Regulation of Thiamine (Vitamin B1)-Dependent Metabolism in Mammals by p53
V. I. Bunik1,2,3,a*, V. A. Aleshin1,2, X. Zhou4, S. Krishnan4, and A. Karlsson4
1Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia2Faculty of Bioengineering and Bioinformatics, Lomonosov Moscow State University, 119991 Moscow, Russia
3Sechenov First Moscow State Medical University of the Ministry of Health of the Russian Federation (Sechenov University), 119991 Moscow, Russia
4Division of Clinical Microbiology, Department of Laboratory Medicine, Karolinska Institute, Karolinska University Hospital, 141 86 Stockholm, Sweden
* To whom correspondence should be addressed.
Received May 25, 2020; Revised June 9, 2020; Accepted June 9, 2020
Transcriptional factor p53 is a master regulator of energy metabolism. Energy metabolism strongly depends on thiamine (vitamin B1) and/or its natural derivatives. Thiamine diphosphate (ThDP), which is a major thiamine derivative, affects p53 binding to DNA. In order to elucidate the mechanism of regulation of thiamine-dependent metabolism by p53, we assessed putative p53-binding sites near transcription starting points in genes coding for transporters and enzymes, whose function is associated with thiamine and/or its derivatives. The predictions were validated by studying cell metabolic response to the p53 inducer cisplatin. Expression of p53 and its known target, p21, has been evaluated in cisplatin-treated and control human lung adenocarcinoma A549 cells that possess functional p53 pathway. We also investigated the activity of enzymes involved in the thiamine-dependent energy metabolism. Along with upregulating the expression of p53 and p21, cisplatin affected the activities of metabolic enzymes, whose genes were predicted as carrying the p53-binding sites. The activity of glutamate dehydrogenase GDH2 isoenzyme strongly decreased, while the activities of NADP+-dependent isocitrate dehydrogenase (IDH) and malic enzymes, as well as the activity of 2-oxoglutarate dehydrogenase complex at its endogenous ThDP level, were elevated. Simultaneously, the activities of NAD+-dependent IDH, mitochondrial aspartate aminotransferase, and two malate dehydrogenase isoenzymes, whose genes were not predicted to have the p53-binding sequences near the transcription starting points, were upregulated by cisplatin. The p53-dependent regulation of the assayed metabolic enzymes correlated with induction of p21 by p53 rather than induction of p53 itself.
KEY WORDS: A549 cells, cisplatin, glutathione, p53, p21, succinyl phosphonate, thiamine-dependent metabolismDOI: 10.1134/S0006297920070081
Abbreviations: GDH1(2), glutamate dehydrogenase 1(2); GOT, aspartate aminotransferase; GSH, glutathione; GSSG, glutathione disulfide; IDH, isocitrate dehydrogenase; MDH, malate dehydrogenase; OGDHC, 2-oxoglutarate dehydrogenase complex; SP, succinyl phosphonate; ThDP, thiamine diphosphate.
INTRODUCTION
DNA binding of p53 may regulate transcription of several hundreds of genes directly, with the list extended to thousands of those regulated indirectly [1]. The p53-dependent transactivation has been studied mostly for the p53 targets involved in the regulation of the cell fate, such as p53-destabilizing HDM2 protein, cell cycle control p21, cell death/survival regulators PUMA and TIGAR, etc. As the cell fate is closely related to the energy status, it is not surprising that p53 also acts as a master regulator of cellular energy metabolism. Direct p53-dependent activation and repression of corresponding transporters and enzymes have been investigated in several studies [2-5]. Remarkably, one of the p53 targets is the thiamine (vitamin B1) transporter [2]. Major intracellular derivative of thiamine, thiamine diphosphate (ThDP), not only serves as coenzyme of central metabolism, but also regulates p53 binding to DNA [6].
Although most studies on the transcriptional regulation by p53 have been focused on the p53 inactivation in tumorigenesis, some cancer cells, such as non-small cell lung adenocarcinoma A549 cells, have a functional p53 pathway [7, 8]. The p53 pathway is linked to changes in the thiamine-dependent metabolism of tumor cells [9, 10] and their response to cisplatin [8], which is a first-line drug in lung cancers. However, cancer cells develop resistance to cisplatin. Therefore, the p53- and thiamine-dependent metabolic regulation in such cells is of particular interest.
The DECODE proprietary database created by SABiosciences, combines text mining of ChIP-seq data and data from the UCSC Genome Browser, predicting the binding sites for more than 200 transcription factors. In this study, we used predictions of the p53-binding sites in genes encoding proteins involved in the thiamine-regulated metabolism in order to assess the role of p53 in the thiamine-dependent metabolism. The predicted p53-binding sites in the genes encoding enzymes of the thiamine-dependent metabolism, were experimentally verified by assaying the activity of these enzymes after p53 upregulation.
MATERIALS AND METHODS
Bioinformatics search for potential p53-binding sites in genomes. The SABiosciences’ proprietary database DECODE (DECipherment of DNA Elements) from QIAGEN was searched for the p53-binding sites in the proximity of gene transcription starting points as in other studies [11, 12]. The DECODE database combines the results of text mining analysis of ChIP-seq data and the UCSC Genome Browser, providing identification of binding sites for more than 200 transcription factors located closely to the transcription starting points of genes. For human genes, the data were compared with the information provided by GeneCards (https://www.genecards.org/) as “Top Transcription factor binding sites by QIAGEN”.
Cell culture and reagents. Adenocarcinoma non-small cell lung cancer A549 cells (ATCC® CCL-185™) were cultured in Dulbecco’s Modified Eagle’s medium (DMEM; Thermo Fischer Scientific, 21855, USA) containing 1 g/liter glucose, 1 mM pyruvate, 4 mM GlutaMAX™, 10% FBS (Gibco, 10270, USA), 100 U/ml penicillin, and 0.1 mg/ml streptomycin (Thermo Fisher Scientific, 15140, USA) at 37°C in 5% CO2 in a humidified atmosphere.
A549 cells were seeded in 6-well plates (2 × 105 cells per well). After 24 h, the medium was replaced with the fresh one supplemented with 5 µM cisplatin or 4.5 mM succinyl phosphonate (SP). SP was synthesized according to the previously published procedure [13]; its identity and purity were confirmed by NMR spectroscopy. After the cells were cultured for 24 h (approximately to 70% confluence), cells lysates were obtained as described previously [14] using protease and phosphatase inhibitor cocktails cOmplete™ (Roche, 04693116001, Switzerland) and PhosSTOP™ (Roche, PHOSS-RO, Switzerland), respectively, and assayed for the enzyme activity on the same day.
Enzymatic activity assays. Enzyme activities were assayed as described previously [14]. The activity of the OGDHC holoenzyme was determined at the endogenous ThDP level by omitting ThDP in the reaction medium. Glutamate dehydrogenase 2 (GDH2) activity was assayed by adding 1 µM GTP [to inhibit glutamate dehydrogenase 1 (GDH1)] and 100 µM ADP (to activate GTP-resistant GDH2), based on different regulatory properties of the two enzymes [15]. The activity of NAD+-dependent isocitrate dehydrogenase (IDH) was determined as described by Cox et al. [16] in HEPES buffer. NADP+-dependent IDHs were assayed as described by Artiukhov et al. [17]. NAD+, NADP+, ADP, GTP, NADH, 2-oxoglutarate, DL-isocitrate, malate, oxaloacetate, CoA, and thiamine pyrophosphate were from Sigma-Aldrich.
Total protein content was determined with a Bio-Rad Protein Assay Kit I, 5000001 (Bio-Rad, USA), according to manufacturer’s instructions, and used for normalization of enzymatic activities.
p53, p21, and GOT2 assays. The levels of p53 and aspartate aminotransferase (GOT2) proteins were determined with anti-p53 (Santa Cruz Biotechnology, sc-126, USA) and anti-GOT2 (Sigma-Aldrich, HPA018139, USA) primary antibodies. Actin and VDAC/porin stained with anti-β-actin (Sigma-Aldrich, A5441) and anti-VDAC/porin (Cell Signaling Technology, 4661, USA) antibodies, respectively, were used as loading controls.
The level of p21 mRNA was analyzed by RT-qPCR using 5′-CAGACCAGCATGACAGATTTC-3′ and 5′-TTAGGGCTTCCTCTTGGAGA-3′ primers (Kapa Biosystems, USA). RNA was isolated with RNeasy mini kit (74106, QIAGEN, The Netherlands). β-Actin and S18 mRNAs were used for normalization.
Assays of glutathione and glutathione disulfide. The concentration of glutathione (GSH) was determined using 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) as described by Boiko et al. [18]. Glutathione disulfide (GSSG) was quantified from the fluorescence of the product of its reaction with o-phthalic aldehyde according to the method of Hissin and Hilf [19] optimized by Senft et al. [20].
Statistical analysis. The data are presented as mean ± SEM. Independent experiments were performed with different batches of cells. Statistical analysis was carried out using the GraphPad Prism program, version 7.0 (GraphPad Software Inc., USA). The data for the control and treatment groups were compared using the two-tailed Student’s t-test with the Holm–Sidak correction for multiple comparisons. All data passed the D’Agostino omnibus normality test.
RESULTS AND DISCUSSION
Enzymes and transporters that are regulated by thiamine and its natural derivatives or require these compounds for their activity, are shown in the table. In addition to the known enzymes that use ThDP (main intracellular derivative of thiamine) as a coenzyme, the table includes metabolic enzymes allosterically or competitively regulated by thiamine and its derivatives [21, 22], as well as metabolic partners of thiamine-dependent enzymes and currently known thiamine-metabolizing enzymes and transporters.
Putative p53-DNA binding sites in the proximity of transcription
starting points of the genes encoding proteins of thiamine-dependent
metabolism in the human, mouse, and rat genomes
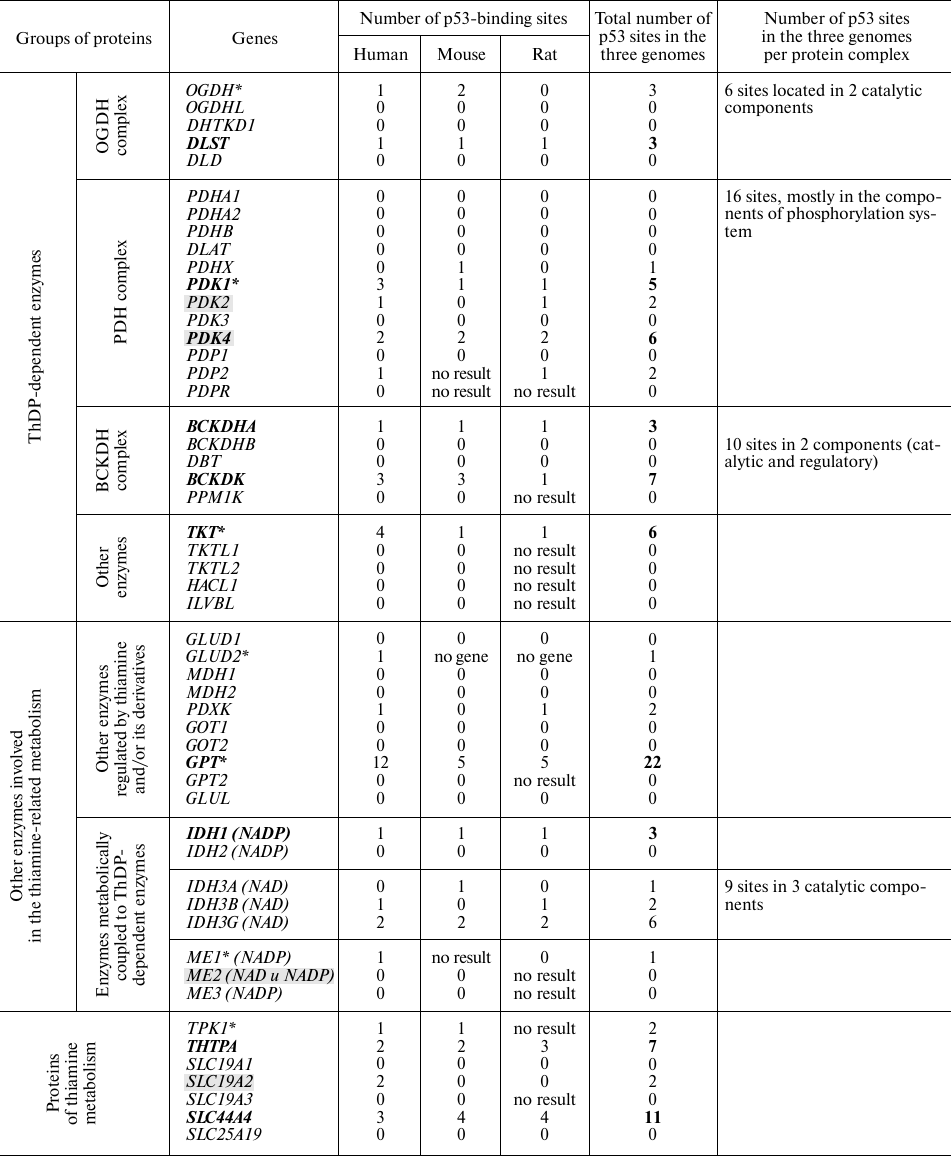
Note. Asterisks denote p53-binding sites according to GeneCards
(https://www.genecards.org/). Genes consistently predicted as
transcriptionally regulated by p53 are shown in bold. Direct
transcriptional targets of p53 (according to published papers) are
marked by grey shading.
Assuming that p53-dependent regulation is conserved in mammalian species, we evaluated the presence of p53-binding sites near the gene transcription starting points in three well-characterized mammalian genomes (human, mouse, and rat) using the DECODE data. Multiple hits for such p53-binding sites were considered as indicators of a higher probability of the p53-mediated gene regulation. The total number of potential p53-binding sites near the transcription starts in the three mammalian genomes (table) was considered as a cumulative marker of the prediction consistency. The numbers of p53-binding sites in the genes encoding for the components of heteroprotein complexes (including regulatory enzymes, such as kinases and phosphatases) in all the three genomes were summed up. For instance, transcription of the first and the second enzymatic components of the OGDH multienzyme complex was predicted as regulated by p53 (table). This increases the probability of p53-dependent regulation for the major form of this multienzyme complex that contains the OGDH gene product, compared to the complexes formed by the OGDH isoenzymes encoded by recently characterized OGDHL and DHTKD1 genes [17]. However, the activities of the OGDHL and DHTKD1 complexes may also depend on the p53-regulated transcription of their second enzymatic component encoded by the DLST gene. The predicted p53-binding sites in the genes of ThDP-dependent multienzyme 2-oxo acid dehydrogenase complexes (table) are in good agreement with the existing data on the p53-induced upregulation of these genes in the chemical models of thiamine deficiency [23].
Compared to the information extracted from the DECODE database, predictions stored in GeneCards exhibit a lower sensitivity. Thus, the top hits revealed by GeneCards did not include known p53 direct targets, such as SLC19A2 [2], PDK2, and PDK4 [4, 5, 24] that have from two to six predicted p53-binding sites according to the DECODE database (table).
To test the bioinformatics prediction on the regulation of thiamine-related genes by p53, we used cisplatin, a well-known p53 inducer, in A549 cells [8].
Incubation of A549 cells with 5 µM cisplatin (IC50 for cisplatin in A549 cells is 18 µM [25]) upregulated p53 expression, leading to the induction of its target p21 (cyclin-dependent kinase inhibitor 1A) (Fig. 1a). Under these conditions, the redox potential of cellular glutathione (GSH/GSSG) decreased, which was in accordance with the molecular mechanism of cisplatin action, since cisplatin is known to induce oxidative stress [26]. However, the levels of both oxidized and reduced glutathione increased, although the former more than the latter (Fig. 1a). Hence, cisplatin activated glutathione biosynthesis, which is controlled by the interaction between the ROS/p53/p21 and Nrf pathways [27]. By assaying the activity of enzymes selected from those listed in the table, we revealed the metabolic targets of p53 involved in such regulation.
Fig. 1. Changes in the p53 and p21 protein levels, cellular glutathione status and activities or expression of enzymes related to the thiamine-dependent metabolism after 24-h incubation with 5 µM cisplatin (a) or 4.5 mM SP (OGDHC inhibitor) (b); * p < 0.05. Abbreviations: GDH, glutamate dehydrogenase; IDH, isocitrate dehydrogenase; ME, malic enzymes (all three isoenzymes); MDH, malate dehydrogenase; OGDHC, 2-oxoglutarate dehydrogenase complex; GOT, aspartate aminotransferase; GSH, glutathione; GSSG, glutathione disulfide. Numbers refer to the isoenzymes as in the table.
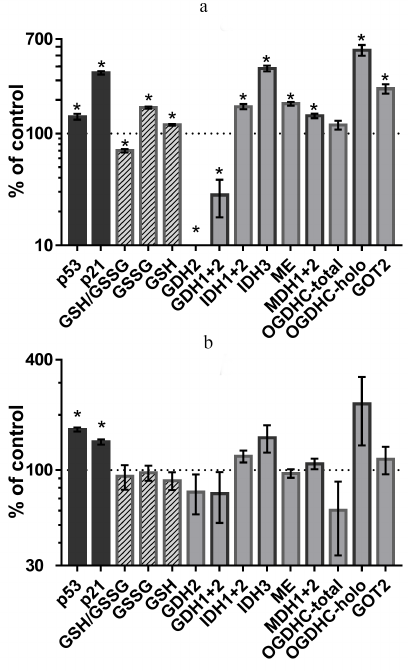
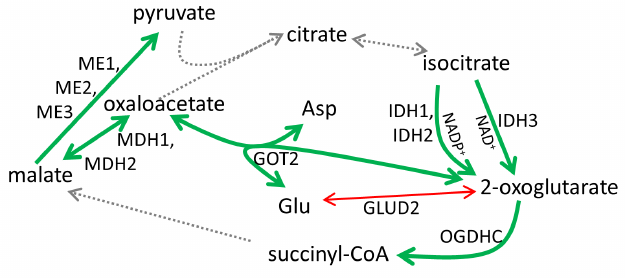
As shown in Figs. 1a and 2, cisplatin induced significant changes in activities of the enzymes of the glutamate/2-oxoglutarate node and affiliated malate metabolism. Cisplatin increased the flux through the OGDHC by increasing its saturation with the coenzyme (ThDP) from 23 ± 9 to 100% (p < 0.05). Considering that no increase in the total activity of the OGDHC was observed, the higher extent of enzyme saturation with endogenous ThDP is in good agreement with the p53-dependent increase in the thiamine influx [2]. Other metabolic enzymes known to be controlled by p53, such as NADP+-dependent IDHs and malic enzymes [28, 29], were also upregulated along with p53 and p21 induction (Fig. 1a). At the same time, the human-specific isoenzyme of glutamate dehydrogenase (GDH2) predicted to have the p53-binding site (table), was strongly downregulated by cisplatin (Fig. 1a). The effect on the total GDH activity was less pronounced, in accordance with the predicted absence of transcriptional regulation of GDH1 by p53 (table). The activity of the OGDHC holoenzyme increased 6-fold (p < 0.02), while the activity of TCA cycle isoenzyme (IDH3) that catalyzes formation of the OGDHC substrate 2-oxoglutarate, increased 4-fold following p53 induction by cisplatin (p < 0.01). At the same time, activities of several enzymes whose expression was not predicted to depend on p53, such as malate dehydrogenase (MDH) isoenzymes (1.4-fold, p < 0.02) and GOT2 (2.5-fold, p < 0.01) (table), increased, too. One of these enzymes, MDH1, is known to be a co-activator of p53 transcriptional activity [30, 31]. The mechanisms of the observed upregulation of these enzymes require further studies. These enzymes may be indirect targets of p53. Alternatively, their genes may have the p53-binding sites beyond the transcription starting points considered in the DECODE predictions (table). Supported by chromatin structural organization, the regulatory p53-binding sites may also be located in other regions of the genome. Overall, the observed cisplatin-induced changes in the homeostasis of A549 cells (Fig. 1a) pointed to the p53-dependent upregulation of the production and oxidation of 2-oxoglutarate in the TCA cycle, simultaneously limiting 2-oxoglutarate generation from glutamate by GDH2. Such changes are in good accord with a higher glutamate provision to the glutathione biosynthesis in the cisplatin-induced oxidative stress.
Fig. 2. Metabolic rearrangements in response to p53 activation by cisplatin in A549 cells. Bold green arrows, upregulation; thin red arrows, downregulation; dashed arrows, not assayed. For enzyme abbreviations, see Fig. 1 and the table.
Depending on the signal, upregulation of p53 expression may result in different outcomes, which may be regulated by post-translational modifications of p53 and its partners proteins involved in DNA binding [1]. Indeed, compared to the action of cisplatin (Fig. 1a), incubation of A549 cells with 4.5 mM SP (specific inhibitor of OGDHC [13]) resulted in a similar level of p53 upregulation, which, however, led to a less pronounced upregulation of p21 (Fig. 1b). This effect was observed in the absence of statistically significant changes in the glutathione homeostasis and levels of studied enzymes. However, the overall patterns of changes in the enzyme activities in the cells treated with cisplatin (Fig. 1a) and SP (Fig. 1b), were similar. Particularly noticeable was induction of the OGDHC holoenzyme and IDH3. OGDHC saturation with endogenous ThDP increased from 23 ± 9% to 78 ± 16% (p = 0.6) after the SP treatment, which was accompanied with a 1.5-fold (although, statistically insignificant) increase in IDH3 activity. Thus, SP induced p53 expression, similar to cisplatin. This in turn affected the activity of the OGDHC holoenzyme, for which the changes were the most pronounced compared to other enzymatic activities tested. The data suggest that the p53-controlled increase in the intracellular thiamine via upregulation of SLC19A2 [2] is the first-line defense against impairments in mitochondrial metabolism caused by the OGDHC inhibition. Indeed, increased thiamine influx was shown to occur upon SP administration to cells or animals [32]. Interestingly, cisplatin is also known to inhibit the OGDHC [33].
Overall, our experimental study on the metabolic changes after the cisplatin induction of p53 in A549 cells (Figs. 1 and 2) shows a validity of predicting the p53-binding sites using the DECODE database (table). The bioinformatics analysis of genome is a valuable tool for both planning and interpretation of results of complex biological experiments. Our data on the p53 regulation of the thiamine-dependent metabolism in A549 cells exposed to cisplatin, which remains the first line drug in lung cancer, provide insights into the metabolic features of cancer cells and molecular mechanisms of the cisplatin action.
Acknowledgements. The study was supported by the Swedish Cancer Society (grant CAN 2016/1342-1345 to Anna Karlsson) and Russian Science Foundation (project No. 18-14-00116 to Victoria I. Bunik).
Ethics declarations. The authors declare no conflict of interest. All international, national, and institutional guidelines for the use of cell cultures were followed. Cell culture experiments presented in this study were performed at the Karolinska Institute and approved by the local authorities.
REFERENCES
1.Beckerman, R., and Prives, C. (2010)
Transcriptional regulation by p53, Cold Spring Harb. Perspect.
Biol., 2, a000935,
doi: 10.1101/cshperspect.a000935.
2.Lo, P. K., Chen, J. Y., Tang, P. P., Lin, J., Lin,
C. H., Su, L. T., Wu, C. H., Chen, T. L., Yang, Y., and Wang, F. F.
(2001) Identification of a mouse thiamine transporter gene as a direct
transcriptional target for p53, J. Biol. Chem., 276,
37186-37193, doi: 10.1074/jbc.M104701200.
3.Itahana, Y., and Itahana, K. (2018) Emerging roles
of p53 family members in glucose metabolism, Int. J. Mol. Sci.,
19, doi: 10.3390/ijms19030776.
4.Janky, R., Verfaillie, A., Imrichova, H., Van de
Sande, B., Standaert, L., Christiaens, V., Hulselmans, G., Herten, K.,
Naval Sanchez, M., Potier, D., Svetlichnyy, D., Kalender Atak, Z.,
Fiers, M., Marine, J. C., and Aerts, S. (2014) iRegulon: from a gene
list to a gene regulatory network using large motif and track
collections, PLoS Comput. Biol., 10, e1003731,
doi: 10.1371/journal.pcbi.1003731.
5.Wang, B., Niu, D., Lam, T. H., Xiao, Z., and Ren,
E. C. (2014) Mapping the p53 transcriptome universe using p53 natural
polymorphs, Cell Death Differ., 21, 521-532,
doi: 10.1038/cdd.2013.132.
6.McLure, K. G., Takagi, M., and Kastan, M. B. (2004)
NAD+ modulates p53 DNA binding specificity and function,
Mol. Cell. Biol., 24, 9958-9967,
doi: 10.1128/MCB.24.22.9958-9967.2004.
7.Breen, L., Heenan, M., Amberger-Murphy, V., and
Clynes, M. (2007) Investigation of the role of p53 in chemotherapy
resistance of lung cancer cell lines, Anticancer Res.,
27, 1361-1364.
8.Guntur, V. P., Waldrep, J. C., Guo, J. J., Selting,
K., and Dhand, R. (2010) Increasing p53 protein sensitizes non-small
cell lung cancer to paclitaxel and cisplatin in vitro,
Anticancer Res., 30, 3557-3564.
9.Jonus, H. C., Hanberry, B. S., Khatu, S., Kim, J.,
Luesch, H., Dang, L. H., Bartlett, M. G., and Zastre, J. A. (2018) The
adaptive regulation of thiamine pyrophosphokinase-1 facilitates
malignant growth during supplemental thiamine conditions,
Oncotarget, 9, 35422-35438,
doi: 10.18632/oncotarget.26259.
10.Duan, L., Perez, R. E., Chen, L., Blatter, L. A.,
and Maki, C. G. (2018) p53 promotes AKT and SP1-dependent metabolism
through the pentose phosphate pathway that inhibits apoptosis in
response to Nutlin-3a, J. Mol. Cell Biol., 10, 331-340,
doi: 10.1093/jmcb/mjx051.
11.Ye, L., Gu, L., Caprioli, J., and Piri, N. (2018)
RNA-binding protein Rbpms is represented in human retinas by isoforms A
and C and its transcriptional regulation involves Sp1-binding site,
Mol. Genet. Genom., 293, 819-830,
doi: 10.1007/s00438-018-1423-8.
12.Sidibe, A., Ropraz, P., Jemelin, S., Emre, Y.,
Poittevin, M., Pocard, M., Bradfield, P. F., and Imhof, B. A. (2018)
Angiogenic factor-driven inflammation promotes extravasation of human
proangiogenic monocytes to tumours, Nat. Commun., 9, 355,
doi: 10.1038/s41467-017-02610-0.
13.Bunik, V. I., Denton, T. T., Xu, H., Thompson, C.
M., Cooper, A. J., and Gibson, G. E. (2005) Phosphonate analogues of
alpha-ketoglutarate inhibit the activity of the alpha-ketoglutarate
dehydrogenase complex isolated from brain and in cultured cells,
Biochemistry, 44, 10552-10561,
doi: 10.1021/bi0503100.
14.Aleshin, V. A., Artiukhov, A. V., Oppermann, H.,
Kazantsev, A. V., Lukashev, N. V., and Bunik, V. I. (2015)
Mitochondrial impairment may increase cellular NAD(P)H: resazurin
oxidoreductase activity, perturbing the NAD(P)H-based viability assays,
Cells, 4, 427-451, doi: 10.3390/cells4030427.
15.Plaitakis, A., Latsoudis, H., and Spanaki, C.
(2011) The human GLUD2 glutamate dehydrogenase and its regulation in
health and disease, Neurochem. Int., 59, 495-509,
doi: 10.1016/j.neuint.2011.03.015.
16.Cox, G. F., and Davies, D. D. (1967)
Nicotinamide-adenine dinucleotide-specific isocitrate dehydrogenase
from pea mitochondria. Purification and properties, Biochem. J.,
105, 729-734, doi: 10.1042/bj1050729.
17.Artiukhov, A. V., Grabarska, A., Gumbarewicz, E.,
Aleshin, V. A., Kahne, T., Obata, T., Kazantsev, A. V., Lukashev, N.
V., Stepulak, A., Fernie, A. R., and Bunik, V. I. (2020) Synthetic
analogues of 2-oxo acids discriminate metabolic contribution of the
2-oxoglutarate and 2-oxoadipate dehydrogenases in mammalian cells and
tissues, Sci. Rep., 10, 1886,
doi: 10.1038/s41598-020-58701-4.
18.Boyko, A., Ksenofontov, A., Ryabov, S., Baratova,
L., Graf, A., and Bunik, V. (2017) Delayed influence of spinal cord
injury on the amino acids of NO(*) metabolism in rat cerebral cortex is
attenuated by thiamine, Front. Med., 4, 249,
doi: 10.3389/fmed.2017.00249.
19.Hissin, P. J., and Hilf, R. (1976) A fluorometric
method for determination of oxidized and reduced glutathione in
tissues, Anal. Biochem., 74, 214-226,
doi: 10.1016/0003-2697(76)90326-2.
20.Senft, A. P., Dalton, T. P., and Shertzer, H. G.
(2000) Determining glutathione and glutathione disulfide using the
fluorescence probe o-phthalaldehyde, Anal. Biochem., 280,
80-86, doi: 10.1006/abio.2000.4498.
21.Mkrtchyan, G., Aleshin, V., Parkhomenko, Y.,
Kaehne, T., Di Salvo, M. L., Parroni, A., Contestabile, R., Vovk, A.,
Bettendorff, L., and Bunik, V. (2015) Molecular mechanisms of the
non-coenzyme action of thiamin in brain: biochemical, structural and
pathway analysis, Sci. Rep., 5, 12583,
doi: 10.1038/srep12583.
22.Aleshin, V. A., Mkrtchyan, G. V., and Bunik, V.
I. (2019) Mechanisms of the non-coenzyme action of thiamin: protein
targets and medical significance, Biochemistry (Moscow),
84, doi: 10.1134/S0320972519080013.
23.Chornyy, S., Parkhomenko, Y., and Chorna, N.
(2017) Thiamine antagonists trigger p53-dependent apoptosis in
differentiated SH-SY5Y cells, Sci. Rep., 7, 10632,
doi: 10.1038/s41598-017-10878-x.
24.Contractor, T., and Harris, C. R. (2012) p53
Negatively regulates transcription of the pyruvate dehydrogenase kinase
Pdk2, Cancer Res., 72, 560-567,
doi: 10.1158/0008-5472.CAN-11-1215.
25.Zhou, X., Paredes, J. A., Krishnan, S., Curbo,
S., and Karlsson, A. (2015) The mitochondrial carrier SLC25A10
regulates cancer cell growth, Oncotarget, 6, 9271-9283,
doi: 10.18632/oncotarget.3375.
26.Chen, B., Shen, Z., Wu, D., Xie, X., Xu, X., Lv,
L., Dai, H., Chen, J., and Gan, X. (2019) Glutathione peroxidase 1
promotes NSCLC resistance to cisplatin via ROS-induced activation of
PI3K/AKT pathway, Biomed Res. Int., 2019, 7640547,
doi: 10.1155/2019/7640547.
27.Jang, D. E., Song, J., Park, J. W., Yoon, S. H.,
and Bae, Y. S. (2020) Protein kinase CK2 activates Nrf2 via autophagic
degradation of Keap1 and activation of AMPK in human cancer cells,
BMB Rep., 53, 272-277.
28.Kil, I. S., Huh, T. L., Lee, Y. S., Lee, Y. M.,
and Park, J. W. (2006) Regulation of replicative senescence by
NADP+-dependent isocitrate dehydrogenase, Free Radic.
Biol. Med., 40, 110-119,
doi: 10.1016/j.freeradbiomed.2005.08.021.
29.Jiang, P., Du, W., Mancuso, A., Wellen, K. E.,
and Yang, X. (2013) Reciprocal regulation of p53 and malic enzymes
modulates metabolism and senescence, Nature, 493,
689-693, doi: 10.1038/nature11776.
30.Lee, S. M., Dho, S. H., Ju, S. K., Maeng, J. S.,
Kim, J. Y., and Kwon, K. S. (2012) Cytosolic malate dehydrogenase
regulates senescence in human fibroblasts, Biogerontology,
13, 525-536, doi: 10.1007/s10522-012-9397-0.
31.Lee, S. M., Kim, J. H., Cho, E. J., and Youn, H.
D. (2009) A nucleocytoplasmic malate dehydrogenase regulates p53
transcriptional activity in response to metabolic stress, Cell Death
Differ., 16, 738-748, doi: 10.1038/cdd.2009.5.
32.Mkrtchyan, G., Graf, A., Bettendorff, L., and
Bunik, V. (2016) Cellular thiamine status is coupled to function of
mitochondrial 2-oxoglutarate dehydrogenase, Neurochem. Int.,
101, 66-75, doi: 10.1016/j.neuint.2016.10.009.
33.Zhang, L., Cooper, A. J., Krasnikov, B. F., Xu,
H., Bubber, P., Pinto, J. T., Gibson, G. E., and Hanigan, M. H. (2006)
Cisplatin-induced toxicity is associated with platinum deposition in
mouse kidney mitochondria in vivo and with selective
inactivation of the alpha-ketoglutarate dehydrogenase complex in
LLC-PK1 cells, Biochemistry, 45, 8959-8971,
doi: 10.1021/bi060027g.