibitor of the angiotensin-converting enzyme, belongs to this group [80]. Among four captopril diastereomers, D-captopril was shown to be the most efficient against MBLs and inhibited MBLs of the B1 and B3 (but not B2) subclusters in vitro [81], which might be related to the differences in the action mechanisms of these enzymes (Fig. 7b). In the complex with the two-zinc MBL NDM-1 (B1 subcluster), the thiol group of D-captopril binds two Zn2+ ions and thus substitutes the nucleophilic hydroxide [82]. In the complex with the one-zinc BL CphA (B2 subcluster), the thiol group of D-captopril interacts with the hydrophobic residues of the enzyme active site, whereas the Zn2+ ion is very poorly coordinated by the D-captopril carboxyl group [83].
Other potential broad-spectrum inhibitors of MBLs are some nitrogen-containing heterocyclic compounds, in particular those with the azolylthioacetamide scaffold [84]. These compounds were shown to inhibit NDM-1 and VIM-2 MBLs.
Phosphonate-containing compounds, such as 6-(phosphonomethyl)pyridine-2-carboxylates, were found to possess a high ability for Zn2+ ion binding and inhibition constants for MBLs in the nanomolar range in vitro [85]. Derivatives of carbonic acids [nitrilotriacetic acid and N-(phosphonomethyl)iminodiacetic acid] have similar binding constants for Zn2+ ion and are active in vitro against enzymes of the NDM, VIM, and IMP types. However, it is unlikely that such strong Zn-binding low-molecular carboxylates will be used as pharmaceuticals; more probably, they will be used as a basis for the development of prodrugs that can be activated after penetration into a bacterial cell.
Zn-independent inhibitors. Zn-independent inhibitors include activated 3-mercaptopropionic acid esters that interact with the conserved Lys224 residue in the active site of subcluster B1 MBLs [86]. The selen-containing inhibitor ebselen efficiently inhibits NDM-1 by forming the covalent Se-S bond with the active site residue Cys221 (Fig. 7c) [87]. Some of the inhibitors of subcluster B1 and B2 MBLs have been found to interact simultaneously with Lys224 and Cys221 [88].
Bismuth sulfate colloid solution has been also proposed as an MBL inhibitor. Its action mechanism is based on the displacement of Zn2+ from the NDM-1 active site with the production of inactive Bi3+ complex [89]. It should be noted that ebselen and colloid bismuth sulfate are used in clinical practice for the therapy of other pathologies, which will facilitate the approval of clinical preparations based on these compounds.
The main advantage of novel non-β-lactam inhibitors is their broad inhibitory activity against BLs of various molecular classes (table). Some of these compounds possess an inherent antibacterial activity and act synergistically with β-lactam antibiotics, which gives the latter “the second life” in the treatment of severe infections caused by multidrug-resistant microorganisms [90].
Notwithstanding the achieved progress, the development of preparations for the inhibition of MBLs in vivo still remains a difficult task, in particular, because of the necessity to decrease their affinity for other Zn-containing metalloenzymes, e.g., the angiotensin-converting enzyme.
NEW TRENDS IN THE DEVELOPMENT OF BL INHIBITORS
The beginning of the antibiotic application in clinical practice was extremely successful: several tens of thousands various antibiotics have been synthesized for combating infectious bacteria, and many of these antibiotics have moved on to being produced industrially. However, within 50 years of active use of antimicrobial preparations, bacteria have changed dramatically and many antibiotic-resistant forms have emerged with significantly higher pathogenicity and virulence toward human and animal organisms as a result of an insufficient knowledge and absence of understanding of biochemical and genetic mechanisms of bacterial adaptation to toxic compounds, including antibiotics. Despite the continuing searches for new antibiotics, bacterial resistance remains unconquered. Its main mechanisms are enzymatic hydrolysis and modification of antibiotics, as well as modification of targets attacked by antibiotics [7]. The use of BL inhibitors as an approach for overcoming the resistance of Gram-negative bacteria to β-lactams was proposed about 50 years ago. Despite initial success of the first-generation inhibitors, their application has become limited because of the narrow spectrum of action and development of bacterial resistance to them. The novel generation of inhibitors was more successful and allowed to return β-lactams to the clinical practice as combinations with the inhibitors. However, both the first and the second generations of the inhibitors were directed against the active sites of BLs and therefore, similarly to antibiotics, were subjected to the pressure of the resistance mechanisms.
The promising approaches in the search for new non-β-lactam inhibitors and their targets are computer simulation and virtual screening. Virtual screening based on the characterization of ligand molecules with altered backbone structure using a set of descriptors has allowed us to find structural analogs of the known allosteric inhibitors of class A BLs [91]. Two new non-β-lactam inhibitors were identified that structurally belonged to other chemical classes, but manifested similar biological activity against the same targets and were more efficient against the TEM-type BLs than the initial molecules [92, 93].
Studying the structural and functional features of BLs using new methods of structural biology, physico-chemical biology, and molecular modeling allows to discover new surface “hot points” of the BL protein globule and to use them as new targets for modification of the enzyme activity and stability. The promising targets are mobile loops, because mutations in their sequences can change the catalytic functions of the enzyme and result in the appearance of new features. In serine BLs, such target can be the Ω-loop, the shape of which is provided by fixation of the terminal amino acids closely to each other. The loops with similar shape have been described in 60 proteins, some of them manifesting allosteric regulatory functions [94]. The Ω-loop of serine BLs is situated at the entrance to the enzyme active site and includes catalytically important conserved Glu166, mutations of which lead to almost complete loss of the enzyme activity. The Ω-loop is involved in all stages of the catalytic cycle, and its role is especially critical in the reactions of deacylation and release of the hydrolyzed antibiotic molecule [95]. Despite the differences in the amino acid sequences of BLs of various types, the spatial folding of their Ω-loops is the same. Considering the structure and functions of the Ω-loop, it may be considered as a “mosaic” model of the targets for the action of allosteric inhibitors.
The search for allosteric inhibitors is underway for MBLs, although no targets have been established accurately in this enzyme family [96]. A number of tricyclic natural compounds from the extract of the Chaetomium funicola fungus have been found to inhibit some MBLs (IMP-1 from Bacillus cereus II and P. aeruginosa, CfiA from Bacteroides fragilis) with rather low (µM) inhibition constants [97]. One of these compounds has an affinity for the loop located near the enzyme active site. Macromolecules (peptides, nanoantibodies, aptamers) directed to the regions distant from the active site are also studied as potential allosteric inhibitors. Using the phage display, a nano-antibody was found with the epitope represented by the L6 loop fragments and α2-helices; this nano-antibody was able to inhibit MBL VIM-4 with the micromolar Ki value [98]. A novel approach to the inhibition is the use of constructs based on DNA self-organization that can inhibit different classes of MBLs (including NDM-1) with the IC50 value ~10 nM [99].
The expected worldwide growth in the commercial production of β-lactam antibiotics will inevitably lead to the appearance of new BLs and further mutations in the existing enzymes. Even now, we observe the increasing diversity of class B MBLs [9] and formation of hybrid molecules from different BLs [23-25]. Other variants of evolution of this enzyme superfamily are also probable, but their direction is difficult to predict. This problem must be solved by studying the genetic mechanisms of synthesis and regulation of the new enzymes. Induction of BL synthesis is a complex process that involves the antibiotics themselves, products of the bacterial wall hydrolysis, and enzymes participating in the signaling pathways and regulating expression of bacterial genes. Analysis of these processes will allow to identify new target proteins, the inhibition of which will affect the synthesis of BLs. Considering the expansion of genetic studies on the regulation of bacterial cell biosynthesis, this trend can be very effective in the creation of novel antibacterial preparations that will target the proteins and nucleic acids strictly specific for microorganisms.
CONCLUSION
The volume of commercial produced β-lactam antibiotics is increasing despite the growth of bacterial resistance to them. According to the worldwide predictions, the consumption of β-lactams can double by 2030, and therefore, the problem of developing BL inhibitors based on new principles and new chemical structures is especially urgent. The search for new targets, including allosteric ones, distant from the enzyme active site is a promising approach to broaden the specificity of the inhibitors against different BLs. Creation of novel allosteric inhibitors and their combined use with antibiotics will prolong the “life” of β-lactams, the therapeutic action of which is well studied and advantageous due to a broad specificity, low toxicity and low allergenicity in comparison with other antibiotics.
Funding. This work was supported by the Russian Science Foundation (project no. 15-14-00014-П, Analysis of Inhibitors of Serine β-Lactamases) and State Task of the Lomonosov Moscow State University (AAAA-A16-116052010081-5, Analysis of Inhibitors of Metallo-β-Lactamases).
Ethics declarations. The authors declare no conflict of interests. The article does not contain descriptions of studies with the participation of humans or animals performed by any of the authors.
REFERENCES
1.Smith, P. W., Watkins, K., and Hewlett, A. (2012)
Infection control through the ages, Am. J. Infect. Control,
40, 35-42, doi: 10.1016/j.ajic.2011.02.019.
2.Zapun, A., Contreras-Martel, C., and Vernet, T.
(2008) Penicillin-binding proteins and β-lactam resistance,
FEMS Microbiol. Rev., 32, 361-385, doi:
10.1111/j.1574-6976.2007.00095.x.
3.Bush, K., and Bradford, P. A. (2016) β-Lactams
and β-lactamase inhibitors: an overview, Cold Spring Harb.
Perspect. Medici., 6, a025247, doi:
10.1101/cshperspect.a025247.
4.Klein, E. Y., Van Boeckel, T. P., Martinez, E. M.,
Pant, S., Gandra, S., Levin, S. A., Goossens, H., and Laxminarayan, R.
(2018) Global increase and geographic convergence in antibiotic
consumption between 2000 and 2015, Proc. Natl. Acad. Sci. USA,
115, E3463-E3470, doi: 10.1073/pnas.1717295115.
5.URL: https://www.who.int/emergencies/ten-threats-to-global-health-in-2019.
6.King, D. T., Sobhanifar, S., and Strynadka, N. C.
J. (2017) The mechanisms of resistance to β-lactam antibiotics, in
Handbook of Antimicrobial Resistance A (Berghuis, A.,
Matlashewski, G., Wainberg, M. A., and Sheppard, D., eds.) Springer New
York, pp. 177-201, doi: 10.1007/978-1-4939-0694-9_10.
7.Egorov, A. M., Ulyashova, M. M., and Rubtsova, M.
Y. (2018) Bacterial enzymes and antibiotic resistance, Acta
Naturae, 10, 33-48.
8.Fishovitz, J., Hermoso, J. A., Chang, M., and
Mobashery, S. (2014) Penicillin-binding protein 2a of
methicillin-resistant Staphylococcus aureus, IUBMB Life,
66, 572-577, doi: 10.1002/iub.1289.
9.Bush, K. (2018) Past and present perspectives on
β-lactamases, Antimicrob. Agents Chemother., 62,
e01076-18, doi: 10.1128/AAC.01076-18.
10.Rozwandowicz, M., Brouwer, M. S. M., Fischer, J.,
Wagenaar, J. A., Gonzalez-Zorn, B., Guerra, B., Mevius, D. J., and
Hordijk, J. (2018) Plasmids carrying antimicrobial resistance genes in
Enterobacteriaceae, J. Antimicrob. Chemother., 73,
1121-1137, doi: 10.1093/jac/dkx488.
11.Magiorakos, A.-P., Srinivasan, A., Carey, R. B.,
Carmeli, Y., Falagas, M. E., et al. (2012) Multidrug-resistant,
extensively drug-resistant and pandrug-resistant bacteria: an
international expert proposal for interim standard definitions for
acquired resistance, Clin. Microbiol. Infect., 18,
268-281, doi: 10.1111/j.1469-0691.2011.03570.x.
12.Stec, B., Holtz, K. M., Wojciechowski, C. L., and
Kantrowitz, E. R. (2005) Structure of the wild-type TEM-1
β-lactamase at 1.55 Å and the mutant enzyme Ser70Ala at 2.1
Å suggest the mode of noncovalent catalysis for the mutant
enzyme, Acta Cryst., 61, 1072-1079, doi:
10.1107/S0907444905014356.
13.Egorov, A., Ulyashova, M., and Rubtsova, M.
(2020) Impact of key and secondary drug resistance mutations on
structure and activity of β-lactamases, in Antibiotic Drug
Resistance (Martinez, J. C., and G. Igrejas, G., eds.) John Wiley
and Sons, Inc., pp. 121-140.
14.Abriata, L. A., Salverda, M. L. M., and Tomatis,
P. E. (2012) Sequence-function-stability relationships in proteins from
datasets of functionally annotated variants: the case of TEM
β-lactamases, FEBS Lett., 586, 3330-3335, doi:
10.1016/j.febslet.2012.07.010.
15.Bush, K. (2013) The ABCD’s of
β-lactamase nomenclature, J. Infect. Chemother., 19,
549-559, doi: 10.1007/s10156-013-0640-7.
16.Tooke, C. L., Hinchliffe, P., Bragginton, E. C.,
Colenso, C. K., Hirvonen, V. H. A., Takebayashi, Y., and Spencer, J.
(2019) β-Lactamases and β-lactamase inhibitors in the 21st
century, J. Mol. Biol., 431, 3472-3500, doi:
10.1016/j.jmb.2019.04.002.
17.Drawz, S. M., and Bonomo, R. A. (2010) Three
decades of β-lactamase inhibitors, Clin. Microbiol. Rev.,
23, 160-201, doi: 10.1128/CMR.00037-09.
18.Liakopoulos, A., Mevius, D., and Ceccarelli, D.
(2016) A review of SHV extended-spectrum β-lactamases: neglected
yet ubiquitous, Front. Microbiol., 7, 1374, doi:
10.3389/fmicb.2016.01374.
19.Arpin, C., Labia, R., Andre, C., Frigo, C., El
Harrif, Z., and Quentin, C. (2001) SHV-16, a β-lactamase with a
pentapeptide duplication in the omega loop, Antimicrob. Agents
Chemother., 45, 2480-2485, doi:
10.1128/AAC.45.9.2480-2485.2001.
20.Shcherbinin, D., Veselovsky, A., Rubtsova, M.,
Grigorenko, V., and Egorov, A. (2020) The impact of long-distance
mutations on the Ω-loop conformation in TEM type
β-lactamases, J. Biomol. Struct. Dyn., 38,
2369-2376, doi: 10.1080/07391102.2019.1634642.
21.D’Andrea, M. M., Arena, F., Pallecchi, L.,
and Rossolini, G. M. (2013) CTX-M-type β-lactamases: a successful
story of antibiotic resistance, Int. J. Med. Microbiol.,
303, 305-317, doi: 10.1016/j.ijmm.2013.02.008.
22.Bonnet, R. (2004) Growing group of
extended-spectrum beta-lactamases: the CTX-M enzymes, Antimicrob.
Agents Chemother., 48, 1-14, doi:
10.1128/aac.48.1.1-14.2004.
23.Cantón, R., González-Alba, J. M.,
and Galán, J. C. (2012) CTX- M enzymes: origin and diffusion,
Front. Microbiol., 3, 110, doi:
10.3389/fmicb.2012.00110.
24.Zhao, W. H., and Hu, Z. Q. (2013) Epidemiology
and genetics of CTX- M extended-spectrum β-lactamases in
gram-negative bacteria, Crit. Rev. Microbiol., 39,
79-101, doi: 10.3109/1040841X.2012.691460.
25.Fursova, N., Pryamchuk, S., Kruglov, A., Abaev,
I., Pecherskikh, E., Kartsev, N., Svetoch, E., and Dyatlov, I. (2013)
The novel CTX-M-116 β-lactamase gene discovered in Proteus
mirabilis is composed of parts of the CTX- M-22 and CTX-M-23 genes,
Antimicrob. Agents Chemother., 57, 1552-1555, doi:
10.1128/AAC.01471-12.
26.Palzkill, T. (2013) Metallo-β-lactamase
structure and function, Ann. N. Y. Acad. Sci., 1277,
91-104, doi: 10.1111/j.1749-6632.2012.06796.x.
27.Malabanan, M. M., Amyes, T. L., and Richard, J.
P. (2010) A role for flexible loops in enzyme catalysis, Curr. Opin.
Struct. Biol., 20, 702-710, doi: 10.1016/j.
sbi.2010.09.005.
28.Boucher, H. W., Talbot, G. H., Bradley, J. S.,
Edwards, J. E., Gilbert, D., et al. (2009) Bad bugs, no drugs: no
ESKAPE! An update from the infectious diseases society of America,
Clin. Infect. Dis., 48, 1-12, doi: 10.1086/595011.
29.Reading, C., and Cole, M. (1977) Clavulanic acid:
a beta-lactamase inhibiting beta-lactam from Streptomyces
clavuligerus, Antimicrob. Agents Chemother., 11,
852-857, doi: 10.1128/AAC.11.5.852.
30.Brown, A. G. (1986) Clavulanic acid, a novel
beta-lactamase inhibitor – a case study in drug discovery and
development, Drug Des. Deliv., 1, 1-21.
31.English, A. R., Retsema, J. A., Girard, A. E.,
Lynch, J. E., and Barth, W. E. (1978) CP-45,899, a beta-lactamase
inhibitor that extends the antibacterial spectrum of beta-lactams:
initial bacteriological characterization, Antimicrob. Agents
Chemother., 14, 414-419, doi: 10.1128/AAC.14.3.414.
32.Fisher, J., Belasco, J. G., Charnas, R. L.,
Khosla, S., and Knowles, J. R. (1980) Beta-lactamase inactivation by
mechanism-based reagents, Philos. Trans. R. Soc. Lond. B Biol.
Sci., 289, 309-319, doi: 10.1098/rstb.1980.0048.
33.Payne, D. J., Cramp, R., Winstanley, D. J., and
Knowles, D. J. C. (1994) Comparative activities of clavulanic acid,
sulbactam, and tazobactam against clinically important
β-lactamases, Antimicrob. Agents Chemother., 38,
767-772, doi: 10.1128/AAC.38.4.767.
34.Papp-Wallace, K. M., Bethel, C. R., Barnes, M.
D., Rutter, J. D., Taracila, M. A., et al. (2017) AAI101, a novel
β-lactamase inhibitor: microbiological and enzymatic profiling,
Open Forum Infect. Dis., 4, S375.
35.Manageiro, V., Ferreira, E., Cougnoux, A.,
Albuquerque, L., Caniça, M., and Bonnet, R. (2012)
Characterization of the inhibitor-resistant SHV β-lactamase
SHV-107 in a clinical Klebsiella pneumoniae strain coproducing
GES-7 enzyme, Antimicrob. Agents Chemother., 56,
1042-1046, doi: 10.1128/AAC.01444-10.
36.Rodkey, E. A., Drawz, S. M., Sampson, J. M.,
Bethel, C. R., Bonomo, R. A., and Van Den Akker, F. (2012) Crystal
structure of a preacylation complex of the β-lactamase inhibitor
sulbactam bound to a sulfenamide bond-containing
thiol-β-lactamase, J. Am. Chem. Soc., 134,
16798-16804, doi: 10.1021/ja3073676.
37.Van den Akker, F., and Bonomo, R. A. (2018)
Exploring additional dimensions of complexity in inhibitor design for
serine β-lactamases: mechanistic and intra- and inter-molecular
chemistry approaches, Front. Microbiol., 9, 1-10, doi:
10.3389/fmicb.2018.00622.
38.Grace, M. E., Fu, K. P., Gregory, F. J., and
Hung, P. P. (1987) Interaction of clavulanic acid, sulbactam and
cephamycin antibiotics with beta-lactamases, Drugs Exp. Clin.
Res., 13, 145-148.
39.Docquier, J.-D., and Mangani, S. (2018) An update
on β-lactamase inhibitor discovery and development, Drug
Resist. Updat., 36, 13-29, doi:
10.1016/j.drup.2017.11.002.
40.Horita, N., Shibata, Y., Watanabe, H., Namkoong,
H., and Kaneko, T. (2017) Comparison of antipseudomonal β-lactams
for febrile neutropenia empiric therapy: systematic review and network
meta-analysis, Clin. Microbiol. Infect., 23, 723-729,
doi: 10.1016/j.cmi.2017.03.024.
41.Nimmich, E. B., Bookstaver, P. B., Kohn, J.,
Justo, J. A., Hammer, K. L., Albrecht, H., and Al-Hasan, M. N. (2017)
Development of institutional guidelines for management of gram-negative
bloodstream infections: incorporating local evidence, Hosp.
Pharm., 52, 691-697, doi: 10.1177/0018578717720506.
42.Zhanel, G. G., Chung, P., Adam, H., Zelenitsky,
S., Denisuik, A., et al. (2014) Ceftolozane/Tazobactam: a novel
cephalosporin/β-lactamase inhibitor combination with activity
against multidrug-resistant gram-negative bacilli, Drugs,
74, 31-51, doi: 10.1007/s40265-013-0168-2.
43.Shortridge, D., Duncan, L. R., Pfaller, M. A.,
and Flamm, R. K. (2019) Activity of ceftolozane-tazobactam and
comparators when tested against gram-negative isolates collected from
paediatric patients in the USA and Europe between 2012 and 2016 as part
of a global surveillance programme, Int. J. Antimicrob. Agents,
53, 637-643, doi: 10.1016/j.ijantimicag.2019.01.015.
44.Walkty, A., Adam, H., Baxter, M.,
Lagacé-Wiens, P., Karlowsky, J. A., Hoban, D. J., and Zhanel, G.
G. (2018) In vitro activity of ceftolozane/tazobactam versus
antimicrobial non-susceptible Pseudomonas aeruginosa clinical
isolates including MDR and XDR isolates obtained from across Canada as
part of the CANWARD study, 2008-16, J. Antimicrob. Chemother.,
73, 703-708, doi: 10.1093/jac/dkx468.
45.Tselepis, L., Langley, G. W., Aboklaish, A. F.,
Widlake, E., Jackson, D. E., et al. (2020) In vitro efficacy of
imipenem-relebactam and cefepime-AAI101 against a global collection of
ESBL-positive and carbapenemase-producing Enterobacteriaceae, Int.
J. Antimicrob. Agents, 56, 105925, doi:
10.1016/j.ijantimicag.2020.105925.
46.Bulik, C. C., and Nicolau, D. P. (2011)
Double-carbapenem therapy for carbapenemase-producing Klebsiella
pneumoniae, Antimicrob. Agents Chemother., 55,
3002-3004, doi: 10.1128/AAC.01420-10.
47.De Pascale, G., Martucci, G., Montini, L.,
Panarello, G., Cutuli, S. L., et al. (2017) Double carbapenem as a
rescue strategy for the treatment of severe carbapenemase-producing
Klebsiella pneumoniae infections: a two-center, matched
case-control study, Crit. Care, 21, 1-10, doi:
10.1186/s13054-017-1769-z.
48.Coleman, K. (2011) Diazabicyclooctanes (DBOs): a
potent new class of non-β-lactam β-lactamase inhibitors,
Curr. Opin. Microbiol., 14, 550-555, doi:
10.1016/j.mib.2011.07.026.
49.King, D. T., King, A. M., Lal, S. M., Wright, G.
D., and Strynadka, N. C. J. (2016) Molecular mechanism of
avibactam-mediated β-lactamase inhibition, ACS Infect.
Dis., 1, 175-184, doi: 10.1021/acsinfecdis.5b00007.
50.Lahiri, S. D., Mangani, S., Jahić, H.,
Benvenuti, M., Durand-Reville, T. F., et al. (2015) Molecular basis of
selective inhibition and slow reversibility of avibactam against class
D carbapenemases: a structure-guided study of OXA-24 and OXA-48, ACS
Chem. Biol., 10, 591-600, doi: 10.1021/cb500703p.
51.Livermore, D. M., Mushtaq, S., Warner, M., Zhang,
J., Maharjan, S., Doumith, M., and Woodford, N. (2011) Activities of
NXL104 combinations with ceftazidime and aztreonam against
carbapenemase-producing Enterobacteriaceae, Antimicrob. Agents
Chemother., 55, 390-394, doi: 10.1128/AAC.00756-10.
52.Levasseur, P., Girard, A. M., Miossec, C., Pace,
J., and Coleman, K. (2015) In vitro antibacterial activity of
the ceftazidime-avibactam combination against Enterobacteriaceae,
including strains with well-characterized β-lactamases,
Antimicrob. Agents Chemother., 59, 1931-1934, doi:
10.1128/AAC.04218-14.
53.Zhanel, G. G., Lawson, C. D., Adam, H.,
Schweizer, F., Zelenitsky, S., et al. (2013) Ceftazidime-avibactam: a
novel cephalosporin/β-lactamase inhibitor combination,
Drugs, 73, 159-177, doi: 10.1007/s40265-013-0013-7.
54.Biedenbach, D. J., Kazmierczak, K., Bouchillon,
S. K., Sahm, D. F., and Bradford, P. A. (2015) In vitro activity
of aztreonam-avibactam against a global collection of gram-negative
pathogens from 2012 and 2013, Antimicrob. Agents Chemother.,
59, 4239-4248, doi: 10.1128/AAC.00206-15.
55.Livermore, D. M., Warner, M., and Mushtaq, S.
(2013) Activity of MK-7655 combined with imipenem against
Enterobacteriaceae and Pseudomonas aeruginosa, J. Antimicrob.
Chemother., 68, 2286-2290, doi: 10.1093/jac/dkt178.
56.Lapuebla, A., Abdallah, M., Olafisoye, O.,
Cortes, C., Urban, C., Landman, D., and Quale, J. (2015) Activity of
imipenem with relebactam against gram-negative pathogens from New York
City, Antimicrob. Agents Chemother., 59, 5029-5031, doi:
10.1128/AAC.00830-15.
57.Morinaka, A., Tsutsumi, Y., Yamada, M., Suzuki,
K., Watanabe, T., et al. (2015) OP0595, a new diazabicyclooctane: mode
of action as a serine β-lactamase inhibitor, antibiotic and
β-lactam “enhancer”, J. Antimicrob. Chemother.,
70, 2779-2786, doi: 10.1093/jac/dkv166.
58.Livermore, D. M., Mushtaq, S., Warner, M.,
Vickers, A., and Woodford, N. (2017) In vitro activity of
cefepime/zidebactam (WCK 5222) against gram-negative bacteria, J.
Antimicrob. Chemother., 72, 1373-1385, doi:
10.1093/jac/dkw593.
59.Durand-Réville, T. F., Guler, S.,
Comita-Prevoir, J., Chen, B., et al. (2017) ETX2514 is a broad-spectrum
β-lactamase inhibitor for the treatment of drug-resistant
gram-negative bacteria including Acinetobacter baumannii,
Nat. Microbiol., 2, 17104, doi:
10.1038/nmicrobiol.2017.104.
60.Higgins, P. G., Wisplinghoff, H., Stefanik, D.,
and Seifert, H. (2004) In vitro activities of the
β-lactamase inhibitors clavulanic acid, sulbactam, and tazobactam
alone or in combination with β-lactams against epidemiologically
characterized multidrug-resistant Acinetobacter baumannii
strains, Antimicrob. Agents Chemother., 48, 1586-1592,
doi: 10.1128/AAC.48.5.1586-1592.2004.
61.Hecker, S. J., Reddy, K. R., Totrov, M., Hirst,
G. C., Lomovskaya, O., et al. (2015) Discovery of a cyclic boronic acid
β-lactamase inhibitor (RPX7009) with utility vs class A serine
carbapenemases, J. Med. Chem., 58, 3682-3692, doi:
10.1021/acs.jmedchem.5b00127.
62.Lomovskaya, O., Sun, D., Rubio-Aparicio, D.,
Nelson, K., Tsivkovski, R., Griffith, D. C., and Dudley, M. N. (2017)
Vaborbactam: spectrum of beta-lactamase inhibition and impact of
resistance mechanisms on activity in Enterobacteriaceae, Antimicrob.
Agents Chemother., 61, 1-15, doi: 10.1128/AAC.01443-17.
63.Rojas, L. J., Taracila, M. A., Papp-Wallace, K.
M., Bethel, C. R., Caselli, E., et al. (2016) Boronic acid transition
state inhibitors active against KPC and other Class A
β-lactamases: structure-activity relationships as a guide to
inhibitor design, Antimicrob. Agents Chemother., 60,
1751-1759, doi: 10.1128/AAC.02641-15.
64.Brem, J., Cain, R., Cahill, S., McDonough, M. A.,
Clifton, I. J., et al. (2016) Structural basis of
metallo-β-lactamase, serine-β-lactamase and
penicillin-binding protein inhibition by cyclic boronates, Nat.
Commun., 7, 1-8, doi: 10.1038/ncomms12406.
65.Cahill, S. T., Cain, R., Wang, D. Y., Lohans, C.
T., Wareham, D. W., et al. (2017) Cyclic boronates inhibit all classes
of β-lactamases, Antimicrob. Agents Chemother., 61,
doi: 10.1128/AAC.02260-16.
66.Hamrick, J. C., Docquier, J. D., Uehara, T.,
Myers, C. L., Six, D. A., et al. (2020) VNRX-5133 (Taniborbactam), a
broad-spectrum inhibitor of serine- and metallo-β-lactamases,
restores activity of cefepime in enterobacterales and Pseudomonas
aeruginosa, Antimicrob. Agents Chemother., 64, doi:
10.1128/AAC.01963-19.
67.Liu, B., Trout, R. E. L., Chu, G. H., Mcgarry,
D., Jackson, R. W., et al. (2020) Discovery of taniborbactam
(VNRX-5133): a broad-spectrum serine- and metallo-β-lactamase
inhibitor for carbapenem-resistant bacterial infections, J. Med.
Chem., 63, 2789-2801, doi: 10.1021/acs.jmedchem.9b01518.
68.Tsivkovski, R., Totrov, M., and Lomovskaya, O.
(2020) Biochemical characterization of QPX7728, a new
ultra-broad-spectrum beta-lactamase inhibitor of serine and
metallo-beta-lactamases, Antimicrob. Agents Chemother.,
64, e00130-20, doi: 10.1128/aac.00130-20.
69.Cheng, Z., Thomas, C. A., Joyner, A. R., Kimble,
R. L., Sturgill, A. M., et al. (2020) MBlinhibitors.com, a website
resource offering information and expertise for the continued
development of metallo-β-lactamase inhibitors,
Biomolecules, 10, doi: 10.3390/biom10030459.
70.Ju, L.-C., Cheng, Z., Fast, W., Bonomo, R. A.,
and Crowder, M. W. (2018) The continuing challenge of
metallo-β-lactamase inhibition: mechanism matters, Trends
Pharmacol. Sci., 39, 635-647, doi:
10.1016/j.tips.2018.03.007.
71.Ma, J., McLeod, S., MacCormack, K., Sriram, S.,
Gao, N., Breeze, A. L., and Hu, J. (2014) Real-time monitoring of New
Delhi metallo-β-lactamase activity in living bacterial cells by
1H NMR spectroscopy, Angew. Chem. Int. Ed. Engl.,
53, 2130-2133, doi: 10.1002/anie.201308636.
72.Falconer, S. B., Reid-Yu, S. A., King, A. M.,
Gehrke, S. S., Wang, W., Britten, J. F., Coombes, B. K., Wright, G. D.,
and Brown, E. D. (2015) Zinc chelation by a small-molecule adjuvant
potentiates meropenem activity in vivo against NDM-1-producing
Klebsiella pneumoniae, ACS Infect. Dis., 1,
533-543, doi: 10.1021/acsinfecdis.5b00033.
73.King, A. M., Reid-Yu, S. A., Wang, W., King, D.
T., De Pascale, G., et al. (2014) Aspergillomarasmine A overcomes
metallo-beta-lactamase antibiotic resistance, Nature,
510, 503-506, doi: 10.1038/nature13445.
74.Bergstrom, A., Katko, A., Adkins, Z., Hill, J.,
Cheng, Z., et al. (2018) Probing the interaction of aspergillomarasmine
A with metallo-β-lactamases NDM-1, VIM-2, and IMP-7, ACS
Infect. Dis., 4, 135-145, doi:
10.1021/acsinfecdis.7b00106.
75.Somboro, A. M., Tiwari, D., Bester, L. A.,
Parboosing, R., Chonco, L., et al. (2015) NOTA: a potent
metallo-β-lactamase inhibitor, J. Antimicrob. Chemother.,
70, 1594-1596, doi: 10.1093/jac/dku538.
76.Zhang, E., Wang, M.-M., Huang, S.-C., Xu, S.-M.,
Cui, D.-Y., et al. (2018) NOTA analogue: A first dithiocarbamate
inhibitor of metallo-β-lactamases, Bioorg. Med. Chem.
Lett., 28, 214-221, doi: 10.1016/j.bmcl.2017.10.074.
77.Abboud, M. I., Kosmopoulou, M., Krismanich, A.
P., Johnson, J. W., Hinchliffe, P., et al. (2018) Cyclobutanone mimics
of intermediates in metallo-β-lactamase catalysis,
Chemistry, 24, 5734-5737, doi:
10.1002/chem.201705886.
78.Büttner, D., Kramer, J. S., Klingler, F. M.,
Wittmann, S. K., Hartmann, M. R., et al. (2018) Challenges in the
development of a thiol-based broad-spectrum inhibitor for
metallo-β-lactamases, ACS Infect. Dis., 4, 360-372,
doi: 10.1021/acsinfecdis.7b00129.
79.Liu, S., Jing, L., Yu, Z.-J., Wu, C., Zheng, Y.,
et al. (2018) ((S)-3-Mercapto-2-methylpropanamido)acetic acid
derivatives as metallo-β-lactamase inhibitors: synthesis, kinetic
and crystallographic studies, Eur. J. Med. Chem., 145,
649-660, doi: 10.1016/j.ejmech.2018.01.032.
80.Klingler, F. M., Wichelhaus, T. A., Frank, D.,
Cuesta-Bernal, J., El-Delik, J., et al. (2015) Approved drugs
containing thiols as inhibitors of metallo-β-lactamases: strategy
to combat multidrug-resistant bacteria, J. Med. Chem.,
58, 3626-3630, doi: 10.1021/jm501844d.
81.Brem, J., van Berkel, S. S., Zollman, D., Lee, S.
Y., Gileadi, O., et al. (2016) Structural basis of
metallo-β-lactamase inhibition by captopril stereoisomers,
Antimicrob. Agents Chemother., 60, 142-150, doi:
10.1128/AAC.01335-15.
82.King, D. T., Worrall, L. J., Gruninger, R., and
Strynadka, N. C. J. (2012) New delhi metallo-β-lactamase:
structural insights into β-lactam recognition and inhibition,
J. Am. Chem. Soc., 134, 11362-11365, doi:
10.1021/ja303579d.
83.Liénard, B. M. R., Garau, G., Horsfall,
L., Karsisiotis, A. I ., Damblon, C., et al. (2008) Structural basis
for the broad-spectrum inhibition of metallo-β-lactamases by
thiols, Org. Biomol. Chem., 6, 2282-2294, doi:
10.1039/b802311e.
84.Xiang, Y., Chang, Y.-N., Ge, Y., Kang, J. S.,
Zhang, Y.-L., et al. (2017) Azolylthioacetamides as a potent scaffold
for the development of metallo-β-lactamase inhibitors, Bioorg.
Med. Chem. Lett., 27, 5225-5229, doi:
10.1016/j.bmcl.2017.10.038.
85.Hinchliffe, P., Tanner, C. A., Krismanich, A. P.,
Labbé, G., Goodfellow, V. J., et al. (2018) Structural and
kinetic studies of the potent inhibition of metallo-β-lactamases
by 6-phosphonomethylpyridine-2-carboxylates, Biochemistry,
57, 1880-1892, doi: 10.1021/acs.biochem.7b01299.
86.Kurosaki, H., Yamaguchi, Y., Higashi, T., Soga,
K., Matsueda, S., et al. (2005) Irreversible inhibition of
metallo-β-lactamase (IMP-1) by
3-(3-mercaptopropionylsulfanyl)-propionic acid pentafluorophenyl ester,
Angewandte Chemie, 44, 3861-3864, doi:
10.1002/anie.200500835.
87.Chiou, J., Wan, S., Chan, K. F., So, P. K., He,
D., et al. (2015) Ebselen as a potent covalent inhibitor of New Delhi
metallo-β-lactamase (NDM-1), Chem. Commun., 51,
9543-9546, doi: 10.1039/c5cc02594j.
88.Chen, C., Xiang, Y., Yang, K. W., Zhang, Y.,
Wang, W. M., Su, J. P., Ge, Y., and Liu, Y. (2018) A protein
structure-guided covalent scaffold selectively targets the B1 and B2
subclass metallo-β-lactamases, Chem. Commun., 54,
4802-4805, doi: 10.1039/c8cc01067f.
89.Wang, R., Lai, T. P., Gao, P., Zhang, H., Ho, P.
L., et al. (2018) Bismuth antimicrobial drugs serve as broad-spectrum
metallo-β-lactamase inhibitors, Nat. Commun., 9,
1-12, doi: 10.1038/s41467-018-02828-6.
90.Papp-Wallace, K. M., Nguyen, N. Q., Jacobs, M.
R., Bethel, C. R., Barnes, M. D., et al. (2018) Strategic approaches to
overcome resistance against gram-negative pathogens using
β-lactamase inhibitors and β-lactam enhancers: activity of
three novel diazabicyclooctanes WCK 5153, Zidebactam (WCK 5107), and
WCK 4234, J. Med. Chem., 61, 4067-4086, doi:
10.1021/acs.jmedchem.8b00091.
91.Beshnova, D. A., Carolan, C., Grigorenko, V. G.,
Rubtsova, M. Y., Gbekor, E., Lewis, J., Lamzin, V. S., and Egorov, A.
M. (2019) Scaffold hopping computational approach for searching novel
β-lactamase inhibitors, Biomed. Khim., 65, 468-476,
doi: 10.18097/pbmc20196506468.
92.Grigorenko, V. G., Andreeva, I. P., Rubtsova, M.
Y., Deygen, I. M., Antipin, R. L., et al. (2017) Novel
non-β-lactam inhibitor of β-lactamase TEM-171 based on
acylated phenoxyaniline, Biochimie, 132, 45-53, doi:
10.1016/j.biochi.2016.10.011.
93.Antipin, R. L., Beshnova, D. A., Petrov, R. A.,
Shiryaeva, A. S., Andreeva, I. P., et al. (2017) Synthesis, SAR and
molecular docking study of novel non-β-lactam inhibitors of TEM
type β-lactamase, Bioorg. Med. Chem. Lett., 27,
1588-1592, doi: 10.1016/j.bmcl.2017.02.025.
94.Papaleo, E., Saladino, G., Lambrughi, M.,
Lindorff-Larsen, K., Gervasio, F. L., and Nussinov, R. (2016) The role
of protein loops and linkers in conformational dynamics and allostery,
Chem. Rev., 116, 6391-6423, doi:
10.1021/acs.chemrev.5b00623.
95.Egorov, A., Rubtsova, M., Grigorenko, V., Uporov,
I., and Veselovsky, A. (2019) The role of the Ω-loop in
regulation of the catalytic activity of TEM-type β-lactamases,
Biomolecules, 9, doi: 10.3390/biom9120854.
96.Fast, W., and Sutton, L. D. (2013)
Metallo-β-lactamase: inhibitors and reporter substrates,
Biochim. Biophys. Acta, 1834, 1648-1659, doi:
10.1016/j.bbapap.2013.04.024.
97.Payne, D. J., Hueso-Rodríguez, J. A.,
Boyd, H., Concha, N. O., Janson, C. A., et al. (2002) Identification of
a series of tricyclic natural products as potent broad-spectrum
inhibitors of metallo-β-lactamases, Antimicrob. Agents
Chemother., 46, 1880-1886, doi:
10.1128/AAC.46.6.1880-1886.2002.
98.Sohier, J. S., Laurent, C., Chevigné, A.,
Pardon, E., Srinivasan, V., et al. (2013) Allosteric inhibition of VIM
metallo-β-lactamases by a camelid nanobody, Biochem. J.,
450, 477-486, doi: 10.1042/BJ20121305.
99.Ouyang, X., Chang, Y. N., Yang, K. W., Wang, W.
M., Bai, J. J., et al. (2017) A DNA nanoribbon as a potent inhibitor of
metallo-β-lactamases, Chem. Commun., 53, 8878-8881,
doi: 10.1039/c7cc04483f.