Catalytically Competent Conformation of the Active Site of Human 8-Oxoguanine-DNA Glycosylase
A. V. Popov1,a*, A. V. Yudkina1,2, Yu. N. Vorobjev1, and D. O. Zharkov1,2,b*
1Institute of Chemical Biology and Fundamental Medicine, Siberian Branch of the Russian Academy of Sciences, 630090 Novosibirsk, Russia2Novosibirsk State University, 630090 Novosibirsk, Russia
* To whom correspondence should be addressed.
Received April 15, 2019; Revised November 9, 2019; Accepted November 9, 2019
8-Oxoguanine-DNA N-glycosylase (OGG1) is a eukaryotic DNA repair enzyme responsible for the removal of 8-oxoguanine (oxoG), one of the most abundant oxidative DNA lesions. OGG1 catalyzes two successive reactions – N-glycosidic bond hydrolysis (glycosylase activity) and DNA strand cleavage on the 3′-side of the lesion by β-elimination (lyase activity). The enzyme also exhibits lyase activity with substrates containing apurinic/apyrimidinic (AP) sites (deoxyribose moieties lacking the nucleobase). OGG1 is highly specific for the base opposite the lesion, efficiently excising oxoG and cleaving AP sites located opposite to C, but not opposite to A. The activity is also profoundly decreased by amino acid changes that sterically interfere with oxoG binding in the active site of the enzyme after the lesion is everted from the DNA duplex. Earlier, the molecular dynamics approach was used to study the conformational dynamics of such human OGG1 mutants in complexes with the oxoG:C-containing substrate DNA, and the population density of certain conformers of two OGG1 catalytic residues, Lys249 and Asp268, was suggested to determine the enzyme activity. Here, we report the study of molecular dynamics of human OGG1 bound to the oxoG:A-containing DNA and OGG1 mutants bound to the AP:C-containing DNA. We showed that the enzyme low activity is associated with a decrease in the populations of Lys249 and Asp268 properly configured for catalysis. The experimentally measured rate constants for the OGG1 mutants show a good agreement with the models. We conclude that the enzymatic activity of OGG1 is determined majorly by the population density of the catalytically competent conformations of the active site residues Lys249 and Asp268.
KEY WORDS: DNA damage, DNA repair, 8-oxoguanine-DNA N-glycosylase, substrate specificityDOI: 10.1134/S0006297920020066
Abbreviations: AP site, apurinic/apyrimidinic site; MD, molecular dynamics; OGG1, 8-oxoguanine-DNA N-glycosylase; oxodG, 8-oxo-2′-deoxyguanosine; oxoG, 8-oxoguanine; WT, wild type.
Oxidation of macromolecules in the cells of aerobic organisms is a
common process caused by reactive oxygen species generated as
byproducts of normal metabolism or as a result of exposure to ionizing
radiation [1, 2]. Oxidative
damage to mitochondrial and nucelar DNA is directly associated with
diseases and aging. Reactive oxygen species can cause breaks in the DNA
sugar-phosphate backbone, formation of apurinic/apyrimidinic (AP) sites
(deoxyribose moieties lacking the nucleobase), and premutagenic lesions
of nucleobases [1-3].
One of the most frequent types of nucleobase damage occurring in nature is formation of 8-oxoguanine (oxoG). Due to the presence of C8 keto group, free 8-oxo-2′-deoxyguanosine (oxodG) nucleoside exists preferentially in the syn-conformation, in contrast to the canonical purine deoxyribonucleosides [4]. This conformation is also stabilized upon the formation of the Hoogsteen base pair with A. When paired with C, oxodG acquires the anti-conformation similar to the canonical G:C pair [5-7]. The formation of the stable oxoG:A pair accounts for the mutagenicity of this lesion resulting in the preferential generation of the G→T transversion [8, 9]. The content of endogenous oxoG in DNA is ~1 oxoG per 106 G; however, it can increase several-fold under the genotoxic stress [10]. Even more numerous AP sites in DNA are formed as a result of spontaneous hydrolysis of the N-glycoside bond and as intermediates during the base excision DNA repair [3]. The content of AP sites in human cells at any time is ~1 per 105 bases [11].
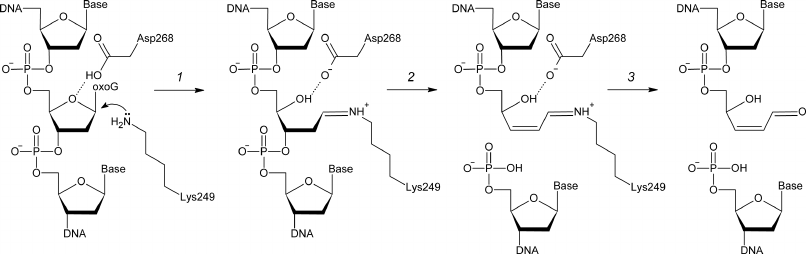
8-Oxoguanine-DNA N-glycosylase (OGG1, EC 3.2.2.23) hydrolyzes the N-glycosidic bond in oxoG nucleosides in DNA and is responsible for the oxoG removal from the human genome and genomes of other eukaryotes [12-15]. OGG1 also catalyzes β-elimination accompanied by the cleavage of the C3′–O3′ bond after the removal of oxoG and in spontaneously formed AP sites (Fig. 1). The Fpg enzyme performs the same function in bacteria; however, its amino acid sequence is not homologous to the OGG1 sequence [16, 17]. OGG1 cleaves the glycosidic bond by the nucleophilic substitution mechanism with the formation of a covalent intermediate (Schiff base) between the ε-amino group of strictly conserved Lys249 residue and C1′ atom of the damaged nucleotide [18, 19]. Another strictly conserved residue, Asp268, likely participates in the coordination or protonation of deoxyribose residue [20, 21] (Fig. 1).
Fig. 1. The mechanism of OGG1-catalyzed cleavage of damaged DNA. Deprotonated ε-amino group of Lys249 attacks C1′ with the formation of a covalent intermediate (Schiff base) (1). DNA backbone is cleaved by the β-elimination mechanism (2), followed by the Schiff base hydrolysis with the release of enzyme and formation of the end product – single-strand DNA break with α,β-unsaturated γ-hydroxy aldehyde at the 3′-end (3).
The structure of human OGG1 has been determined for the protein in its free state, as well as in complexes with DNA containing oxoG, AP site, and undamaged G base and complexes simulating conformers formed in process of reaction [20-29] (PDB: 1EBM, 1FN7, 1HU0, 1KO9, 1LWW, 1LWV, 1LWY, 1M3H, 1M3Q, 1N39, 1N3A, 1N3C, 1YQK, 1YQL, 1YQM, 1YQR, 2I5W, 2NOB, 2NOE, 2NOF, 2NOH, 2NOI, 2NOL, 2NOZ). However, none of the static structures provides a comprehensive idea on the conformational ensembles of the intermediate DNA–protein complexes formed during the lesion recognition. Such information can be obtained by the method of molecular dynamics (MD) simulation. This method has been previously used to study the process of everting the damaged base from DNA by OGG1 [26, 30, 31], the behavior of the base in the enzyme active site after the N-glycosidic bond hydrolysis [24], and distortions in the precatalytic complex structure caused by amino acid substitutions sterically blocking the oxoG-binding site in the enzyme molecule [32]. The latter study has shown the possibility of four stable active site conformations the with different positions and orientations of catalytic amino acid residues Lys249 and Asp268. One of the conformations is prevalent in the complex of the wild-type OGG1 with DNA containing the oxoG:C pair, while the rest mostly occur in inactive or low-active mutant variants of the protein. In this work, the catalytic competence of different active site conformations was analyzed by simulating the complex of wild-type OGG1 with DNA containing oxoG:A. The removal of oxoG from this structure was approximately two orders of magnitude less efficient compared to the optimal oxoG:C substrate. We also simulated the complexes with DNA containing AP:C for the wild-type enzyme and its mutant variants with substitutions sterically blocking the oxoG-binding site. In these cases, the activity of the enzyme correlated with the population density of one of the conformations, which therefore can be considered as catalytically competent.
MATERIALS AND METHODS
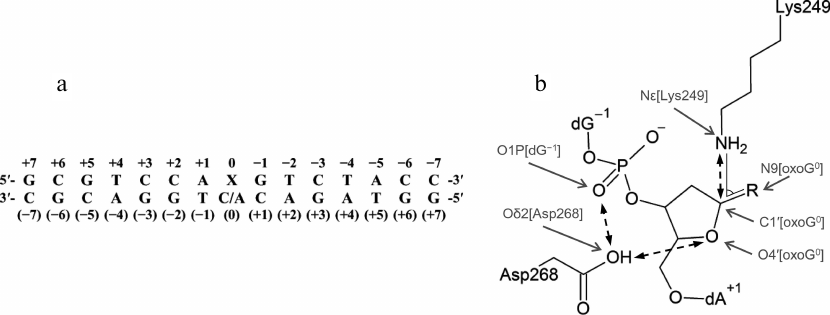
Model preparation. The tertiary structure of the complex between human OGG1 and DNA (PDB 1EBM) includes a double-stranded DNA of 15 bp (Fig. 2a) with the oxoG:C pair. The oxoG base in the anti-conformation is everted to the enzyme active site [20]. The original structure lacks eight N-terminal amino acid residues not included in the model and the internal region Gly80-Lys82, which was taken from the structure of free OGG1 (PDB 1KO9 [23]) to obtain the WT-oxoG:C model. To build the WT-oxoG:A model, the atomic structure of dA nucleoside was aligned with the structure of dC at O4′, C1′, and N1/N9 atoms using the Kabsch algorithm [33] and incorporated into the model instead of dC. Analogous procedure was used for the following amino acid substitutions: C253I, C253L, Q315W, and inverse substitution Q249K instead of the inactivating mutation K249Q [20]. Some amino acid residues were modeled in nonstandard charge states in accordance with the known reaction mechanism and published QM/MM (quantum mechanics/molecular mechanics) simulation data: Cys253 as a thiolate anion, Lys249 with neutral ε-amino group, and Asp268 with neutral carboxylic group [20, 21, 26]. The AP site was built from oxoG by removing base atoms and substituting the nitrogen atom involved in the formation of glycosidic bond with oxygen. Reconstruction of missing hydrogen atoms and side chains of amino acid residues, as well as verification of the models for the absence of errors, were performed automatically with BioPASED [34].
Fig. 2. a) The sequence of DNA substrate used in the simulation (X0 is oxoG or AP site) with numbered nucleotide bases; b) critical distances and angles (between the marked atoms) in the OGG1 active site formed by the catalytic residues Lys249 and Asp268. R is oxoG for the substrate containing oxoG, or OH for the substrate containing AP site.
Molecular dynamics and trajectory analysis. The point charges and other parameters of the force field for the amino acid residues in nonstandard charge states were based on the respective parameters from AMBER 99 [35]. The parameters for oxoG were taken from the study by Perlow-Poehnelt et al. [36]; the AP site parameters were kindly provided by Professor C. Simmerling (State University of New York at Stony Brook, USA). All prepared structures were optimized in 500 steps in BioPASED by the conjugate gradient method (Fletcher algorithm) until reaching the values of the potential energy gradient of about 10−1 kcal/mol and changes of coordinates about 10−4 Å and then for 150 ps by the MD method with gradual heating from 50 to 300 K. The integration step was 1 fs. Simulation was performed in the canonical NVT ensemble. The implicit solvent model was used for more efficient sampling of the conformational space due to the absence of dynamic friction of explicit water molecules [37]. The structures were simulated with BioPASED using the force field derived from ff99SB [34] for 10 ns, taking into account hydrogen bonds (scale multiplier, 2.5), with artificial restraints on the potential of the heavy-atom force field introduced for stabilization of the protein–DNA complex: 0.001 kcal/Å2 for protein atoms, 0.25 kcal/Å2 for the atoms of terminal DNA nucleotides, and 0.0025 kcal/Å2 for other DNA atoms. The negative charge of the system was neutralized by scaling the charges of phosphate groups with the factor 0.2 [38, 39]. The system coordinates were recorded every 2 ps.
The trajectories were analyzed with MDTRA [40]. The trajectories and the structures were visualized by VMD [41], RasMol [42], and PyMol (Schrödinger, USA). The trajectory for the WT-oxoG:C model was obtained previously under identical conditions [32] and used for comparative analysis without modifications. Analysis of the trajectory stabilization time by the reverse cumulative averaging method [43] and model clustering were performed using R scripts; the dendrograms were constructed with iTOL [44].
Oligonucleotides and enzymes. T4 polynucleotide kinase (Biosan, Russia) and uracil-DNA glycosylase from E. coli (New England Biolabs, USA) were used in the work. Purification of recombinant wild-type OGG1 and its mutant variants and determination of the active enzyme concentration were performed as described previously [32]. The enzymatic activity was studied using oligodeoxyribonucleotides synthesized at the Laboratory of Medical Chemistry of the Institute of Chemical Biology and Fundamental Medicine (Siberian Branch of the Russian Academy of Sciences): OG23 5′-CTCTCCCTTCXCTCCTTTCCTCT-3′ (X = oxoG); U23 5′-CTCTCCCTTCXCTCCTTTCCTCT-3′ (X = Ura); and C23 5′-AGAGGAAAGGAGCGAAGGGAGAG-3′. The substrates were modified by introducing radioactive label into OG23 and U23 by the standard procedure using γ[32P]ATP and polynucleotide kinase [45], followed by annealing with a twofold molar excess of C23. The substrate containing the U23 nucleotide was then treated for 30 min at 37°C with uracil-DNA glycosylase in the buffer containing 50 mM Tris-HCl (pH 7.5), 100 mM NaCl, 1 mM EDTA and 1 mM dithiothreitol (DTT) and immediately used in the reaction.
Kinetic parameters of the OGG1-catalyzed reactions were determined in a standard reaction mixture containing 50 mM Tris-HCl (pH 7.5), 100 mM NaCl, 1 mM EDTA, 1 mM DTT, 0.1 mg/ml BSA, 50 nM substrate and 1-1000 nM of OGG1 or its mutant variants; the reaction was carried out for 10 min at 37°C. In the case of oxoG:C substrate, the reaction was stopped by adding NaOH up to 100 mM, followed by sample heating for 2 min at 95°C, neutralization with an equimolar amount of HCl, and addition of an equal volume of quenching buffer containing 80% (v/v) formamide, 20 mM EDTA, 0.1% (w/v) xylene cyanol, and 0.1% (w/v) bromophenol blue. In the case of AP:C substrate, the reaction was stopped by adding an equal volume of quenching buffer. The reaction products were analyzed by electrophoresis in 20% polyacrylamide gel in the presence of 8 M urea [45]. The reaction rate constants kex and kAP were determined under the conditions of the single-turnover kinetics at 13°C at the substrate and enzyme concentrations of 10 and 200 nM, respectively; the constant kcl was determined under the burst-phase kinetics conditions at 37°C at the substrate and enzyme concentrations of 100 and 5 nM, respectively, as described earlier [46, 47]. Statistical significance of the differences between the apparent rate constants for different protein variants was assessed using the Student’s t-test.
RESULTS AND DISCUSSION
Conformational dynamics of the OGG1 active site in a complex with the oxoG:A substrate. Previous MD simulations of the OGG1–DNA complex have shown that the wild-type enzyme differs from its low-active or inactive mutant variants C253I, C253L and Q315W in the population density of particular active site conformations [32] (Fig. 2b). The active site of OGG1 contains catalytic residues Lys249 (for the nucleophilic attack at C1′) and Asp268 (for restraining O4′ of the damaged nucleotide). As it has been shown by the QM/MM simulation, both residues exist preferentially in the neutral charge state [20, 21, 26]. The active site of the wild-type enzyme is characterized by the Nζ[Lys249]–C1′[oxoG]–N9[oxoG] angle around 75° optimal for the nucleophilic attack in the C1′→N9 direction, as well as by the presence of protonated carboxylic group Asp268 close to O4′ and the absence of its alternative bond with phosphate at the 3′-side from the damaged nucleotide [32]. The C253I, C253L, and Q315W substitutions reduce the volume of the active site and cause its distortion during oxoG binding. As a result, the population of conformers typical for the wild-type protein considerably decreases (C253I) or completely disappears (C253L, Q315W), with the preferential population of conformations with the alternative Asp268 bond and the turn of Lys249 by ~35° away from the optimal angle of attack [32]. Similar changes were found in the static structures and in the MD simulations of other mutant variants (R46Q, R131Q, R154H, Q315F) with sterically blocked active site [28, 48], as well as in the MD simulations for the oxoG binding in the catalytically unfavorable syn-conformation [49].
It is still unclear to what extent conformational changes of the active site in the three mutant enzymes reflect the features of the decrease in the OGG1 activity in other cases. To answer this question, we studied the MD of the wild-type OGG1 complex with DNA containing oxoG:A (WT-oxoG:A) in comparison to the previously constructed model with oxoG:C (WT-oxoG:C). OGG1 demonstrates high specificity toward the base opposite to the lesion; it preferably removes oxoG from the oxoG:C pair but not from oxoG:A [19, 50]. The dA nucleoside was simulated in the anti-conformation, in which it exists in the oxoG:A pair in DNA [5, 6] because, due to the close engulfment by the protein of the base opposite to oxoG, its rotation around the N-glycosidic bond is sterically hindered. Analysis of the heavy-atom root mean square deviation (RMSD) by the reverse cumulative averaging method [43] has shown that both trajectories are stabilized within ~5 ns; therefore, the last 5 ns were further used to analyze conformations in the active site.
The WT-oxoG:A model showed only insignificant deviation (less than 1.6 Å) of heavy atom positions (Fig. 3, a and b). Similar to the WT-oxoG:C model, the oxoG base was everted to the pocket formed by Phe319 and Cys253 residues and retained by the hydrogen bonds with Gln315 and Gly42. The WT-oxoG:A and WT-oxoG:C models were similar in the interaction with the oxoG base (Fig. 4a).
Fig. 3. RMSD of the atom positions during simulation. The calculation was made for all heavy atoms (C, N, O and P) of the polypeptide and nucleotide backbone (a-f) or for all heavy atoms of Lys249 and Asp268 residues and AP0 (g-j). The names of the models are given on the plots; light lines show moving average of 50 points.
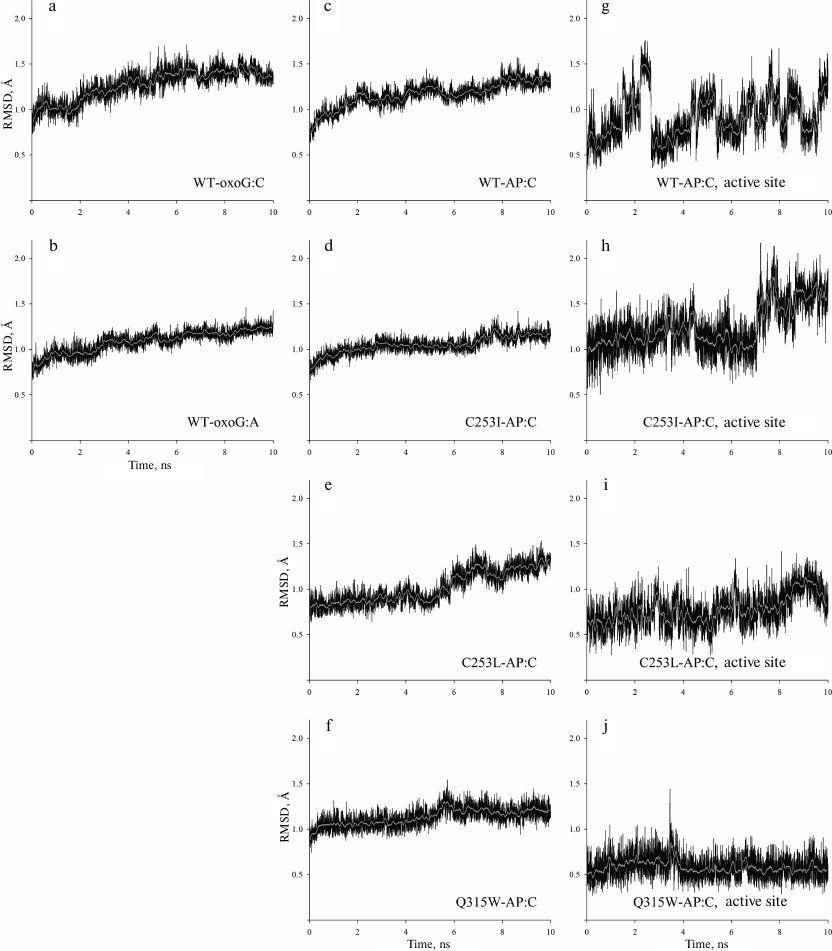
Fig. 4. The structure and dynamics of the active site of OGG1 in the complex with the oxoG:A substrate. a) Interactions between OGG1 amino acid residues and the oxoG base. The average distances between N and O atoms forming the hydrogen bond during MD simulation are indicated. b) Interactions between OGG1 and the C base opposite to the lesion revealed by X-ray structure analysis in the 1EBM structure [20]. c) Interactions between OGG1 and the A base opposite to the lesion revealed during MD simulation. The distances corresponding to potential formation of the hydrogen bond are given in angstroms. d) Representative snapshots of WT-oxoG:C (carbon atoms are marked in black) and WT-oxoG:A (carbon atoms are marked in white) trajectories, illustrating mutual positions of the catalytic residues Lys249 and Asp268 and damaged nucleotide oxoG0. The arrow shows the turn of Lys249 ε-amino group. e) The values of the Nζ[Lys249]–C1′[oxoG0] distance and the Nζ[Lys249]–C1′[oxoG0]–N9[oxoG0] angle for the models of OGG1 complexes with oxoG:C (black dots) and oxoG:A (gray dots). The median values of the distributions are shown with lines of respective colors.
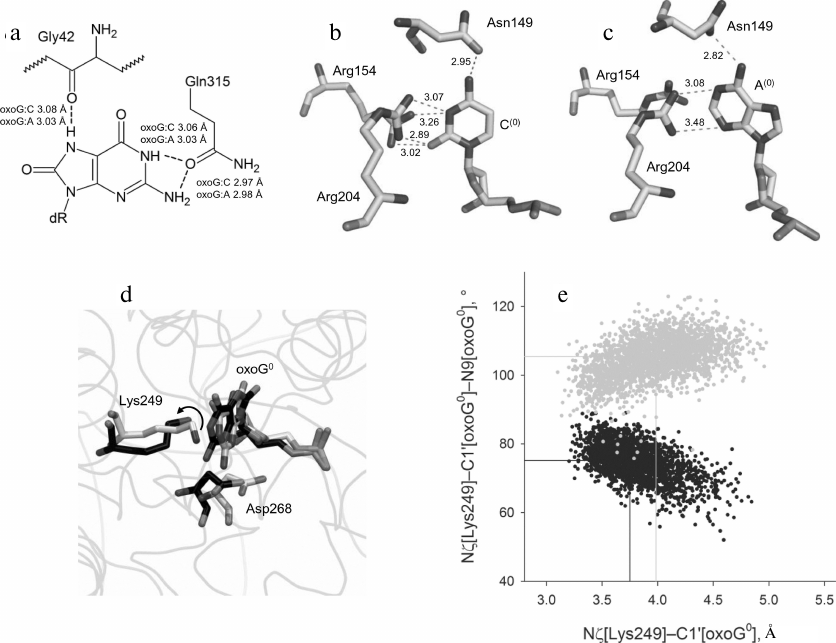
The contacts between Asn149, Arg154, and Arg204 and the opposite DNA strand were similar in the WT-oxoG:C and WT-oxoG:A models (Fig. 4, b and c). In the WT-oxoG:C model, complementary cytosine C(0) formed the hydrogen bond Oδ1[Asn149]…N4[C(0)], while positions of the Nη1 and Nη2 atoms of Arg154 and Arg204 allowed formation of hydrogen bonds with O2 and N3 of C(0) in different combinations (Fig. 4b). In the WT-oxoG:A model, the Oδ1[Asn149]…N6[A(0)] bond was observed along the entire trajectory, while the Arg204 guanidine group interacted with N3 and N1 of the A(0) base either with the formation of hydrogen bonds or through cation–π interactions (Fig. 4c). The side chain of Arg154 in the WT-oxoG:A model turned away from A(0) and formed bonds with DNA at its 5′-side. Therefore, OGG1 is capable of forming stable interactions with both C and A bases opposite to oxoG. It is unlikely that destabilization of interactions with the base opposite to the lesion causes the low activity of OGG1 on oxoG:A substrates.
Analysis of conformations of the catalytic residues in the active site yielded different results (Fig. 4, d and e). The Nζ[Lys249]–C1′[oxoG] distance in the WT-oxoG:A model was by ~0.25 Å greater than in the oxoG:C model. The Nζ[Lys249]–C1′[oxoG]–N9[oxoG] angle was also stably different (105° in the WT-oxoG:A model vs. 75° in the WT-oxoG:C model). The Asp268 carboxylic group in both models was close to O4′. Overall, we can conclude that the decisive parameter in the reduction of the OGG1 activity in the presence of A opposite to the lesion is the unfavorable orientation of Lys249 amino group.
Conformational dynamics of the OGG1 active site in complexes with substrates containing the AP site. Amino acid substitutions resulting in steric hindrance of oxoG binding in the lesion recognition pocket in the OGG1 structure cause distortion of the active site around the catalytic Lys249 and Asp268 residues and enzyme inactivation [28, 32, 48]. The presence of the AP site instead of oxoG should lead to the appearance of extra free space for the bulky side chains forming the walls of the active site. It can be expected that the activity of the mutant variants with this substrate should decrease to a lesser extent. To verify this hypothesis, we simulated the MD of the complexes of such DNA substrates with the wild-type enzyme (WT-AP model) and its mutants (C253I-AP, C253L-AP, and Q315W-AP models). In all the cases, the base opposite to the lesion was C. All models equilibrated within 7-7.5 ns (Fig. 3, c-j). The RMSD of positions of heavy atoms in the protein backbone and DNA was no more than 1.8 Å. However, due to the absence of oxoG, the active site of the enzyme was more mobile (Fig. 3, g-j).
In the WT-oxoG:C complex and MD models on its basis, the oxoG base everted from DNA was sandwiched between the aromatic ring of Phe319 and the side chain of Cys253 and stabilized by hydrogen bonds O[Gly42]…N7[oxoG0], Oε1[Gln315]…N1[oxoG0], and Oε1[Gln315]…N2[oxoG0] (Fig. 4a). In all models with the AP site, these interactions were absent, and the active site residues exhibited much greater mobility. Leu253 and Ile253 in the C253L-AP and C253I-AP models protruded to the active site but did not cause its deformation, as they did not encounter an obstacle in the form of oxoG (Fig. 5a). The contacts with the opposite DNA strand without the AP site generally corresponded to those observed in the WT-oxoG:C model.
Fig. 5. The structure and dynamics of the OGG1 active site complex with the AP:C substrate. a) Representative trajectories of WT-AP (carbon atoms are marked in white) and C253I-AP (carbon atoms are marked in black) illustrating mutual positions of the catalytic residues Lys249 and Asp268 and the AP site. b) Clustering of the models by the Nζ[Lys249]–C1′[AP0], Oδ2[Asp268]–O4′[AP0], and Oδ2[Asp268]–O1P[dG–1] distances in the Mahalanobis space. The numbers correspond to the lengths of dendrogram branches.
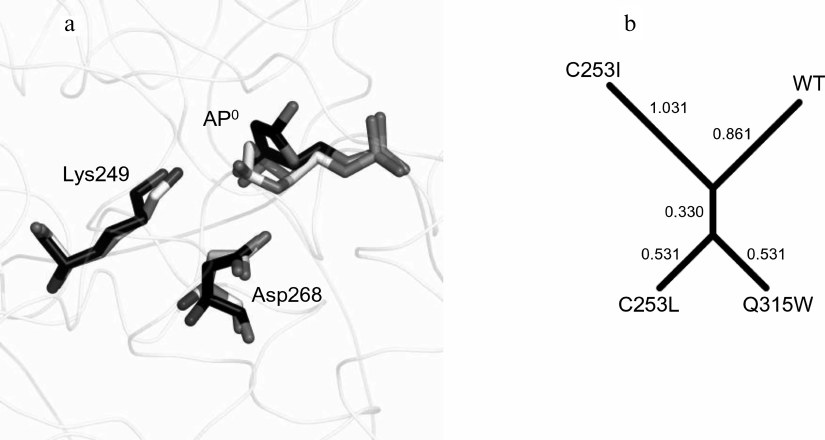
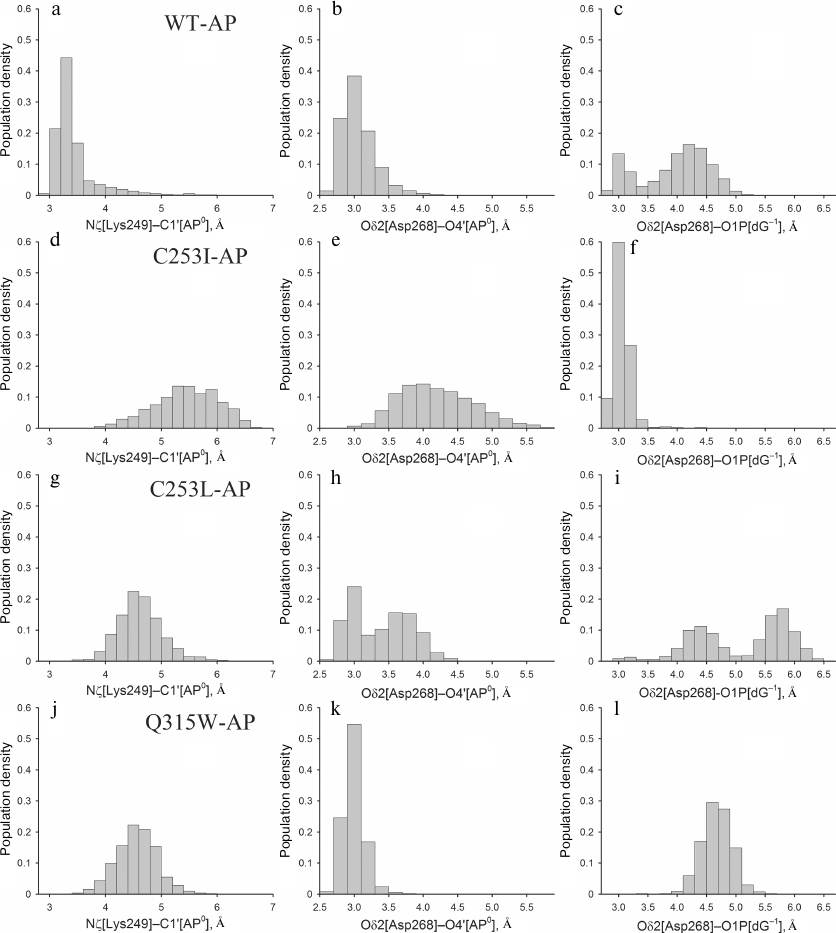
Despite the absence of active site deformation, catalytically important amino acid residues Lys249 and Asp268 in the models with the AP site were fixed near the damaged nucleotide less tightly than in the WT-oxoG:C model, which is in agreement with lower enzyme activity against the AP substrates [19]. This was more evident for the mutant enzymes: in the C253I-AP and C253L-AP models, Asp268 often interacted with O1P[dG–1], losing the contact with O4′ of the AP site during the last 2-2.5 ns. The Q315W-AP model demonstrated more stable dynamics, generally corresponding to the behavior of the wild-type model. The angle of attack of the Lys249 amino group in the case of AP site does not play a critical role because, due to epimerization, the hemiacetal hydroxyl group at C1′ has no definite orientation. However, the Nζ[Lys249]–C1′[AP0] distance is still catalytically significant [32]; it strongly fluctuated as a result of enhanced active site mobility in all models with the AP site. Thus, the following three catalytically significant distances were chosen for analysis: Nζ[Lys249]–C1′[AP0], Oδ2[Asp268]–O4′[AP0], and Oδ2[Asp268]–O1P[dG–1]. The last more stable 2.5 ns of the dynamics were analyzed to minimize the effects of excessive mobility of residues in the absence of nitrogenous base. The distribution of the populations of these distances has shown that the catalytically favorable conformation is formed mainly in the wild-type model (Fig. 6, a-c), where the distance between the C1′ atom of the AP site and the Lys249 amino group is <3.5 Å. In the C253I-AP model (Fig. 6, d-f), an increase in this distance (~5.5 Å) was accompanied with the stable hydrogen bond between Asp268 and dG–1 phosphate group; therefore, the activity of this mutant on the substrates with the AP site should supposedly decrease to the greatest extent. The population of conformations broke up into two marked clusters with respect to the Asp268 orientation in the C253L-AP model (Fig. 6, g-i), but, on the contrary, was rather compact in the Q315W-AP model (Fig. 6, j-l). The average Nζ(Lys249)–C1′[AP0] distance in both cases was 4.5 Å. Thus, the position of the catalytically important Lys249 amino group in the OGG1 active size proved to be decisive also in case of substrates with the AP site.
Fig. 6. The histograms of distribution of the values of distances Nζ[Lys249]–C1′[AP0] (a, d, g, j). Oδ2[Asp268]–O4′[AP0] (b, e, h, k) and Oδ2[Asp268]–O1P[dG–1] (c, f, i, l) for WT-AP (a-c), C253I-AP (d-f), C253L-AP (g-i) and Q315W-AP (j-l).
These three physical distances were used for model clustering to determine the similarity between the models (Fig. 5b). The distance between the models was assessed based on the distance between the centers of the distributions in Euclidean space, the Mahalanobis distance (the distance between the centers of distributions, taking into account the correlation between the variables), and the Hodges–Lehmann estimator (the median of pairwise distances between all points of two samples). In all cases, the C253L-AP and Q315W-AP models were the most similar to each other, while the C253I-AP model was the most different from them and from the wild-type model. In general, the results of simulation suggest that the activity of the mutant variants against the substrates containing the AP site will be higher than against the substrates with oxoG, and the activity of the C253L and Q315W enzymes will be higher than the activity of C253I.
Kinetic parameters of the reactions catalyzed by the mutant variants of OGG1. To assess the effects of amino acid substitutions in the OGG1 active site on the enzyme activity, mutant proteins carrying the C253I, C253L, and Q315W substitutions were overproduced and purified. As expected, they had much lower activity in the oxoG removal from DNA (Fig. 7a and Table 1).
Fig. 7. The activity of OGG1 mutants. a) Cleavage of substrate with the oxoG:C pair; b) cleavage of substrate with the AP:C pair. The plots show the data from three independent experiments.
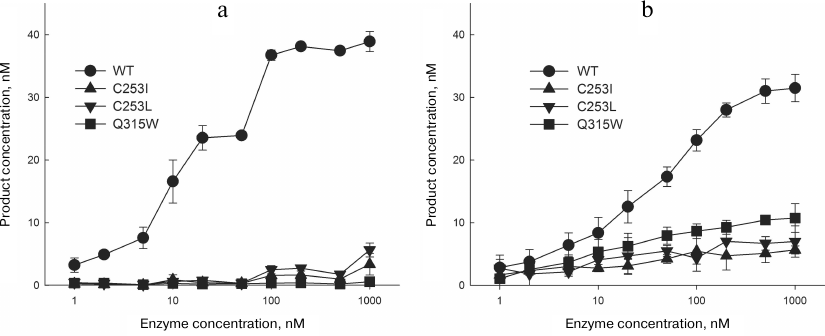
Table 1. The apparent rate constants of
successive stages of cleavage of DNA substrate with the oxoG:C pair by
OGG1

OGG1 successively catalyzes the reaction of N-glycosidic bond hydrolysis and β-elimination; the second stage of this reaction is much slower than the first one [19, 46, 47, 51]. Hence, the kinetic mechanism of the OGG1 action is usually described by a two-stage scheme, where the reaction rate is limited by the last stage:

The kex constant in this case represents the rate of N-glycosidic bond hydrolysis, while the kcl constant is a combined parameter of the rate of β-elimination and dissociation of the enzyme–substrate complex. For the wild type enzyme, the measured kex and kcl values were (9.7 ± 0.9)·10−2 and (0.5 ± 0.1)·10−2 min−1, respectively, which is in agreement with the published data. The C253I and C253L substitutions decreased kex by about an order of magnitude (Table 1), while the Q315W substitution resulted in complete enzyme inactivation. Such consequences are quite expected for the mutations interfering with oxoG binding in the active site. The impact of the substitutions on kcl were less pronounced; however, this constant also decreased approximately 5-fold (Table 1). Obviously, conformational distortions appearing as a result of steric interactions between oxoG and bulky groups also influence the efficiency of β-elimination. Interestingly, some authors consider the oxoG base, which is retained in the active site in anionic form after the first reaction stage, to be a potential acceptor of the C2′ proton removed in the β-elimination reaction [24]. In this case, the efficiency of β-elimination can decrease not only due to the conformational distortions of deoxyribose in the active site, but also due to a decrease in the affinity of free oxoG for OGG1.
The activity of mutant variants with the DNA substrate containing the AP also decreased but to a lesser extent than with the oxoG substrate (Fig. 7b and Table 2). In this case, the reaction is confined only to β-elimination; at the same time, it is obvious that the cleaved oxoG base cannot be a proton acceptor. The apparent constant kAP determined by the single-turnover kinetics method in this case has the physical meaning of the rate constant of β-elimination not complicated by the product release. The C253I, C253L, and Q315W substitutions produced a slight effect on the kAP value by decreasing it approximately twofold (Table 2), which is in agreement with the absence of the base binding in the active site. For the wild type enzyme and the C253I and C253L mutants, these values were 2.4-5.0-fold higher than kcl for the oxoG substrates, demonstrating the contribution of both β-elimination and dissociation of the enzyme–substrate complex to the combined constant kcl in the presence of the damaged base in the substrate.
Table 2. The apparent rate constants of the
cleavage of DNA substrate with the AP:C pair by OGG1

As one can see from Fig. 7 and Tables 1 and 2, the activities of OGG1 mutant variants on AP substrates were higher than on oxoG substrates. This observation is in agreement with the data on the activity of other bifunctional DNA glycosylases, in which substitutions in the active site often completely inhibit the DNA glycosylase activity but do not affect the AP lyase activity, despite the involvement of the same amino acid residues in catalysis [52, 53]. Such difference in the concequences of amino acid substitutions is usually explained by partially extrahelical location of the AP site and by the plasticity of the active site. It should be also noted that the relative activities of different mutant variants of OGG1 against oxoG and AP substrates are different; the activity of the mutant variants decrease in the C253L ≥ C253I >> Q315W order for the oxoG substrates and in the Q315W > C253L ≥ C253I order for the AP substrates. For both substrates, the rate constants for C253L and C253I were not statistically significantly different from each other. The kAP value with the AP substrate was significantly higher for Q315W than for C253I (p < 0.01). The relative activity is in good agreement with the population density of catalytically favorable conformers, which can be considered as experimental conformation of the MD simulation results.
In conclusion, the MD study of OGG1 in complexes with different DNA substrates suggests that the population density of catalytically competent conformers of Lys249 and Asp268 is one of the main factors determining the enzymatic activity of OGG1. In this respect, human OGG1 as DNA glycosylase is similar to the bacterial 8-oxoguanine-DNA N-glycosylase Fpg, although there is no homology between the primary and tertiary structures of these proteins. In the case of Fpg, the main parameters of the active site promoting effective catalysis are conformations of Pro1 and Glu2, which perform the same functions as Lys249 and Asp268 in OGG1 [54, 55]. The results of our work are in agreement with the publihed data, which show that mutations preventing productive oxoG binding in the pocket inside the OGG1 protein globule, as well as oxoG binding in the unfavorable orientation, cause conformational changes that extend to more than 10 Å and reach the catalytic amino acid residues [48, 49]. Determination of distances and angles critical for catalysis makes it possible to use the active site geometry to predict the activity of natural OGG1 variants, which may be important for the prognosis of individual risks of developing cancer and the response of tumors to DNA-damaging chemo- and radiotherapy. The studies in this field are currently underway.
Funding. The study was supported by the Russian Science Foundation (project 18-74-00052; simulation, enzyme kinetics), Program for Fundamental Scientific Research of the State Academies of Sciences for 2013-2020 (project AAAA-A17-117020210023-1; oligonucleotide synthesis, protein purification), and Ministry of Education and Science of the Russian Federation (project 6.5773.2017/VU, data analysis).
Acknowledgements. The calculations were performed at the supercomputer NKS-30T cluster of the Siberian Supercomputer Center, Siberian Branch of the Russian Academy of Sciences.
Conflict of interest. The authors declare no conflict of interest.
Compliance with ethical standard.This article does not contain any studies involving animals or human participants performed by any of the authors.
REFERENCES
1.Von Sonntag, C. (2006) Free-Radical-Induced DNA
Damage and Its Repair: A Chemical Perspective, Springer,
Berlin-Heidelberg.
2.Halliwell, B., and Gutteridge, J. M. C. (2007)
Free Radicals in Biology and Medicine, 4th Edn., Oxford
University Press, Oxford.
3.Friedberg, E. C., Walker, G. C., Siede, W., Wood,
R. D., Schultz, R. A., and Ellenberger, T. (2006) DNA Repair and
Mutagenesis, ASM Press, Washington, D. C.
4.Culp, S. J., Cho, B. P., Kadlubar, F. F., and
Evans, F. E. (1989) Structural and conformational analyses of
8-hydroxy-2′-deoxyguanosine, Chem. Res. Toxicol.,
2, 416-422, doi: 10.1021/tx00012a010.
5.Kouchakdjian, M., Bodepudi, V., Shibutani, S.,
Eisenberg, M., Johnson, F., Grollman, A. P., and Patel, D. J. (1991)
NMR structural studies of the ionizing radiation adduct
7-hydro-8-oxodeoxyguanosine (8-oxo-7H-dG) opposite
deoxyadenosine in a DNA duplex.
8-Oxo-7H-dG(syn)•dA(anti) alignment at lesion
site, Biochemistry, 30, 1403-1412, doi:
10.1021/bi00219a034.
6.McAuley-Hecht, K. E., Leonard, G. A., Gibson, N.
J., Thomson, J. B., Watson, W. P., Hunter, W. N., and Brown, T. (1994)
Crystal structure of a DNA duplex containing
8-hydroxydeoxyguanine-adenine base pairs, Biochemistry,
33, 10266-10270, doi: 10.1021/bi00200a006.
7.Lipscomb, L. A., Peek, M. E., Morningstar, M. L.,
Verghis, S. M., Miller, E. M., Rich, A., Essigmann, J. M., and
Williams, L. D. (1995) X-Ray structure of a DNA decamer containing
7,8-dihydro-8-oxoguanine, Proc. Natl. Acad. Sci. USA, 92,
719-723, doi: 10.1073/pnas.92.3.719.
8.Shibutani, S., Takeshita, M., and Grollman, A. P.
(1991) Insertion of specific bases during DNA synthesis past the
oxidation-damaged base 8-oxodG, Nature, 349, 431-434,
doi: 10.1038/349431a0.
9.Grollman, A. P., and Moriya, M. (1993) Mutagenesis
by 8-oxoguanine: an enemy within, Trends Genet., 9,
246-249, doi: 10.1016/0168-9525(93)90089-Z.
10.ESCODD (European Standards Committee on Oxidative
DNA Damage), Gedik, C. M., and Collins, A. (2005) Establishing the
background level of base oxidation in human lymphocyte DNA: results of
an interlaboratory validation study, FASEB J., 19, 82-84,
doi: 10.1096/fj.04-1767fje.
11.Atamna, H., Cheung, I., and Ames, B. N. (2000) A
method for detecting abasic sites in living cells: age-dependent
changes in base excision repair, Proc. Natl. Acad. Sci. USA,
97, 686-691, doi: 10.1073/pnas.97.2.686.
12.Auffret van der Kemp, P., Thomas, D., Barbey, R.,
de Oliveira, R., and Boiteux, S. (1996) Cloning and expression in
Escherichia coli of the OGG1 gene of Saccharomyces
cerevisiae, which codes for a DNA glycosylase that excises
7,8-dihydro-8-oxoguanine and
2,6-diamino-4-hydroxy-5-N-methylformamidopyrimidine, Proc.
Natl. Acad. Sci. USA, 93, 5197-5202, doi:
10.1073/pnas.93.11.5197.
13.Rosenquist, T. A., Zharkov, D. O., and Grollman,
A. P. (1997) Cloning and characterization of a mammalian 8-oxoguanine
DNA glycosylase, Proc. Natl. Acad. Sci. USA, 94,
7429-7434, doi: 10.1073/pnas.94.14.7429.
14.Radicella, J. P., Dherin, C., Desmaze, C., Fox,
M. S., and Boiteux, S. (1997) Cloning and characterization of
hOGG1, a human homolog of the OGG1 gene of
Saccharomyces cerevisiae, Proc. Natl. Acad. Sci. USA,
94, 8010-8015, doi: 10.1073/pnas.94.15.8010.
15.Roldan-Arjona, T., Wei, Y.-F., Carter, K. C.,
Klungland, A., Anselmino, C., Wang, R.-P., Augustus, M., and Lindahl,
T. (1997) Molecular cloning and functional expression of a human cDNA
encoding the antimutator enzyme 8-hydroxyguanine-DNA glycosylase,
Proc. Natl. Acad. Sci. USA, 94, 8016-8020, doi:
10.1073/pnas.94.15.8016.
16.Tchou, J., Kasai, H., Shibutani, S., Chung,
M.-H., Laval, J., Grollman, A. P., and Nishimura, S. (1991)
8-Oxoguanine (8-hydroxyguanine) DNA glycosylase and its substrate
specificity, Proc. Natl. Acad. Sci. USA, 88, 4690-4694,
doi: 10.1073/pnas.88.11.4690.
17.Boiteux, S., Gajewski, E., Laval, J., and
Dizdaroglu, M. (1992) Substrate specificity of the Escherichia
coli Fpg protein (formamidopyrimidine-DNA glycosylase): excision of
purine lesions in DNA produced by ionizing radiation or
photosensitization, Biochemistry, 31, 106-110, doi:
10.1021/bi00116a016.
18.Nash, H. M., Lu, R., Lane, W. S., and Verdine, G.
L. (1997) The critical active-site amine of the human 8-oxoguanine DNA
glycosylase, hOgg1: direct identification, ablation and chemical
reconstitution, Chem. Biol., 4, 693-702, doi:
10.1016/S1074-5521(97)90225-8.
19.Zharkov, D. O., Rosenquist, T. A., Gerchman, S.
E., and Grollman, A. P. (2000) Substrate specificity and reaction
mechanism of murine 8-oxoguanine-DNA glycosylase, J. Biol.
Chem., 275, 28607-28617, doi: 10.1074/jbc.M002441200.
20.Bruner, S. D., Norman, D. P. G., and Verdine, G.
L. (2000) Structural basis for recognition and repair of the endogenous
mutagen 8-oxoguanine in DNA, Nature, 403, 859-866, doi:
10.1038/35002510.
21.Norman, D. P. G., Chung, S. J., and Verdine, G.
L. (2003) Structural and biochemical exploration of a critical amino
acid in human 8-oxoguanine glycosylase, Biochemistry, 42,
1564-1572, doi: 10.1021/bi026823d.
22.Norman, D. P. G., Bruner, S. D., and Verdine, G.
L. (2001) Coupling of substrate recognition and catalysis by a human
base-excision DNA repair protein, J. Am. Chem. Soc., 123,
359-360, doi: 10.1021/ja003144m.
23.Bjoras, M., Seeberg, E., Luna, L., Pearl, L. H.,
and Barrett, T. E. (2002) Reciprocal “flipping” underlies
substrate recognition and catalytic activation by the human
8-oxoguanine DNA glycosylase, J. Mol. Biol., 317,
171-177, doi: 10.1006/jmbi.2002.5400.
24.Fromme, J. C., Bruner, S. D., Yang, W., Karplus,
M., and Verdine, G. L. (2003) Product-assisted catalysis in
base-excision DNA repair, Nat. Struct. Biol., 10,
204-211, doi: 10.1038/nsb902.
25.Chung, S. J., and Verdine, G. L. (2004)
Structures of end products resulting from lesion processing by a DNA
glycosylase/lyase, Chem. Biol., 11, 1643-1649, doi:
10.1016/j.chembiol.2004.09.014.
26.Banerjee, A., Yang, W., Karplus, M., and Verdine,
G. L. (2005) Structure of a repair enzyme interrogating undamaged DNA
elucidates recognition of damaged DNA, Nature, 434,
612-618, doi: 10.1038/nature03458.
27.Banerjee, A., and Verdine, G. L. (2006) A
nucleobase lesion remodels the interaction of its normal neighbor in a
DNA glycosylase complex, Proc. Natl. Acad. Sci. USA, 103,
15020-15025, doi: 10.1073/pnas.0603644103.
28.Radom, C. T., Banerjee, A., and Verdine, G. L.
(2007) Structural characterization of human 8-oxoguanine DNA
glycosylase variants bearing active site mutations, J. Biol.
Chem., 282, 9182-9194, doi: 10.1074/jbc.M608989200.
29.Lee, S., Radom, C. T., and Verdine, G. L. (2008)
Trapping and structural elucidation of a very advanced intermediate in
the lesion-extrusion pathway of hOGG1, J. Am. Chem. Soc.,
130, 7784-7785, doi: 10.1021/ja800821t.
30.Crenshaw, C. M., Nam, K., Oo, K., Kutchukian, P.
S., Bowman, B. R., Karplus, M., and Verdine, G. L. (2012) Enforced
presentation of an extrahelical guanine to the lesion recognition
pocket of human 8-oxoguanine glycosylase, hOGG1, J. Biol. Chem.,
287, 24916-24928, doi: 10.1074/jbc.M111.316497.
31.Li, H., Endutkin, A. V., Bergonzo, C., Fu, L.,
Grollman, A. P., Zharkov, D. O., and Simmerling, C. (2017) DNA
deformation-coupled recognition of 8-oxoguanine: conformational kinetic
gating in human DNA glycosylase, J. Am. Chem. Soc., 139,
2682-2692, doi: 10.1021/jacs.6b11433.
32.Lukina, M. V., Popov, A. V., Koval, V. V.,
Vorobjev, Y. N., Fedorova, O. S., and Zharkov, D. O. (2013) DNA damage
processing by human 8-oxoguanine-DNA glycosylase mutants with the
occluded active site, J. Biol. Chem., 288, 28936-28947,
doi: 10.1074/jbc.M113.487322.
33.Kabsch, W. (1976) A solution for the best
rotation to relate two sets of vectors, Acta Crystallogr. A,
32, 922-923, doi: 10.1107/S0567739476001873.
34.Popov, A. V., and Vorobjev, Y. N (2010)
GUI-BioPASED program for molecular dynamics simulations of biopolymers
with a graphical user interface, Mol. Biol., 44, 735-742,
doi: 10.1134/S0026893310040217.
35.Case, D. A., Darden, T. A., Cheatham, T. E., III,
Simmerling, C. L., Wang, J., et al. (2012) AMBER 12, University
of California, San Francisco.
36.Perlow-Poehnelt, R. A., Zharkov, D. O., Grollman,
A. P., and Broyde, S. (2004) Substrate discrimination by
formamidopyrimidine-DNA glycosylase: distinguishing interactions within
the active site, Biochemistry, 43, 16092-16105, doi:
10.1021/bi048747f.
37.Vorobjev, Y. N. (2011) Advances in implicit
models of water solvent to compute conformational free energy and
molecular dynamics of proteins at constant pH, Adv. Protein Chem.
Struct. Biol., 85, 281-322, doi:
10.1016/B978-0-12-386485-7.00008-9.
38.Manning, G. S. (1978) The molecular theory of
polyelectrolyte solutions with applications to the electrostatic
properties of polynucleotides, Q. Rev. Biophys., 11,
179-246, doi: 10.1017/s0033583500002031.
39.Ravishanker, G., Auffinger, P., Langley, D. R.,
Jayaram, B., Young, M. A., and Beveridge, D. L. (1997) Treatment of
counterions in computer simulations of DNA, Rev. Comput. Chem.,
11, 317-372, doi: 10.1002/9780470125885.ch6.
40.Popov, A. V., Vorobjev, Y. N., and Zharkov, D. O.
(2013) MDTRA: a molecular dynamics trajectory analyzer with a graphical
user interface, J. Comput. Chem., 34, 319-325, doi:
10.1002/jcc.23135.
41.Humphrey, W., Dalke, A., and Schulten, K. (1996)
VMD: visual molecular dynamics, J. Mol. Graph., 14,
33-38, doi: 10.1016/0263-7855(96)00018-5.
42.Sayle, R. A., and Milner-White, E. J. (1995)
RASMOL: biomolecular graphics for all, Trends Biochem. Sci.,
20, 374-376, doi: 10.1016/S0968-0004(00)89080-5.
43.Yang, W., Bitetti-Putzer, R., and Karplus, M.
(2004) Free energy simulations: use of reverse cumulative averaging to
determine the equilibrated region and the time required for
convergence, J. Chem. Phys., 120, 2618-2628, doi:
10.1063/1.1638996.
44.Letunic, I., and Bork, P. (2016) Interactive tree
of life (iTOL) v3: an online tool for the display and annotation of
phylogenetic and other trees, Nucleic Acids Res., 44,
W242-W245, doi: 10.1093/nar/gkw290.
45.Sambrook, J., and Russell, D. W. (2001)
Molecular Cloning: a Laboratory Manual, 3rd Edn.,
Cold Spring Harbor Laboratory Press, Cold Spring Harbor.
46.Sidorenko, V. S., Nevinsky, G. A., and Zharkov,
D. O. (2007) Mechanism of interaction between human 8-oxoguanine-DNA
glycosylase and AP endonuclease, DNA Repair, 6, 317-328,
doi: 10.1016/j.dnarep.2006.10.022.
47.Sidorenko, V. S., Mechetin, G. V., Nevinsky, G.
A., and Zharkov, D. O. (2008) Ionic strength and magnesium affect the
specificity of Escherichia coli and human 8-oxoguanine-DNA
glycosylases, FEBS J., 275, 3747-3760, doi:
10.1111/j.1742-4658.2008.06521.x.
48.Anderson, P. C., and Daggett, V. (2009) The R46Q,
R131Q and R154H polymorphs of human DNA glycosylase/β-lyase hOgg1
severely distort the active site and DNA recognition site but do not
cause unfolding, J. Am. Chem. Soc., 131, 9506-9515, doi:
10.1021/ja809726e.
49.Sowlati-Hashjin, S., and Wetmore, S. D. (2018)
Structural insight into the discrimination between 8-oxoguanine
glycosidic conformers by DNA repair enzymes: a molecular dynamics study
of human oxoguanine glycosylase 1 and formamidopyrimidine-DNA
glycosylase, Biochemistry, 57, 1144-1154, doi:
10.1021/acs.biochem.7b01292.
50.Bjoras, M., Luna, L., Johnsen, B., Hoff, E.,
Haug, T., Rognes, T., and Seeberg, E. (1997) Opposite base-dependent
reactions of a human base excision repair enzyme on DNA containing
7,8-dihydro-8-oxoguanine and abasic sites, EMBO J., 16,
6314-6322, doi: 10.1093/emboj/16.20.6314.
51.Kuznetsov, N. A., Koval, V. V., Zharkov, D. O.,
Nevinsky, G. A., Douglas, K. T., and Fedorova, O. S. (2005) Kinetics of
substrate recognition and cleavage by human 8-oxoguanine-DNA
glycosylase, Nucleic Acids Res., 33, 3919-3931, doi:
10.1093/nar/gki694.
52.Zharkov, D. O., Golan, G., Gilboa, R., Fernandes,
A. S., Gerchman, S. E., Kycia, J. H., Rieger, R. A., Grollman, A. P.,
and Shoham, G. (2002) Structural analysis of an Escherichia coli
endonuclease VIII covalent reaction intermediate, EMBO J.,
21, 789-800, doi: 10.1093/emboj/21.4.789.
53.Li, H., Endutkin, A. V., Bergonzo, C., Campbell,
A. J., de los Santos, C., Grollman, A., Zharkov, D. O., and Simmerling,
C. (2016) A dynamic checkpoint in oxidative lesion discrimination by
formamidopyrimidine–DNA glycosylase, Nucleic Acids Res.,
44, 683-694, doi: 10.1093/nar/gkv1092.
54.Sowlati-Hashjin, S., and Wetmore, S. D. (2014)
Computational investigation of glycosylase and β-lyase activity
facilitated by proline: applications to FPG and comparisons to hOgg1,
J. Phys. Chem. B, 118, 14566-14577, doi:
10.1021/jp507783d.
55.Popov, A. V., Endutkin, A. V., Vorobjev, Y. N.,
and Zharkov, D. O. (2017) Molecular dynamics simulation of the
opposite-base preference and interactions in the active site of
formamidopyrimidine-DNA glycosylase, BMC Struct. Biol.,
17, 5, doi: 10.1186/s12900-017-0075-y.