PTPN11 Knockdown Prevents Changes in the Expression of Genes Controlling Cell Cycle, Chemotherapy Resistance, and Oncogene-Induced Senescence in Human Thyroid Cells Overexpressing BRAF V600E Oncogenic Protein
L. V. Putlyaeva1,2, D. E. Demin1,3, A. N. Uvarova1,4, L. S. Zinevich5, M. M. Prokofjeva1, G. R. Gazizova6, E. I. Shagimardanova6, and A. M. Schwartz1,a*
1Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, 119991 Moscow, Russia2Center of Life Sciences, Skolkovo Institute of Science and Technology, 121205 Moscow, Russia
3Moscow Institute of Physics and Technology, 141701 Dolgoprudnyi, Moscow Region, Russia
4Lomonosov Moscow State University, Faculty of Biology, 119234 Moscow, Russia
5Koltzov Institute of Developmental Biology, Russian Academy of Sciences, 119334 Moscow, Russia
6Institute of Fundamental Medicine and Biology, Kazan Federal University, 420008 Kazan, Russia
* To whom correspondence should be addressed.
Received September 21, 2019; Revised October 20, 2019; Accepted October 20, 2019
The MAPK (RAS/BRAF/MEK/ERK) signaling pathway is a kinase cascade involved in the regulation of cell proliferation, differentiation, and survival in response to external stimuli. The V600E mutation in the BRAF gene has been detected in various tumors, resulting in a 500-fold increase in BRAF kinase activity. However, monotherapy with selective BRAF V600E inhibitors often leads to reactivation of MAPK signaling cascade and emergence of drug resistance. Therefore, new targets are being developed for the inhibition of components of the aberrantly activated cascade. It was recently discovered that resistance to BRAF V600E inhibitors may be associated with the activity of the tyrosine phosphatase SHP-2 encoded by the PTPN11 gene. In this paper, we analyzed transcriptional effects of PTPN11 gene knockdown and selective suppression of BRAF V600E in a model of thyroid follicular epithelium. We found that the siRNA-mediated knockdown of PTPN11 after vemurafenib treatment prevented an increase in the expression CCNA1 and NOTCH4 genes involved in the formation of drug resistance of tumors. On the other hand, downregulation of PTPN11 expression blocked the transcriptional activation of genes (p21, p15, p16, RB1, and IGFBP7) involved in cell cycle regulation and oncogene-induced senescence in response to BRAF V600E expression. Therefore, it can be assumed that SHP-2 participates not only in emergence of drug resistance in cancer cells, but also in oncogene-induced cell senescence.
KEY WORDS: SHP-2, BRAF V600E, thyroid tumor, oncogene-induced senescenceDOI: 10.1134/S0006297920010101
Abbreviations: EGFR, epidermal growth factor receptor; OIS, oncogene-induced senescence; RT-qPCR, reverse transcription/quantitative PCR.
Aberrantly activation of RAS/RAF/MEK/ERK signaling (MAPK cascade)
contributing to cell survival and metastasis has been described as a
common mechanism in cancer emergence and progression [1]. The V600E mutation in the BRAF gene
(BRAF V600E) identified in multiple cancer types (e.g., thyroid
cancer) is one of the most common driver mutations affecting components
of the MAPK-cascade [2]. Cells expressing the BRAF
V600E protein exhibit elevated chromosomal instability and mitotic
activity and possess higher invasion potential due to the MEK-dependent
upregulation of the expression of matrix metalloproteases (MMP-3,
MMP-9, and MMP-13). BRAF V600E-bearing tumors display more invasive
behavior [3] associated with poor therapeutic
response to radioiodine (131I) therapy and higher mortality
[4]. Taking into account the aforementioned data,
BRAF V600E has been recently viewed as a major therapeutic target in
these types of cancer.
SHP-2 encoded by the PTPN11 gene was the first protein phosphatase identified as an oncogenic protein [5-7]. It is expressed in diverse cell types and participates in oncogenic events in leukemia and breast and lung cancers by regulating invasion, metastasis, apoptosis, oncogene-induced senescence (OIS), DNA damage, cell proliferation, cell cycle progression, and drug resistance [8, 9]. It is believed that SHP-2 plays an important role in the activation of Ras family proteins [10, 11], as well as JAK/STAT, PI3K/Akt, and other signaling pathways [12].
Inactivation of the PTPN11 gene results in embryonic death and multiple congenital anomalies occurring at gastrulation stage in different species (mouse, fly, and worm) [13]. In addition, SHP-2 regulates the insulin signaling [14] and development of central nervous system and heart [15, 16]. The role of this phosphatase in the etiology of various tumors differs depending on the disease stage and anatomical location of the tumor. SHP-2 acts as a tumor suppressor in hepatic carcinoma [17] and metachondromatosis [18] and, on contrary, functions as an oncogenic protein in leukemia, lung and breast cancers, and melanoma [19]. PTPN11-activating mutations have been found in samples collected from patients with acute myeloid leukemia, gastric cancer, glioblastoma, and anaplastic large-cell lymphoma, which allowed to define PTPN11 as a protooncogene [20]. PTPN11 expression is upregulated in thyroid cancer and correlates with the low-grade differentiation, high malignancy, and increased rate of lymph node metastasis [21]. Upregulated PTPN11 expression was detected in thyroid cancer cell lines SW579, IHH-4, FTC-133, TPC-1, DRO, TA-K, and ML-1 [21]. It was found that SHP-2 downregulation may prevent reactivation of the RAS/RAF/MEK/ERK cascade mediated by the activated epidermal growth factor receptors (EGFRs) in murine lung adenocarcinoma model and in colon cancer cell lines [22, 23]. The PTPN11 knockdown by antisense oligonucleotides slows down the growth of cancer cells and induces apoptosis in thyroid cancer cell line SW579 [21]. There are data that point at both positive and negative influence of PTPN11-activating mutations on the Ras/MAPK cascade in patients with the Noonan syndrome [14]. However, other data prove that the shRNA-mediated knockdown of PTPN11 has no effect on the growth and proliferation of cancer A2058 cells bearing the BRAF V600E mutation [24].
Recently, it was demonstrated that the tyrosine phosphatase SHP-2 is often activated in human melanoma biopsy samples and melanoma cell lines. SHP-2 was found to participate in the activation of RAS/RAF/MAPK cascade in melanoma cells expressing mutant NRAS protein and cells bearing wild-type BRAF, whereas PTPN11 knockdown resulted in the suppression of cell proliferation and ERK activation in WM3211 and MeWo human melanoma cell lines [19].
Vemurafenib (PLX4032) is a low-molecular-weight inhibitor of BRAF V600E that was approved by the FDA in 2011 for the therapy of metastatic melanoma [25]. When used for the treatment of melanoma and thyroid carcinoma, vemurafenib was able to activate signaling via receptor tyrosine kinases (RTKs) resulting in rapid reactivation and stimulation of the EGFR signaling pathway [26, 27]. SHP-2 is involved in the signal transduction from EGFRs to p21Ras (RASA1) and other signaling molecules, so SHP-2 downregulation may reduce reactivation of the MAPK cascade. Some data indicate that vemurafenib causes SHP-2 activation and reactivates signaling via MEK/ERK in the case of hepatocyte growth factor (HGF) expression in vemurafenib-resistant cells [22].
Our study was aimed at assessing the impact of downregulated PTPN11 transcription on the expression of genes involved in regulation of cell proliferation, chemotherapy resistance, and OIS after selective BRAF V600E inhibition. Transcriptome analysis was performed to identify genes displaying noticeable changes in gene expression. Based on the results of transcriptome analysis, we selected a number of genes involved in the cycle control and cell senescence for further studies by quantitative PCR (qPCR). As a result, we found that PTPN11 knockdown with small interfering RNAs (siRNAs) minimizes effects of the BRAF V600E on transcriptional levels of genes involved in the cell cycle regulation. In particular, downregulation of PTPN11 expression prevented activation of expression of genes participating in the OIS, such as RB1, p21, p15, p16, and IGFBP, in response to BRAF V600E and/or vemurafenib. This suggested that SHP-2 takes part in the induction of OIS, which might account for its tumor-suppressing activity.
MATERIALS AND METHODS
BRAF V600E-bearing lentiviral vectors. For cloning of the BRAF gene containing the oncogenic V600E mutation, the 5′-fragment of the BRAF gene was excised from the commercially available plasmid pBABE-Puro-BRAF(V600E) (Addgene, USA) [28] by the BamHI and MunI restriction sites; the 3′-fragment was amplified by PCR with BRAF-in-Mun-F and BRAF-Not-R primers (table). The BamHI (5′-end) and NotI (3′-end) sites were introduced into commercially available LeGO-iPuro2 plasmid carrying the puromycin resistance gene (Lentiviral Gene Ontology Vectors, Germany) that was then used as a vector for BRAF cloning. Primer sequences are shown in the table.
Primer sequences

Cell culture and lentiviral vector transduction. Immortalized thyroid follicular epithelial Nthy-ori 3-1 cells (Sigma, USA) were grown in RPMI1640 culture medium (PanEco, Russia) supplemented with 10% fetal bovine serum (Biosera, France), 2 mM L-glutamine, penicillin (100 U/ml)/streptomycin (100 µg/ml), and 1 mM sodium pyruvate. The cells were used for obtaining two cell lines transduced with the original LeGO-iPuro2 vector and iPuro2-BRAF V600E carrying the human BRAF gene with the driver oncogenic mutation V600E, respectively. Viral particles were obtained as described by Prokofjeva et al. [29]. Transduced cells were selected on a puromycin-containing medium (final puromycin concentration, 1 µg/ml) for 7 days. Modification of the cell transcriptional program after insertion of the target genes was additionally confirmed by qPCR with the CCND1 F/R, PAPSS2 F/R, and FBLN1 F/R primers (table) and by analysis of the expression profile.
PTPN11 knockdown and selective BRAF V600E inhibition. BRAF V600E was suppressed using selective inhibitor vemurafenib (PLX4032; Selleck, USA) at the concentration of 1 µM. The PTPN11 gene was knocked down in Nthy-ori 3.1 and Nthy-ori 3.1/BRAF V600E cells using the corresponding siRNAs and RNAiMAX transfection reagent (Thermo Fisher Scientific, USA) according to the manufacturer’s recommendations. For the combined effect, siRNAs were added 72 h after incubation with vemurafenib. As a control of PTPN11 knockdown, we used scrambled RNA (scRNA) as described before [30] [see Table S1 in the Supplement to this paper on the web sites of Biochemistry (Moscow) (http://protein.bio.msu.ru/biokhimiya/) and Springer Publishing (https://link.springer.com/journal/10541)].
RNA isolation, cDNA synthesis, and qPCR. Total RNA was isolated from Nthy-ori 3-1 and Nthy-ori 3-1/BRAF V600E cells using Trizol reagent (Invitrogen, USA) according to manufacturer’s recommendations. Reverse transcription reaction was carried out using of 2 µg total RNA and MMLV RT Kit (Evrogen, Russia). qPCR was performed according to Schwartz et al. [31]. Primer sequences are shown in the table.
Western blotting. Western blotting was performed according to Afanasyeva et al. [32]. Cell lysates were mixed with 4× Laemmli buffer at a 3 to 1 ratio, incubated for 5 min at 95°C, and loaded on 10% polyacrylamide gel (30 µg protein per well). Analysis of Erk1/2 phosphorylation was carried out using phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) antibodies (dilution 1 : 2000; Cell Signaling, USA) followed by incubation with secondary goat anti-rabbit IgG horseradish peroxidase-conjugated antibodies (1 : 30,000 dilution; Thermo Scientific, USA). The total amount of Erk1/2 was determined with anti-p44/42 MAPK (Erk1/2) antibodies (dilution, 1 : 2000; Cell Signaling) followed by incubation with secondary goat anti-mouse IgG horseradish peroxidase-conjugated antibodies (dilution, 1 : 30,000; Thermo Scientific).
Cell transcriptome analysis. Total RNA was isolated from Nthy-ori 3-1 and Nthy-ori 3-1/BRAF V600E cells as described above. Next, mRNA was purified from 400 ng of total RNA using NebNext PolyA mRNA Isolation Module (New England Biolabs, UK) fragmented (14 min, 94°C) in the presence of random primers and used as a template in the reverse transcription reaction. The resulting cDNA was isolated using AMPure XP paramagnetic beads (Beckman Coulter, USA) and ligated with adaptors. This was followed by the introduction of barcodes, amplification, and another round of purification on paramagnetic beads. All reactions were performed using NEBNext Ultra II Directional RNA Library preparation kit for Illumina (New England Biolabs). Library quantification was performed with a Qubit 3.0 fluorometer (Invitrogen); library quality was assessed with a Bioanalyser 2100 (Agilent Technologies, USA). Prior to sequencing, library quantification was validated by qPCR with 2.5× EVA Green reaction mix for qPCR (Syntol, Russia) and adaptor primers from Illumina (Evrogen). Based on the effective concentration, the library was diluted to 2 nM and pooled in accordance with expected depth of sequencing. Sequencing was carried out on the Illumina HiSeq 2500 platform (Illumina, Germany) in the 57-bp single-end read mode using HiSeq SR Cluster Kit v4 cBot and HiSeq SBS V4 50-Cycle Kit (Illumina). Raw data are available from the SRA database (ncbi.nlm.nih.gov/sra/; project no. PRJNA529086). Illumina sequencing data were processed with the Trimmomatic software for read trimming [33], STAR software for read mapping to the human reference sequence (release GRCh38) [34], and FeatureCounts algorithm for read counting [35]. The calculations were carried out at the Common Use Genome Center (http://www. eimb.ru/rus/ckp/ccu_genome_c.php). The number of reads in each sample was normalized using the DESeq2 algorithm [36]. The correlation coefficient was defined as the linear Pearson’s correlation coefficient (r-Pearson’s). Genes whose expression changed 2-fold or more were used for analyzing differential expression. Functional enrichment for differentially expressed genes was calculated using the web-based portal Metascape [37].
RESULTS
Expression of BRAF V600E in Nthy-ori 3-1 cells activates intracellular RAS/RAF/MEK/ERK signaling. The effects of phosphatase SHP-2 on the transcriptional program in thyroid cells carrying the oncogenic BRAF V600E mutation were investigated using the cell line stably expressing BRAF V600E that was derived from the human follicular epithelial cell line Nthy-ori 3-1 by transduction with the lentiviral vector LeGO iPuro2-BRAF V600E (see “Materials and Methods”). Expression of the lentiviral mRNA for BRAF V600E was confirmed by RT-qPCR (Fig. 1a).
Fig. 1. a) Expression of lentiviral mRNA carrying BRAF V600E normalized to the GAPDH expression level (y-axis) in parenteral Nthy-ori-3.1 cell line and Nthy-ori-3.1/BRAF V600E cells. b) Expression of CCND1, PAPSS2, and FBLN1 genes in normal thyroid tissue and thyroid tumor bearing the BRAF V600E mutation based on transcriptome analysis (GEO dataset ID, GSE27155 [37]). Expression levels (y-axis) are normalized to the mean values of the expression levels of the same genes in normal thyroid tissue. c) Expression of CCND1, PAPSS2, and FBLN1 in nontransformed Nthy-ori-3.1 cells and Nthy-ori-3.1/BRAF V600E cells overexpressing BRAF V600E (y-axis) normalized to the GAPDH expression level and expression of mRNAs for the same genes in nontransformed Nthy-ori-3.1 cells. d) Western blot analysis of Erk1/2 (p44/42 MAPK) phosphorylation. Right panel, phosphorylated Erk1/2; left panel, total Erk1/2. Loading control is shown on both panels (membrane staining with Ponceau S dye). Relative increase in the total and phosphorylated Erk1/2 in BRAF V600E-bearing cells vs. parental Nthy-ori-3.1 cells is shown. All data (a-d) are shown as means ± SEM from three independent repeats; *, significant difference between the compared groups (p-value < 0.05 according to the Student’s t-test).
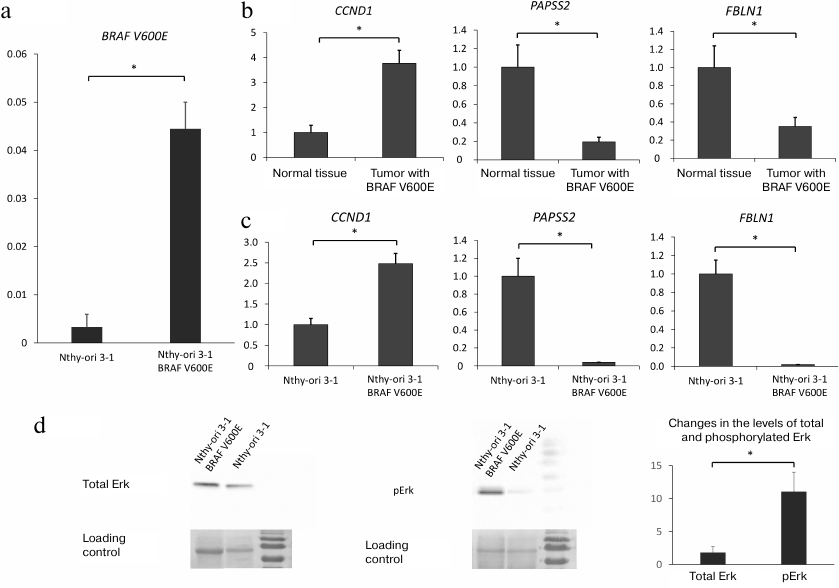
The Nthy-ori/BRAF V600E cell line was analyzed for the expression of cyclin D1 (CCND1), a key effector in the MAPK and PI3K-AKT signaling pathways [38-40], because cyclin D1 expression is known to be activated in BRAF V600E-bearing tumors. We also assessed expression of 3′-phosphoadenosine 5′-phosphosulfate synthase 2 (PAPSS2) and fibulin 1 (FBLN1) that were shown to be profoundly downregulated in BRAF V600E-positive thyroid tumors (according to publicly available transcriptome profiling data [39]) (Fig. 1b). It was found that expression of mutant BRAF V600E resulted in modest upregulation of CCND1 expression (~2.0 to 2.5-fold compared to nontransformed cells) and significant downregulation of the of PAPSS2 and FBLN1 expression (Fig. 1c). In addition, we determined the amounts of Erk1/2 (p44/42 MAPK) [41] and its phosphorylated form pErk (a key component in the MAPK cascade) in Nthy-ori 3-1 and Nthy-ori 3-1/BRAF V600E cells and found that the BRAF V600E-bearing cells contained elevated amounts of pErk (Fig. 1d).
The results of transcriptome analysis of Nthy-ori 3-1 and Nthy-ori 3-1/BRAF V600E cells were compared with similar data published earlier [42]. The correlation coefficient for the earlier published data and the results we received for the Nthy-ori/BRAF V600E cell line was 0.76, demonstrating a relatively high similarity between the data.
Therefore, the Nthy-ori/BRAF V600E cell line stably expressing BRAF V600E displayed all features of the oncogene-mediated changes in the cell transcriptional program.
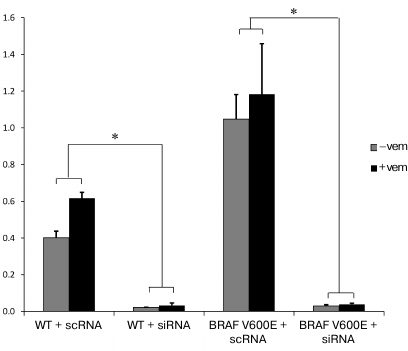
Downregulation of PTPN11 suppresses expression of genes involved in OIS and activation of MAPK and PI3K-AKT pathways in cells expressing BRAF V600E. In order to compare the effects of BRAF V600E selective inhibitor vemurafenib alone or in combination with PTPN11 suppression on gene expression in thyroid epithelial cells carrying the BRAF V600E mutation, the PTPN11 gene was knocked down in Nthy-ori 3-1 and Nthy-ori 3-1/BRAF V600E cells using siRNAs. We measured the efficiency of PTPN11 suppression by RT-qPCR and found that SHP-2 expression was strongly downregulated in both cell lines (Fig. 2).
Fig. 2. PTPN11 knockdown with siRNAs in nontransformed Nthy-ori 3-1 (WT) and Nthy-ori 3-1/BRAF V600E cells (BRAF V600E) without (–vem) and after vemurafenib treatment (+vem). The cells were transfected with siRNA (SI) or scRNA (SC). PTPN11 expression was normalized to the GAPDH expression (y-axis). The data are shown as mean ± SEM from two independent repeats; *, significant difference with the cells transfected with scRNA (p-value < 0.05 according to the Student’s t-test).
To investigate the role of SHP-2 in the modulation of cell response to vemurafenib, we analyzed cell transcriptome in the cells with normal and suppressed PTPN11 expression with and without vemurafenib treatment (see Table S1 in Supplement) to identify the groups of genes whose expression was most affected by the experimental conditions. In particular, significant changes in the mRNA levels of BRAF V600E-expressing cells treated by vemurafenib were described for genes which can be classified into the following groups: “extracellular matrix” (group 1 and 3), “calcium-ion-induced cell response” (group 4) and “phase I xenobiotic response” (group 5) (Fig. S1a in Supplement). Downregulation of PTPN11 expression resulted in altered expression of genes associated with “chronic inflammatory response” (group 2), “corticosteroid-induce cell response” (group 3), and “positive regulation of IL-8 secretion” (group 4) (Fig. S1b in Supplement). Unfortunately, the decrease in the PTPN11 expression after combined use of vemurafenib and PTPN11 siRNA was insufficient for assessing their full impact on the cell transcriptome. In that regard, based on the results obtained from other samples, we selected a set of genes involved in cell growth, survival, and sensitivity to chemotherapy for further analysis. First of all, we noticed a marked influence of PTPN11 downregulation on the levels of IL-8 and some other pro-inflammatory and growth factors. Changes in the mRNA levels for these genes are typical for cells undergoing OIS [43]. Moreover, PTPN11 downregulation resulted in altered expression of some key factors linked to OIS development (IL-8, p15, p16, p21 and IGFBP7). For instance, factor p21 causes cell cycle arrest at the G1 and G2 phases via inhibiting cyclin-dependent kinase 1A [41, 42]. Factor p16 exhibits its effects via pRb signaling pathway by suppressing activity of cyclin-dependent kinases, resulting in the cell cycle arrest at the G1 phase [44], whereas factor p15 acts via the TGF-β pathway and targets cyclin-dependent kinases CDK4 and CDK6 [45]. IGFBP7 plays a pivotal role in the inhibition of BRAF-MEK-ERK pathway and OIS induction in nevus cells bearing the BRAF V600E mutation [46].
Therefore, transcriptome analysis allowed us to find changes in the expression of these and other genes directly involved in the regulation of cell proliferation and survival. For some of these genes, altered expression was verified by RT-qPCR (Fig. 3).
Fig. 3. Relative expression of genes involved in the regulation of cell proliferation and survival after siRNA-mediated PTPN11 knockdown and/or selective BRAF V600E inhibition with vemurafenib. WT, Nthy-ori 3-1 cells; BRAF V600E, Nthy-ori 3-1/BRAF V600E cells; +vem, vemurafenib treatment; SI, cells transfected anti-PTPN11 siRNA; SC, cells transfected with scRNA. Gene expression (y-axis) was normalized to expression; * significant difference with cells transfected with scRNA (p-value < 0.05 according to the Student’s t-test).
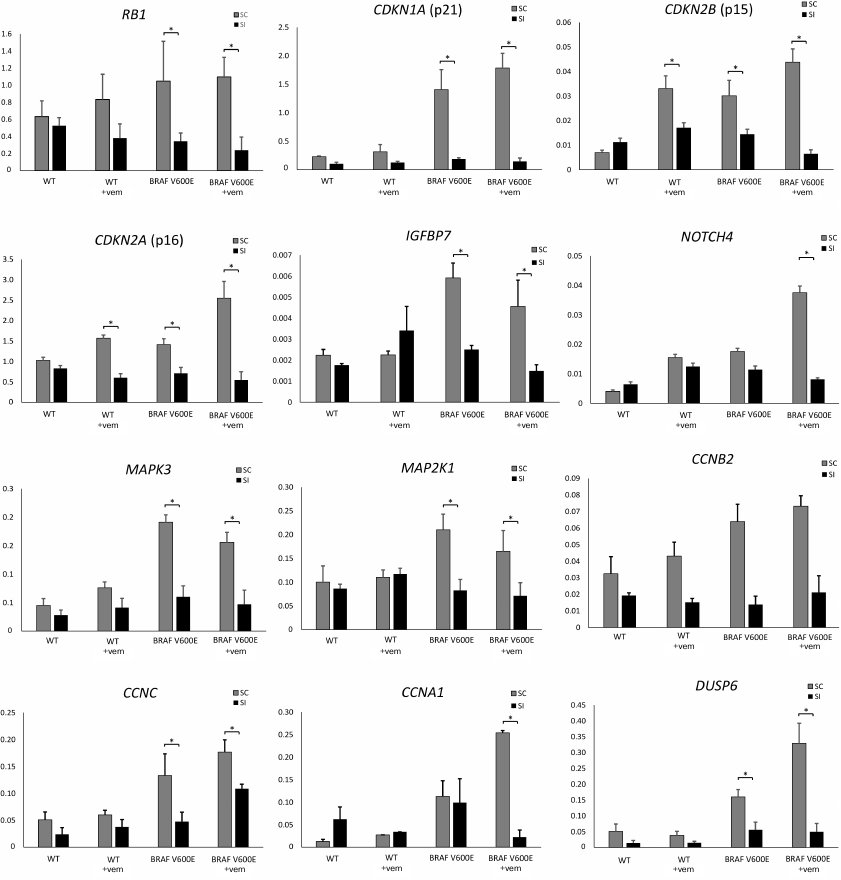
Expression of several genes involved in the OIS development (p15, p16, p21, and IGFBP7) was upregulated in Nthy-ori 3-1/BRAF V600E cells compared to the parental Nthy-ori 3-1 cells (Fig. 3). Besides, expression of the BRAF V600E resulted in the upregulation of mRNAs for IL-6, IL-8, and VEGFA, whose secretion is activated in cells during OIS [43] (Fig. S2 in Supplement). These data confirm our hypothesis that expression of these genes was activated by the presence of the BRAF V600E. We also demonstrated that PTPN11 knockdown significantly decreased expression of OIS regulators (RB1, p15, p16, p21, and IGFBP7) and genes of senescence-associated secretory phenotype (IL6, IL8, and VEGFA), lowering their expression in Nthy-ori 3-1/BRAF V600E cells to the level observed in the parental cell line (Fig. 3 and Supplementary Fig. S2). Hence, we suggest that tyrosine phosphatase SHP-2 functions as a pivotal component in the OIS regulation. Interestingly, treatment by vemurafenib in itself upregulates expression of p15 and p16 genes even in the BRAF V600E-free cells, but this effect was also abolished by the PTPN11 knockdown (Fig. 3).
We also showed that introduction of the BRAF V600E mutation upregulated expression of genes involved in the activation of MAPK and PI3K-AKT signaling in tumors with aggressive phenotype, such as Notch4 [44], MAPK3 (ERK1), MEK1, and multiple cyclin family members (genes CCNB2, CCNC, and CCNA1). Expression of DUSP6 as a negative regulator of BRAF overexpression was upregulated as well [45, 46]. It should be noted that for the aforementioned genes (Notch4, CCNA1, DUSP6, etc.), vemurafenib was often able to augment the effect caused by the introduction of BRAF V600E that was then abrogated by the PTPN11 suppression. It was shown earlier that Notch4 and CCNA1 participate in the development of drug resistance in breast [47] and ovarian [48] cancers. Moreover, upregulated expression of CCNA1 is associated with the increased rate of acute lymphoblastic leukemia development in the mouse model of this disease [49], whereas high levels of CCNB2 and CCNC expression may be observed in various growing tumors in colorectal adenocarcinoma, breast cancer [50], and pituitary adenoma [51]. On the contrary, downregulation of PTPN11 expression attenuates the influence of BRAF V600E on the expression of cyclin genes CCNA1, CCNB2, and CCNC and Notch4 gene, thereby pointing at the ambiguous role of SHP-2 in the development of tumors with the BRAF V600E mutation.
DISCUSSION
Impairments in the RAS/RAF/MEK/ERK signaling have been described in various human tumors. The control of MAPK signaling is based on the negative feedback mechanism involving proteins participating in the cascade. In normal functioning, the components of MAPK cascade are activated sequentially; however, the emergence of constitutively activated elements (e.g., BRAF V600E) or inhibition of some cascade steps might result in signaling blockade or activation, respectively, by the feedback mechanism. We believe that SHP-2 can play an important role in such regulation, on one hand, by reactivating cell proliferation in the case of suppression of some effector molecules and, on the other hand, by suppressing proliferation of cells constitutively expressing active cascade components. Thus, it was shown that the monotherapy aimed at inhibiting a single component of the MAPK cascade (e.g., MEK or RAF) results in the upregulated expression of receptor tyrosine kinases followed by the emergence of the adaptive resistance to the inhibitor in diverse tumor types [52-55]. However, combined use of inhibitors targeting SHP-2 and ALK or MEK efficiently suppressed the growth of cancer cells with altered ALK [56] and RAS [57] regulation. Our data on the SHP-2 role in the regulation of transcription of genes (Notch4, CCNA1) involved in the activation of tumor growth in response to the BRAF V600E expression after vemurafenib treatment also point at the SHP-2 participation in the restoration of cell proliferation in response to the inhibition of the RAS/RAF/MEK/ERK signaling. On the other hand, the same data indicate that SHP-2 plays an important role in the activation of transcription of p15, p16, p21, RB1, and IGFBP7 genes involved in the OIS induced by BRAF V600E expression. Vemurafenib may double the inhibitory effect of PTPN11 downregulation for the p15 and IGFBP7 genes.
In conclusion, we believe that phosphatase SHP-2 is important in the regulation of the MAPK cascade both as a component that transmits the signal from EGFR, HER2, and other proteins and as a factor involved in the negative feedback regulation in the case of cascade disturbance. This may explain the dual effect exhibited by SHP-2 on oncogenesis, which might depend on the presence/absence of mutant components of the MAPK cascade and OIS, as well as the type of therapeutic intervention. Our data point at potential ambiguous consequences of SHP-2 suppression in cells constitutively expressing effector molecules of the RAS/RAF/MEK/ERK cascade. On one hand, inhibition of this phosphatase may prevent reactivation of cell proliferation after exposure to inhibitors targeting some components of this signaling pathway; on the other hand, SHP-2 suppression may result in the activation of cell proliferation due to impaired OIS.
Funding. This study was supported by the Russian Foundation for Basic Research (project no. 18-315-00171 mol_a) and the Russian Science Foundation (grant no. 16-15-10423; construction of the BRAF V600E-expressing lentiviral vector and data presented in Fig. 1).
Conflict of interest. The authors declare no conflict of interest.
Compliance with ethical standards. The article does not contain description of experiments with the participation of humans or animals performed by any of the authors.
REFERENCES
1.Burotto, M., Chiou, V. L., Lee, J. M., and Kohn, E.
C. (2014) The MAPK pathway across different malignancies: a new
perspective, Cancer, 120, 3446-3456, doi:
10.1002/cncr.28864.
2.Davies, H., Bignell, G. R., Cox, C., Stephens, P.,
Edkins, S., et al. (2002) Mutations of the BRAF gene in human
cancer, Nature, 417, 949-954, doi:
10.1038/nature00766.
3.Mesa, C., Jr., Mirza, M., Mitsutake, N., Sartor,
M., Medvedovic, M., Tomlinson, C., Knauf, J. A., Weber, G. F., and
Fagin, J. A. (2006) Conditional activation of RET/PTC3 and BRAFV600E in
thyroid cells is associated with gene expression profiles that predict
a preferential role of BRAF in extracellular matrix remodeling,
Cancer Res., 66, 6521-6529, doi:
10.1158/0008-5472.CAN-06-0739.
4.Long, G. V., Menzies, A. M., Nagrial, A. M., Haydu,
L. E., Hamilton, A. L., Mann, G. J., Hughes, T. M., Thompson, J. F.,
Scolyer, R. A., and Kefford, R. F. (2011) Prognostic and
clinicopathologic associations of oncogenic BRAF in metastatic
melanoma, J. Clin. Oncol., 29, 1239-1246, doi:
10.1200/JCO.2010.32.4327.
5.Loh, M. L., Vattikuti, S., Schubbert, S., Reynolds,
M. G., Carlson, E., Lieuw, K. H., Cheng, J. W., Lee, C. M., Stokoe, D.,
Bonifas, J. M., Curtiss, N. P., Gotlib, J., Meshinchi, S., Le Beau, M.
M., Emanuel, P. D., and Shannon, K. M. (2004) Mutations in
PTPN11 implicate the SHP-2 phosphatase in leukemogenesis,
Blood, 103, 2325-2331, doi:
10.1182/blood-2003-09-3287.
6.Mohi, M. G., Williams, I. R., Dearolf, C. R., Chan,
G., Kutok, J. L., Cohen, S., Morgan, K., Boulton, C., Shigematsu, H.,
Keilhack, H., Akashi, K., Gilliland, D. G., and Neel, B. G. (2005)
Prognostic, therapeutic, and mechanistic implications of a mouse model
of leukemia evoked by Shp2 (PTPN11) mutations, Cancer
Cell, 7, 179-191, doi: 10.1016/j.ccr.2005.01.010.
7.Tartaglia, M., Mehler, E. L., Goldberg, R.,
Zampino, G., Brunner, H. G., Kremer, H., van der Burgt, I., Crosby, A.
H., Ion, A., Jeffery, S., Kalidas, K., Patton, M. A., Kucherlapati, R.
S., and Gelb, B. D. (2001) Mutations in PTPN11, encoding the
protein tyrosine phosphatase SHP-2, cause Noonan syndrome, Nat.
Genet., 29, 465-468, doi: 10.1038/ng772.
8.Chan, G., Kalaitzidis, D., and Neel, B. G. (2008)
The tyrosine phosphatase Shp2 (PTPN11) in cancer, Cancer
Metastasis Rev., 27, 179-192, doi:
10.1007/s10555-008-9126-y.
9.Chan, R. J., and Feng, G. S. (2007) PTPN11
is the first identified proto-oncogene that encodes a tyrosine
phosphatase, Blood, 109, 862-867, doi:
10.1182/blood-2006-07-028829.
10.Li, S. M. (2016) The biological function of SHP2
in human disease, Mol. Biol. (Moscow), 50, 27-33, doi:
10.7868/S0026898416010110.
11.Matozaki, T., Murata, Y., Saito, Y., Okazawa, H.,
and Ohnishi, H. (2009) Protein tyrosine phosphatase SHP-2: a
proto-oncogene product that promotes Ras activation, Cancer
Sci., 100, 1786-1793, doi:
10.1111/j.1349-7006.2009.01257.x.
12.Zhang, S. Q., Tsiaras, W. G., Araki, T., Wen, G.,
Minichiello, L., Klein, R., and Neel, B. G. (2002) Receptor-specific
regulation of phosphatidylinositol 3′-kinase activation by the
protein tyrosine phosphatase Shp2, Mol. Cell. Biol., 22,
4062-4072.
13.Saxton, T. M., Henkemeyer, M., Gasca, S., Shen,
R., Rossi, D. J., Shalaby, F., Feng, G. S., and Pawson, T. (1997)
Abnormal mesoderm patterning in mouse embryos mutant for the SH2
tyrosine phosphatase Shp-2, EMBO J., 16, 2352-2364, doi:
10.1093/emboj/16.9.2352.
14.Tajan, M., Batut, A., Cadoudal, T., Deleruyelle,
S., Le, Gonidec, S., Saint Laurent, C., Vomscheid, M., Wanecq, E.,
Treguer, K., De Rocca Serra-Nedelec, A., Vinel, C., Marques, M. A.,
Pozzo, J., Kunduzova, O., Salles, J. P., Tauber, M., Raynal, P., Cave,
H., Edouard, T., Valet, P., and Yart, A. (2014) LEOPARD
syndrome-associated SHP2 mutation confers leanness and protection from
diet-induced obesity, Proc. Natl. Acad. Sci. USA, 111,
E4494-E4503, doi: 10.1073/pnas.1406107111.
15.Chen, L., Chen, W., Mysliwski, M., Serio, J.,
Ropa, J., Abulwerdi, F. A., Chan, R. J., Patel, J. P., Tallman, M. S.,
Paietta, E., Melnick, A., Levine, R. L., Abdel-Wahab, O.,
Nikolovska-Coleska, Z., and Muntean, A. G. (2015) Mutated Ptpn11
alters leukemic stem cell frequency and reduces the sensitivity of
acute myeloid leukemia cells to Mcl1 inhibition, Leukemia,
29, 1290-1300, doi: 10.1038/leu.2015.18.
16.Qu, C. K., Shi, Z. Q., Shen, R., Tsai, F. Y.,
Orkin, S. H., and Feng, G. S. (1997) A deletion mutation in the SH2-N
domain of Shp-2 severely suppresses hematopoietic cell development,
Mol. Cell. Biol., 17, 5499-5507.
17.Bard-Chapeau, E. A., Li, S., Ding, J., Zhang, S.
S., Zhu, H. H., Princen, F., Fang, D. D., Han, T., Bailly-Maitre, B.,
Poli, V., Varki, N. M., Wang, H., and Feng, G. S. (2011)
Ptpn11/Shp2 acts as a tumor suppressor in hepatocellular
carcinogenesis, Cancer Cell, 19, 629-639, doi:
10.1016/j.ccr.2011.03.023.
18.Yang, W., Wang, J., Moore, D. C., Liang, H.,
Dooner, M., Wu, Q., Terek, R., Chen, Q., Ehrlich, M. G., Quesenberry,
P. J., and Neel, B. G. (2013) Ptpn11 deletion in a novel
progenitor causes metachondromatosis by inducing hedgehog signalling,
Nature, 499, 491-495, doi: 10.1038/nature12396.
19.Hill, K. S., Roberts, E. R., Wang, X., Marin, E.,
Park, T. D., Son, S., Ren, Y., Fang, B., Yoder, S., Kim, S., Wan, L.,
Sarnaik, A. A., Koomen, J. M., Messina, J. L., Teer, J. K., Kim, Y.,
Wu, J., Chalfant, C. E., and Kim, M. (2019) PTPN11 plays
oncogenic roles and is a therapeutic target for BRAF wild-type
melanomas, Mol. Cancer Res., 17, 583-593, doi:
10.1158/1541-7786.MCR-18-0777.
20.Zhan, Y., Counelis, G. J., and O’Rourke, D.
M. (2009) The protein tyrosine phosphatase SHP-2 is required for
EGFRvIII oncogenic transformation in human glioblastoma cells, Exp.
Cell Res., 315, 2343-2357, doi:
10.1016/j.yexcr.2009.05.001.
21.Hu, Z. Q., Ma, R., Zhang, C. M., Li, J., Li, L.,
Hu, Z. T., Gao, Q. I., and Li, W. M. (2015) Expression and clinical
significance of tyrosine phosphatase SHP2 in thyroid carcinoma,
Oncol. Lett., 10, 1507-1512, doi:
10.3892/ol.2015.3479.
22.Prahallad, A., Heynen, G. J., Germano, G.,
Willems, S. M., Evers, B., Vecchione, L., Gambino, V., Lieftink, C.,
Beijersbergen, R. L., Di Nicolantonio, F., Bardelli, A., and Bernards,
R. (2015) PTPN11 is a central node in intrinsic and acquired resistance
to targeted cancer drugs, Cell Rep., 12, 1978-1985, doi:
10.1016/j.celrep.2015.08.037.
23.Schneeberger, V. E., Ren, Y., Luetteke, N.,
Huang, Q., Chen, L., Lawrence, H. R., Lawrence, N. J., Haura, E. B.,
Koomen, J. M., Coppola, D., and Wu, J. (2015) Inhibition of Shp2
suppresses mutant EGFR-induced lung tumors in transgenic mouse model of
lung adenocarcinoma, Oncotarget, 6, 6191-6202, doi:
10.18632/oncotarget.3356.
24.Chen, Y. N., LaMarche, M. J., Chan, H. M.,
Fekkes, P., Garcia-Fortanet, J., et al. (2016) Allosteric inhibition of
SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases,
Nature, 535, 148-152, doi: 10.1038/nature18621.
25.Chapman, P. B., Hauschild, A., Robert, C.,
Haanen, J. B., Ascierto, P., et al. (2011) Improved survival with
vemurafenib in melanoma with BRAF V600E mutation, N. Engl. J.
Med., 364, 2507-2516, doi: 10.1056/NEJMoa1103782.
26.Montero-Conde, C., Ruiz-Llorente, S., Dominguez,
J. M., Knauf, J. A., Viale, A., Sherman, E. J., Ryder, M., Ghossein, R.
A., Rosen, N., and Fagin, J. A. (2013) Relief of feedback inhibition of
HER3 transcription by RAF and MEK inhibitors attenuates their antitumor
effects in BRAF-mutant thyroid carcinomas, Cancer Discov.,
3, 520-533, doi: 10.1158/2159-8290.CD-12-0531.
27.Nazarian, R., Shi, H., Wang, Q., Kong, X., Koya,
R. C., Lee, H., Chen, Z., Lee, M. K., Attar, N., Sazegar, H., Chodon,
T., Nelson, S. F., McArthur, G., Sosman, J. A., Ribas, A., and Lo, R.
S. (2010) Melanomas acquire resistance to B-RAF(V600E) inhibition by
RTK or N-RAS upregulation, Nature, 468, 973-977, doi:
10.1038/nature09626.
28.Boehm, J. S., Zhao, J. J., Yao, J., Kim, S. Y.,
Firestein, R., Dunn, I. F., Sjostrom, S. K., Garraway, L. A.,
Weremowicz, S., Richardson, A. L., Greulich, H., Stewart, C. J.,
Mulvey, L. A., Shen, R. R., Ambrogio, L., Hirozane-Kishikawa, T., Hill,
D. E., Vidal, M., Meyerson, M., Grenier, J. K., Hinkle, G., Root, D.
E., Roberts, T. M., Lander, E. S., Polyak, K., and Hahn, W. C. (2007)
Integrative genomic approaches identify IKBKE as a breast cancer
oncogene, Cell, 129, 1065-1079, doi:
10.1016/j.cell.2007.03.052.
29.Prokofjeva, M. M., Proshkina, G. M., Lebedev, T.
D., Shulgin, A. A., Spirin, P. V., Prassolov, V. S., and Deyev, S. M.
(2017) Lentiviral gene delivery to plasmolipin-expressing cells using
Mus caroli endogenous retrovirus envelope protein, Biochimie,
142, 226-233, doi: 10.1016/j.biochi.2017.09.004.
30.Liu, Z., Zhao, Y., Fang, J., Cui, R., Xiao, Y.,
and Xu, Q. (2017) SHP2 negatively regulates HLA-ABC and PD-L1
expression via STAT1 phosphorylation in prostate cancer cells,
Oncotarget, 8, 53518-53530, doi:
10.18632/oncotarget.18591.
31.Schwartz, A. M., Putlyaeva, L. V., Covich, M.,
Klepikova, A. V., Akulich, K. A., Vorontsov, I. E., Korneev, K. V.,
Dmitriev, S. E., Polanovsky, O. L., Sidorenko, S. P., Kulakovskiy, I.
V., and Kuprash, D. V. (2016) Early B-cell factor 1 (EBF1) is critical
for transcriptional control of SLAMF1 gene in human B cells,
Biochim. Biophys. Acta, 1859, 1259-1268, doi:
10.1016/j.bbagrm.2016.07.004.
32.Afanasyeva, M. A., Britanova, L. V., Korneev, K.
V., Mitkin, N. A., Kuchmiy, A. A., and Kuprash, D. V. (2014) Clusterin
is a potential lymphotoxin beta receptor target that is upregulated and
accumulates in germinal centers of mouse spleen during immune response,
PLoS One, 9, e98349, doi:
10.1371/journal.pone.0098349.
33.Bolger, A. M., Lohse, M., and Usadel, B. (2014)
Trimmomatic: a flexible trimmer for Illumina sequence data,
Bioinformatics, 30, 2114-2120, doi:
10.1093/bioinformatics/btu170.
34.Dobin, A., Davis, C. A., Schlesinger, F.,
Drenkow, J., Zaleski, C., Jha, S., Batut, P., Chaisson, M., and
Gingeras, T. R. (2013) STAR: ultrafast universal RNA-seq aligner,
Bioinformatics, 29, 15-21, doi:
10.1093/bioinformatics/bts635.
35.Liao, Y., Smyth, G. K., and Shi, W. (2014)
featureCounts: an efficient general purpose program for assigning
sequence reads to genomic features, Bioinformatics, 30,
923-930, doi: 10.1093/bioinformatics/btt656.
36.Love, M. I., Huber, W., and Anders, S. (2014)
Moderated estimation of fold change and dispersion for RNA-seq data
with DESeq2, Genome Biol., 15, 550, doi:
10.1186/s13059-014-0550-8.
37.Zhou, Y., Zhou, B., Pache, L., Chang, M.,
Khodabakhshi, A. H., Tanaseichuk, O., Benner, C., and Chanda, S. K.
(2019) Metascape provides a biologist-oriented resource for the
analysis of systems-level datasets, Nat. Commun., 10,
1523, doi: 10.1038/s41467-019-09234-6.
38.Castellone, M. D., De Falco, V., Rao, D. M.,
Bellelli, R., Muthu, M., Basolo, F., Fusco, A., Gutkind, J. S., and
Santoro, M. (2009) The beta-catenin axis integrates multiple signals
downstream from RET/papillary thyroid carcinoma leading to cell
proliferation, Cancer Res., 69, 1867-1876, doi:
10.1158/0008-5472.CAN-08-1982.
39.Giordano, T. J., Kuick, R., Thomas, D. G., Misek,
D. E., Vinco, M., Sanders, D., Zhu, Z., Ciampi, R., Roh, M., Shedden,
K., Gauger, P., Doherty, G., Thompson, N. W., Hanash, S., Koenig, R.
J., and Nikiforov, Y. E. (2005) Molecular classification of papillary
thyroid carcinoma: distinct BRAF, RAS, and RET/PTC mutation-specific
gene expression profiles discovered by DNA microarray analysis,
Oncogene, 24, 6646-6656, doi: 10.1038/sj.onc.1208822.
40.Nucera, C., Porrello, A., Antonello, Z. A.,
Mekel, M., Nehs, M. A., Giordano, T. J., Gerald, D., Benjamin, L. E.,
Priolo, C., Puxeddu, E., Finn, S., Jarzab, B., Hodin, R. A.,
Pontecorvi, A., Nose, V., Lawler, J., and Parangi, S. (2010)
B-Raf(V600E) and thrombospondin-1 promote thyroid cancer progression,
Proc. Natl. Acad. Sci. USA, 107, 10649-10654, doi:
10.1073/pnas.1004934107.
41.Roskoski, R., Jr. (2012) ERK1/2 MAP kinases:
structure, function, and regulation, Pharmacol. Res., 66,
105-143, doi: 10.1016/j.phrs.2012.04.005.
42.Kim, B. A., Jee, H. G., Yi, J. W., Kim, S. J.,
Chai, Y. J., Choi, J. Y., and Lee, K. E. (2017) Expression profiling of
a human thyroid cell line stably expressing the BRAF V600E mutation,
Cancer Genom. Proteomics, 14, 53-67, doi:
10.21873/cgp.20018.
43.Coppe, J. P., Desprez, P. Y., Krtolica, A., and
Campisi, J. (2010) The senescence-associated secretory phenotype: the
dark side of tumor suppression, Annu. Rev. Pathol., 5,
99-118, doi: 10.1146/annurev-pathol-121808-102144.
44.Hardy, K. M., Kirschmann, D. A., Seftor, E. A.,
Margaryan, N. V., Postovit, L. M., Strizzi, L., and Hendrix, M. J.
(2010) Regulation of the embryonic morphogen Nodal by Notch4
facilitates manifestation of the aggressive melanoma phenotype,
Cancer Res., 70, 10340-10350, doi:
10.1158/0008-5472.CAN-10-0705.
45.Kim, Y. H., Choi, Y. W., Han, J. H., Lee, J.,
Soh, E. Y., Park, S. H., Kim, J. H., and Park, T. J. (2014) TSH
signaling overcomes B-RafV600E-induced senescence in papillary thyroid
carcinogenesis through regulation of DUSP6, Neoplasia,
16, 1107-1120, doi: 10.1016/j.neo.2014.10.005.
46.Moulana, F. I., Priyani, A. A. H., de Silva, M.
V. C., and Dassanayake, R. S. (2018) BRAF-oncogene-induced senescence
and the role of thyroid-stimulating hormone signaling in the
progression of papillary thyroid carcinoma, Horm. Cancer,
9, 1-11, doi: 10.1007/s12672-017-0315-4.
47.Simoes, B. M., O’Brien, C. S., Eyre, R.,
Silva, A., Yu, L., Sarmiento-Castro, A., Alferez, D. G., Spence, K.,
Santiago-Gomez A., Chemi, F., Acar, A., Gandhi, A., Howell, A.,
Brennan, K., Ryden, L., Catalano, S., Ando, S., Gee, J., Ucar, A.,
Sims, A. H., Marangoni, E., Farnie, G., Landberg, G., Howell, S. J.,
and Clarke, R. B. (2015) Anti-estrogen resistance in human breast
tumors is driven by JAG1-NOTCH4-dependent cancer stem cell activity,
Cell Rep., 12, 1968-1977, doi:
10.1016/j.celrep.2015.08.050.
48.Huang, K. C., Yang, J., Ng, M. C., Ng, S. K.,
Welch, W. R., Muto, M. G., Berkowitz, R. S., and Ng, S. W. (2016)
Cyclin A1 expression and paclitaxel resistance in human ovarian cancer
cells, Eur. J. Cancer, 67, 152-163, doi:
10.1016/j.ejca.2016.08.007.
49.Liao, C., Wang, X. Y., Wei, H. Q., Li, S. Q.,
Merghoub, T., Pandolfi, P. P., and Wolgemuth, D. J. (2001) Altered
myelopoiesis and the development of acute myeloid leukemia in
transgenic mice overexpressing cyclin A1, Proc. Natl. Acad. Sci.
USA, 98, 6853-6858, doi: 10.1073/pnas.121540098.
50.Valladares, A., Hernandez, N. G., Gomez, F. S.,
Curiel-Quezada, E., Madrigal-Bujaidar, E., Vergara, M. D., Martinez, M.
S., and Arenas Aranda, D. J. (2006) Genetic expression profiles and
chromosomal alterations in sporadic breast cancer in Mexican women,
Cancer Genet. Cytogenet., 170, 147-151, doi:
10.1016/j.cancergencyto.2006.06.002.
51.Takashima, S., Saito, H., Takahashi, N., Imai,
K., Kudo, S., Atari, M., Saito, Y., Motoyama, S., and Minamiya, Y.
(2014) Strong expression of cyclin B2 mRNA correlates with a poor
prognosis in patients with non-small cell lung cancer, Tumour
Biol., 35, 4257-4265, doi: 10.1007/s13277-013-1556-7.
52.Ahmed, T. A., Adamopoulos, C., Karoulia, Z., Wu,
X., Sachidanandam, R., Aaronson, S. A., and Poulikakos, P. I. (2019)
SHP2 drives adaptive resistance to ERK signaling inhibition in
molecularly defined subsets of ERK-dependent tumors, Cell Rep.,
26, 65-78 e65, doi: 10.1016/j.celrep.2018.12.013.
53.Corcoran, R. B., Ebi, H., Turke, A. B., Coffee,
E. M., Nishino, M., Cogdill, A. P., Brown, R. D., Della Pelle, P.,
Dias-Santagata, D., Hung, K. E., Flaherty, K. T., Piris, A., Wargo, J.
A., Settleman, J., Mino-Kenudson, M., and Engelman, J. A. (2012)
EGFR-mediated re-activation of MAPK signaling contributes to
insensitivity of BRAF mutant colorectal cancers to RAF inhibition with
vemurafenib, Cancer Discov., 2, 227-235, doi:
10.1158/2159-8290.CD-11-0341.
54.Duncan, J. S., Whittle, M. C., Nakamura, K.,
Abell, A. N., Midland, A. A., Zawistowski, J. S., Johnson, N. L.,
Granger, D. A., Jordan, N. V., Darr, D. B., Usary, J., Kuan, P. F.,
Smalley, D. M., Major, B., He, X., Hoadley, K. A., Zhou, B., Sharpless,
N. E., Perou, C. M., Kim, W. Y., Gomez, S. M., Chen, X., Jin, J., Frye,
S. V., Earp, H. S., Graves, L. M., and Johnson, G. L. (2012) Dynamic
reprogramming of the kinome in response to targeted MEK inhibition in
triple-negative breast cancer, Cell, 149, 307-321, doi:
10.1016/j.cell.2012.02.053.
55.Karoulia, Z., Wu, Y., Ahmed, T. A., Xin, Q.,
Bollard, J., Krepler, C., Wu, X., Zhang, C., Bollag, G., Herlyn, M.,
Fagin, J. A., Lujambio, A., Gavathiotis, E., and Poulikakos, P. I.
(2016) An integrated model of RAF inhibitor action predicts inhibitor
activity against oncogenic BRAF signaling, Cancer Cell,
30, 485-498, doi: 10.1016/j.ccell.2016.06.024.
56.Dardaei, L., Wang, H. Q., Singh, M., Fordjour,
P., Shaw, K. X., et al. (2018) SHP2 inhibition restores sensitivity in
ALK-rearranged non-small-cell lung cancer resistant to ALK inhibitors,
Nat. Med., 24, 512-517, doi: 10.1038/nm.4497.
57.Mainardi, S., Mulero-Sanchez, A., Prahallad, A.,
Germano, G., Bosma, A., Krimpenfort, P., Lieftink, C., Steinberg, J.
D., de Wit, N., Goncalves-Ribeiro, S., Nadal, E., Bardelli, A.,
Villanueva, A., and Bernards, R. (2018) SHP2 is required for growth of
KRAS-mutant non-small-cell lung cancer in vivo, Nat.
Med., 24, 961-967, doi: 10.1038/s41591-018-0023-9.
Supplementary Figures S1 & S2 (PDF)
Supplementary Table S1 (MS Excel)