Structural Organization and Dynamic Characteristics of the Binding Site for Conformational Rearrangement Inhibitors in Hemagglutinins from H3N2 and H7N9 Influenza Viruses
D. D. Podshivalov1,a*, E. M. Kirilin1,2, S. I. Konnov1, and V. K. Švedas1,2,b*
1Lomonosov Moscow State University, Faculty of Bioengineering and Bioinformatics, 119991 Moscow, Russia2Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received February 4, 2020; Revised March 2, 2020; Accepted March 2, 2020
Computer models of hemagglutinins from the H3N2 and H7N9 influenza viruses were developed to study structural organization and dynamic characteristics of the binding site for the conformational rearrangement inhibitors. The metadynamics was used to map the binding site free energy and to define the volume of its most energetically favorable states. It was demonstrated by simulation of the umifenovir (Arbidol) interaction with hemagglutinin that ligand binding requires an increase in the binding site volume and deformation of its most energetically favorable state. We also identified amino acid residues directly involved in the ligand binding that determine the binding efficiency, as well as the dynamic behavior of the binding site. The revealed features of the binding site structural organization of the influenza virus hemagglutinin should be taken into account when searching for new antiviral drugs capable to modulate its functional properties.
KEY WORDS: hemagglutinin, influenza virus, molecular dynamics, metadynamicsDOI: 10.1134/S0006297920040100
Abbreviations: CV, collective variable; HA, hemagglutinin.
Hemagglutinin (НA) is the important glycoprotein on the surface of
influenza virus that plays an essential role in the infection, as it
recognizes receptors on the host cell surface, undergoes significant
structural rearrangements during binding, and facilitates the fusion of
viral and cellular membranes and penetration of viral
ribonucleoproteins into a healthy cell [1].
Inhibition of the early stages of infection is an important step in
fighting the influenza virus; hence, HA represents an attractive target
searching for novel antiviral preparations. The primary target of
antiviral therapeutics is the binding site recognized by
oligosaccharide receptors. It is located in the HA head region and
oriented away from the viral membrane towards the environment, which
makes it accessible to the immune system. Influenza vaccines often
target this particular site [2]. Unfortunately,
because of such location, this protein region has acquired an extremely
high variability due to mutations [3-6]. That is why the search for the antiviral drugs
targeting other, more conserved binding sites in HA is required.
Recently, a considerable research interest has been focused on a site
located in the highly conserved stem region [7]
near the fusion peptide responsible for the protein structural
rearrangement. Using X-ray diffraction techniques, it was demonstrated
that this site can bind tertiary butylhydroquinone [7], N-cyclohexyltaurine [8], and umifenovir [9]. According
to the authors, when bound at this site in the HA molecule, these
compounds can induce conformational changes of the adjacent residues,
thus forming a network of interactions that stabilize the entire HA
subunit structure and prevent structural rearrangement of HA. This
binding site for the structural rearrangement inhibitors could be the
previously unrecognized target of antiviral drugs and an alternative
target searching for novel anti-influenza preparations.
Docking is a widely used computational technique searching for potential protein inhibitors. In order to use this technique efficiently, one needs to create a library of potential low-molecular weight ligands and to define their binding site. However, when the binding site is not well defined in the protein structure, it is worthwhile to design an adequate computer model of the molecular target, the quality of which would determine the success in the identification of new inhibitors. The dynamic characteristics of an inadequately described site must be also examined in addition to the static three-dimensional structure. As demonstrated for neuraminidase (another influenza virus protein), ligand binding sites could assume several possible conformations, and their volume could depend on a virus strain [10]. Investigating dynamic behavior of the binding site and searching for its most energetically favorable states are the key steps in molecular modeling that precede computational design of novel compounds. In the case of HA structural rearrangement inhibitors, this approach is even more appropriate because the binding site is located in the region most affected by the structural changes occurring during virion fusion with healthy host cells, and this feature must be taken into consideration searching for the complementary inhibitors.
In this study, we developed the computer models of HAs from the H3N2 and H7N9B influenza viruses in order to investigate structural organization and functional features of the binding site for conformational rearrangement inhibitors. The molecular modeling methods were used to examine the dynamic characteristics of the binding sites in order to identify amino acid residues participating in binding and to determine the most energetically favorable states of the site, as well as to elucidate the effect of dynamic properties on the efficiency of interaction with potential inhibitors using umifenovir as a model component. The obtained data could be useful elucidating the mechanism of action of the known preparations or searching for novel antiviral preparations capable of modulation of the influenza virus HA functional properties.
MATERIALS AND METHODS
Molecular dynamics. The structures of HAs from H3N2 (PDB ID 5t6n) and H7N9 (PDB ID 5t6s) influenza viruses were used in the investigation of the umifenovir-binding site. Amber ff14SB force field was used for protein atoms [11]. Each protein was placed into a rectangular parallelepiped-shaped cell with the dimensions of 100 × 100 × 163 Å. The minimal distance between the protein and cell boundary was 12 Å; the limit of non-valent interaction radius was 10 Å. Long-distance electrostatic interactions were calculated using the PME algorithm for periodic boundary conditions; the grid step was 1 Å. The TIP3P type water molecules and Na+ and Cl– ions at the concentration of 0.1 M were added to the system to create solution ionic strength and to neutralize charges. The initial models were optimized in the course of 5000 energy minimization cycles. The limits were applied to the protein atom mobility followed by gradual heating of the system from 0 to 300 K at constant volume. In the next step, the pressure of 1 atm was established at constant temperature. The final HA molecular dynamic simulation at constant volume and temperature was carried out with the pmemd.cuda program of the Amber14 software package [12]. The duration of the obtained trajectories was 100 ns; 5000 images were selected from the trajectories.
Metadynamics. The metadynamics was used to characterize the energy state of the binding sites in HAs of the H3N2 and H7N9 viruses resulting from the changes in their volume [13]. This technique allows to conduct accelerated accumulation of statistically rare events while controlling various parameters of the system using the so-called collective variables (CVs). Accelerated statistics accumulation (or sampling) occurs due to gradual addition of the supplementary set of prescribed Gaussian hills that depend on CVs to the general potential of the system. In the process, these CVs can be selected in a very wide range, from simple distances between atoms and dihedral or torsion angles to complex CVs, for example, for groups of atoms. The free energy surface can be reconstructed from the results of modeling depending on CVs, thus defining the energy characteristics of the investigated process.
Three general principles were used for CV selection: CVs should define initial, transitional, and final states of the investigated process; CVs should be slow for this system; their number should be as low as possible, because introduction of any additional variable significantly slows down the calculations and complicates analysis of the results. In order to investigate the binding site in the H3N2 HA, we selected the volume of the umifenovir-binding site set by the CAVITY function in the metadynamics modeling program Plumed 2.4 [14] as a CV. This function allows to define the volume based on the spatial location of four protein atoms. The vector r1 is drawn from the first atom to the second one. Next, the r2 vector is drawn perpendicular to the plane containing the first, the second, and the third atoms, as well as the vector r3 orthogonal to the vectors r1 and r2. The first atom and projection of the fourth atom on the plane formed by the pairs of created vectors define the final rectangular parallelepiped in which the value of certain parameter is calculated (the number of water molecules in our case). The Cα atoms of amino acid residues adjacent to the binding site (Ala304 and Leu316 from the A chain, Tpr92 from the B chain, and Lys310 from the C chain) were selected as base atoms; the volume of the rectangular parallelepiped was calculated by subtracting the volume of protein fragment falling in it (Fig. 1a). Additional Gaussians with the height of 4 kJ/mol and width of 0.1 were applied based on the created CV.
Fig. 1. a) The binding region for the conformational rearrangement inhibitors on the surface of H3N2 HA selected for examination with the metadynamics: red, the outer boundary of the region; green, elements of the protein secondary structure and some amino acid residues; blue, the region whose volume is changed and measured in the course of the experiment. b) RMSF graph for H7N9 HA. Three protein subunits can be recognized from the graph shape. Red dots, amino acid residues Glu97 from chain F and Asn27 and Arg32 from chain E that were selected for creation of additional CV. These residues have low RMSF values and are located near the examined region. Blue dashed line represents RMSF value (~0.96 Å) for the center of masses of heavy atoms in these residues.
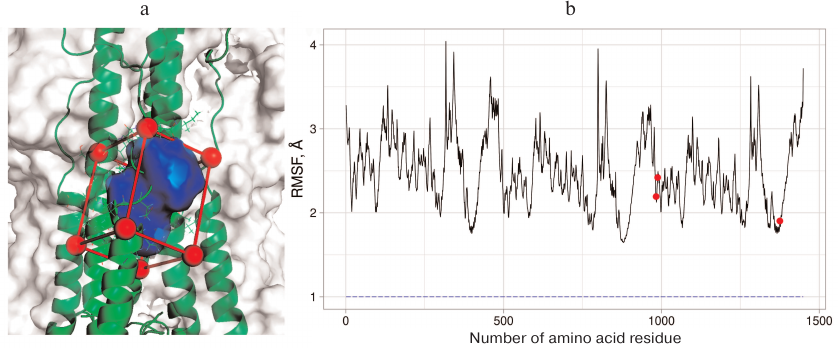
Unlike the examined region in the H3N2 HA, the same region in H7N9 HA can assume open and closed conformations; hence, an additional CV was introduced to develop a comprehensive picture of free energy changes during the modeling of its dynamic behavior. Considering that the open and closed conformations are determined mainly by the location of Arg54 from chain B, the CV used was the difference in the distances d1 and d2, where d1 is the distance between carbon atom in the guanidine group of Arg54 and center of masses of three stable residues, e.g., Glu97 from chain F, Asn27 and Arg32 from chain E (Fig. 1b); d2 is the distance between carbon atom of the guanidine group of the same arginine residue and carbon atom from the carboxyl group of Glu103 from chain D. This CV was selected to measure the energy of the Arg54 residue shift without adding more artificial limitation on the mobility of the rest of the residues. The height of the supplementary Gaussians was 4 kJ/mol; the width was 0.1 and 0.2 for the site volume and distance, respectively.
Docking. The molecular dynamics images of H3N2 HA were aligned with the original structure and then exported as separate pdb files. The pdbqt files of HA and its inhibitor (umifenovir) were prepared using PMV [15, 16]. The docking of the obtained structures was performed with the help of SMINA using the Vina program [17] for docking. SMINA was initiated with the exhaustiveness parameter of 20; the point inside the investigated ligand binding site was selected as a docking center. Further analysis was conducted for 10 docking results that demonstrated the lowest value of the criterion function. A table containing pairwise RMSDs was compiled for these structures and the structures resolved by X-ray diffraction analysis. The final data were clustered using the DBSCAN method [18]. The minPts value was set at 4; the ε value was increased until the crystal conformation of umifenovir reached one of the forming clusters from multiple outliers. Next, the silhouette value for ε was evaluated, starting from the found ε value and finishing with the ε value that had the maximum silhouette value. The obtained clusters were visualized by the principal component analysis using Rstudio and the fviz cluster function. The same procedure was applied to the metadynamics trajectories.
RESULTS AND DISCUSSION
The structure of proteins in aqueous solution experiences natural fluctuations as well as the volume of the HA binding site for conformational rearrangement inhibitors due to the motion of residues that comprise the site. We suggested that the binding site volume can be determined as the number of accommodated water molecules, which allows to characterize this site without using complex algorithms usually applied in various programs [19]. This approach seemed promising for modeling the conformational states and energy characteristics of the binding site using supplementary external effects (as it is done in metadynamics [13]), since the force distribution in the investigated volume occurs uniformly over all water molecules rather than over individual protein atoms (which could result in the distortion of site structure and sampling of highly energetic insignificant conformations). To investigate the binding sites for the structural rearrangement inhibitors in HAs of the H3N2 and H7N9 influenza viruses, we used the number of contained water molecules as a CV. It was demonstrated by the X-ray diffraction analysis, that unlike the binding site in H3N2 HA, the binding site in the H7N9 HA can assume open and closed conformations, determined by the conformation of Arg54 residue of chain B [9]. Hence, we studied the metadynamics of the structural organization and dynamic behavior of the binding site in H7N9 HA using an additional CV characterizing Arg54 position in chain B.
Dynamic properties of the binding in H3N2 HA. The free-energy surface of the binding site in the H3N2 HA was determined by integration of all Gaussian hills added to the system in the course of metadynamics over the selected CV (the number of water molecules) (Fig. 2). It can be seen that there was only one energy minimum, which corresponded to the volume containing from 23 to 34 water molecules; moreover, large additional energy (>4 kcal/mol) is required to surpass this interval. It was important to identify amino acid residues that form the binding site and to determine its dynamic behavior and specific interactions with the inhibitors.
Fig. 2. Dependence of the free-energy surface of the binding site for the conformational rearrangement inhibitors in H3N2 HA on the site volume (number of included water molecules) as determined by modeling using metadynamics.
In order to obtain this information, we selected the images from the entire dynamic trajectory in which the site volume corresponded to the most favorable states. Based on the selected images, an averaged H3N2 HA structure was constructed using the functional from the VMD program [20] that corresponded to the most energetically favorable state of the binding site. Next, the deviations from the averaged structure were calculated for the amino acid residues located at the distance of 8 Å from the site center for the entire dynamics trajectory. The map of the RMSD value dependence for the selected residues in the site volume is presented in Fig. 3.
Fig. 3. Map of the RMSD dependence for amino acid residues forming the binding site in H3N2 HA on the site volume. The region corresponding to the most energetically favorable states of the site is marked with black frame. Blue, the lowest RMSD value; red, the highest RMSD value for the amino acid residue at the indicated state of the site. The graph allows determining the residues that affect the binding site volume the most – their RMSD remains low for the most energetically favorable states of the site (inside the black frame) and increases with the site destabilization (volume increases or decreases relative to the optimal one), while RMSDs of the rest of amino acid residues change independently on the site volume.
The RMSD values for the residues Phe294 in chain A; Lys51, Glu57, Asn95, Ala96, Leu99, and Glu103 in chain B; Lys27, Thr28, Ile29, Asp32, Val309, Lys310, Gln311, and Leu314 in chain C; and Ile89, Asp90, Leu91, Trp92, Ser93, Tyr94, Asn95, Ala96, Glu97, and Leu98 in chain D were low at the optimal binding site volume (i.e., positions of these residues were stable in the most energetically favorable state) and increased significantly outside its boundaries. This implies that the number of water molecules in the binding site depends on the above residues. For other residues, this parameter did not display pronounced minima depending on the volume; hence, they did not affect the number of included water molecules in this site. The RMSD values for Arg54, Asn60, Glu103 in chain B and Lys310 in chain C remained most stable under fluctuations in the site volume; therefore, these residues are of significant interest as points of interactions with potential inhibitors. Three residues from chain D – Ile89, Leu91, and Asn95 – also deserve special attention, as the RMSD values for these residues increase fast with the increase in the volume and then dropped abruptly. This indicates complex dependence of the site volume on the conformational mobility and relaxation rearrangements of these residues.
Dynamic properties of the binding site in H7N9 HA. The free-energy surface of the binding site (Fig. 4a) was determined by integration of all Gaussian hills added to the system in the course of metadynamics over the two selected CVs: (i) number of water molecules in the site and (ii) position of Arg54 residue. Analysis of the surface allowed to determine the difference between the open and closed structures of the binding site from the energy and volume value. The closed conformation, which can be characterized by the volume of 30-39 water molecules, corresponds to the optimal state, while the energy of the less favorable open conformation is higher by 8 kcal/mol. The open state is also characterized by the increased volume (up to 43 water molecules) and shift in the position of Arg54 in chain B (Fig. 4b).
Fig. 4. Dynamic characteristics of the binding site in H7N9 НA at different energy states. a) Free energy surface (values are color-coded according to the given scale) with changes of the site volume (the number of enclosed water molecules). Vertical axis shows the difference in the distances d1 and d2, where d1 is the distance between the carbon atom in guanidine group of Arg54 and center of masses of three stable residues: Glu97 in chain F, sAsn27 and Arg32 in chain E (not shown in the figure), d2 is the distance between the carbon atom in guanidine group of the same arginine and carbon atom in the carboxyl group of Glu103 residue in chain D (see “Materials and Methods”). b) Position of the key amino acid residue Arg54 in the closed conformation of the binding site (upper panel) and changes in its position during transition from the closed to the open state (lower panel); green, elements of HA secondary structure; yellow dashed line, the distance between carbon atoms in guanidine and carboxyl groups of Arg54 and Glu103.
Umifenovir binding to HAs from H3N2 and H7N9 viruses. A wide range of protein conformations obtained using metadynamics can be used to investigate the inhibitor binding to HA. When evaluating interactions in the protein–inhibitor complexes based on X-ray diffraction data, the energy state of the binding site itself is usually not taken into account, as in the case of crystal structures of HA complexes with umifenovir. Umifenovir docking was performed using the most energetically favorable states of the binding site selected from the trajectories of classic molecular dynamics and metadynamics of H3N2 HA. Clustering of the docking results into selected ensemble of HA conformational states (Fig. 5a) revealed no isolated cluster with positions corresponding to the crystal structure of the complex [9]. Complex formation was detected only during clustering of the docking results into HA structures with the expanded state of the binding site. Such isolated cluster (ε = 2.6) corresponding to the X-ray diffraction data is presented in Fig. 5b. This is also corroborated by the fact that all amino acid residues directly interacting with umifenovir (Phe294 and Lys307 from chain A and Leu98 from chain D) are involved in the destabilization of the most energetically favorable state of the site and increase in its volume (see Fig. 3).
Fig. 5. Examination of position of umifenovir molecule in the binding site of H3N2 HA from: a) Clustering of docking results into HA structures of the classic molecular dynamics trajectory: no isolated cluster corresponding to the crystal structure is formed; b) Clustering of docking results into HA structures with the expanded state of the binding site from metadynamics: cluster corresponding to umifenovir position in the crystal structure is marked with red line. Clusters are color-coded, Dim1 and Dim2 form the space of two principle components calculated for the conformation space and positions of umifenovir in the docking results.
The functioning of the binding site for structural rearrangement inhibitors in H7N9 HA differs from that in H3N2 HA. In H7N9 HA, the site exists in two conformational states – open and closed. Docking demonstrated that H7N9 HA bound umifenovir only in the open state, and unlike H3N2 HA, the binding required no significant increase in the site volume. The position of umifenovir corresponded to its position in the crystal structure. The open state of the site was not the most energetically favorable, and its energy was by 8 kcal/mol higher in comparison with the closed state (Fig. 6). Hence, in this case, umifenovir binding required site transition into the less energetically favorable state associated with the shift in the position of Arg54 residue and corresponding changes in the site volume.
Fig. 6. Position of umifenovir molecule in the binding site of H3N2 HA. Umifenovir molecule is shown with thick orange lines. Residues that display RMSD values increasing with the increase of volume are shown with thick lines; residues directly responsible for umifenovir binding are shown in green [9].
The use of molecular modeling techniques allowed us to characterize the most energetically favorable states of the binding site for the conformational rearrangement inhibitors of HAs from two influenza virus strains (H3N2 and H7N9) and to establish that in both cases significant rearrangements of the binding sites are required for the interaction with umifenovir. We also identified amino acid residues that form the binding site, participate directly in the ligand binding, and define the binding efficiency. In both proteins, the binding site rearrangements require energy costs, but proceed via different mechanisms. In H3N2 HA, the requested increase in the binding site volume occurs via shift in the positions of amino acid residues, while in H7N9 HA, the shift in the position of Arg54 in chain B is sufficient for transition to the less energetically favorable state. The obtained data could be used for elucidating the mechanism of action of known antiviral preparations, as well as identifying novel lead compounds capable of modulating the functional properties of the influenza virus HA.
Funding. This work was supported by the Russian Foundation for Basic Research (project No. 18-315-00390).
Acknowledgements. The study was performed using the equipment of the Center of Collective Use of super high-performance computational resources of the Lomonosov Moscow State University [21].
Conflict of interest. The authors declare that they have no conflict of interest.
Compliance with ethical standards. This article does not contain studies with human participants or animals performed by any of the authors.
REFERENCES
1.Skehel, J. J., and Wiley, D. C. (2000) Receptor
binding and membrane fusion in virus entry: the influenza
hemagglutinin, Ann. Rev. Biochem., 69, 531-569,
doi: 10.1146/annurev.biochem.69.1.531.
2.Hensley, S. E. (2014) Challenges of selecting
seasonal influenza vaccine strains for humans with diverse pre-exposure
histories, Curr. Opin. Virol., 8, 85-89,
doi: 10.1016/j.coviro.2014.07.007.
3.Hensley, S. E., Das, S. R., Bailey, A. L., Schmidt,
L. M., Hickman, H. D., Jayaraman, A., Viswanathan, K., Raman, R.,
Sasisekharan, R., Bennink, J. R., and Yewdell, J. W. (2009)
Hemagglutinin receptor binding avidity drives influenza A virus
antigenic drift, Science, 326, 734-736,
doi: 10.1126/science.1178258.
4.Heider, A., Mochalova, L., Harder, T., Tuzikov, A.,
Bovin, N., Wolff, T., Matrosovich, M., and Schweiger, B. (2015)
Alterations in hemagglutinin receptor-binding specificity accompany the
emergence of highly pathogenic avian influenza viruses, J.
Virol., 89, 5395-5405, doi: 10.1128/JVI.03304-14.
5.Tharakaraman, K., Raman, R., Viswanathan, K.,
Stebbins, N. W., Jayaraman, A., Krishnan, A., Sasisekharan, V., and
Sasisekharan, R. (2013) Structural determinants for naturally evolving
H5N1 hemagglutinin to switch its receptor specificity, Cell,
153, 1475-1485, doi: 10.1016/j.cell.2013.05.035.
6.Lvov, D. K., Bogdanova, V. S., Kirillov, I. M.,
Shchelkanov, M. Yu., Burtseva, E. I., Bovin, N. V., Fedyakina, I. T.,
Prilipov, A. G., Alhovsky, S. V., Samokhvalova, E. I., Proshina, E. S.,
Kirillova, E. S., and Syroeshkin, A. V. (2019) Evolution of pandemic
influenza virus A(H1N1)pdm09 in 2009-2016: dynamics of receptor
specificity of the first hemagglutinin subunit (НA1), Vopr.
Virusol., 64, 63-72,
doi: 10.18821/0507-4088-2019-64-2-63-72.
7.Russell, R. J., Kerry, P. S., Stevens, D. J.,
Steinhauer, D. A., Martin, S. R., Gamblin, S. J., and Skehel, J. J.
(2008) Structure of influenza hemagglutinin in complex with an
inhibitor of membrane fusion, Proc. Natl. Acad. Sci. USA,
105, 17736-17741, doi: 10.1073/pnas.0807142105.
8.Kadam, R. U., and Wilson, I. A. (2018) A
small-molecule fragment that emulates binding of receptor and broadly
neutralizing antibodies to influenza A hemagglutinin, Proc. Natl.
Acad. Sci. USA, 115, 4240-4245,
doi: 10.1073/pnas.1801999115.
9.Kadam, R. U., and Wilson, I. A. (2017) Structural
basis of influenza virus fusion inhibition by the antiviral drug
Arbidol, Proc. Natl. Acad. Sci. USA, 114, 206-214,
doi: 10.1073/pnas.1617020114.
10.Han, N., Mu, Y., Miao, H., Yang, Y., Wu, Q., Li,
J., Ding, J., Xu, B., and Huang, Z. (2016) The 340-cavity in
neuraminidase provides new opportunities for influenza drug
development: a molecular dynamics simulation study, Biochem.
Biophys. Res. Commun., 470, 130-136,
doi: 10.1016/j.bbrc.2016.01.007.
11.Maier, J. A., Martinez, C., Kasavajhala, K.,
Wickstrom, L., Hauser, K. E., and Simmerling, C. (2015) ff14SB:
improving the accuracy of protein side chain and backbone parameters
from ff99SB, J. Chem. Theory Comput., 11, 3696-3713,
doi: 10.1021/acs.jctc.5b00255.
12.Case, D. A., Babin, V., Berryman, J., Betz, R.
M., Cai, Q., et al. (2014) Amber 14, University of California,
San Francisco.
13.Barducci, A., Bonomi, M., and Parrinello, M.
(2011) Metadynamics, WIREs Comput. Mol. Sci., 1, 826-843,
doi: 10.1002/wcms.31.
14.Tribello, G. A., Bonomi, M., Branduardi, D.,
Camilloni, C., and Bussi, G. (2014) PLUMED 2: new feathers for an old
bird, Com. Phys. Commun., 185, 604-613,
doi: 10.1016/j.cpc.2013.09.018.
15.Sanner, M. (1999) Python: a programming language
for software integration and development, J. Mol. Graph. Model.,
17, 57-61.
16.Morris, G. M., Huey, R., Lindstrom, W., Sanner,
M. F., Belew, R. K., Goodsell, D. S., and Olson, A. J. (2009) AutoDock4
and autoDocktools4: automated docking with selective receptor
flexibility, J. Comput. Chem., 16, 2785-2791,
doi: 10.1002/jcc.21256.
17.Trott, O., and Olson, A. J. (2010) AutoDock Vina:
improving the speed and accuracy of docking with a new scoring
function, efficient optimization, and multithreading, J. Comput.
Chem., 31, 455-461, doi: 10.1002/jcc.21334.
18.Ester, M., Kriegel, H.-P., Sander, J., and Xu, X.
(1996) A density-based algorithm for discovering clusters in large
spatial databases with noise, Proc. 2nd Int. Conf. Knowl.
Discov. Data Mining, AAAI Press, Portland, Oregon, pp. 226-231.
19.Durrant, J. D., Votapka, L., Sørensen, J.,
and Amaro, R. E. (2014) POVME 2.0: an enhanced tool for determining
pocket shape and volume characteristics, J. Chem. Theory
Comput., 10, 5047-5056, doi: 10.1021/ct500381c.
20.Humphrey, W., Dalke, A., and Schulten, K. (1996)
VMD: visual molecular dynamics, J. Mol. Graph. Model.,
14, 33-38, doi: 10.1016/0263-7855(96)00018-5.
21.Sadovnichy, V., Tikhonravov, A., Voevodin, V.,
and Opanasenko, V. (2013) “Lomonosov”: supercomputing at
Moscow State University, in Contem. High Perform. Comp., Boca
Raton, USA, pp. 283-307, doi: 10.1201/9781351104005-11.