Modeling of the Enzyme–Substrate Complexes of Human Poly(ADP-Ribose) Polymerase 1
D. K. Nilov1#, S. V. Pushkarev2#, I. V. Gushchina2, G. A. Manasaryan3, K. I. Kirsanov4, and V. K. Švedas1,2,a*
1Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia2Lomonosov Moscow State University, Faculty of Bioengineering and Bioinformatics, 119991 Moscow, Russia
3Lomonosov Moscow State University, Faculty of Fundamental Medicine, 119991 Moscow, Russia
4Blokhin National Medical Research Center of Oncology, Institute of Carcinogenesis, 115478 Moscow, Russia
# These authors contributed equally to this paper.
* To whom correspondence should be addressed.
Received August 19, 2019; Revised October 16, 2019; Accepted October 16, 2019
Poly(ADP-ribose) polymerase 1 (PARP-1) is a key DNA repair enzyme and an important target in cancer treatment. Conventional methods of studying the reaction mechanism of PARP-1 have limitations because of the complex structure of PARP-1 substrates; however, the necessary data can be obtained by molecular modeling. In this work, a molecular dynamics model for the PARP-1 enzyme–substrate complex containing NAD+ molecule and the end of the poly(ADP-ribose) chain in the form of ADP molecule was obtained for the first time. Interactions with the active site residues have been characterized where Gly863, Lys903, Glu988 play a crucial role, and the SN1-like mechanism for the enzymatic ADP-ribosylation reaction has been proposed. Models of PARP-1 complexes with more sophisticated two-unit fragments of the growing polymer chain as well as competitive inhibitors 3-aminobenzamide and 7-methylguanine have been obtained by molecular docking.
KEY WORDS: DNA repair, molecular dynamics, docking, substrates, inhibitorsDOI: 10.1134/S0006297920010095
Abbreviations: MD, molecular dynamics; PAR, poly(ADP-ribose); PARP-1, poly(ADP-ribose) polymerase 1.
Human poly(ADP-ribose) polymerase 1 (PARP-1; EC 2.4.2.30) is an
ADP-ribosyltransferase superfamily enzyme which has the DNA-dependent
activity and catalyzes the synthesis of poly(ADP-ribose) (PAR; Fig. 1) from NAD+ molecules [1-5]. PARP-1 transfers ADP-ribose
to an acceptor protein with the release of nicotinamide (side chains of
glutamate and aspartate residues, as well as lysine residues, may
undergo modification) [6, 7].
Next, PAR polyanion is synthesized through sequential attachment of new
ADP-ribose units with the formation of α(1→2)
glycosidic bonds [8, 9].
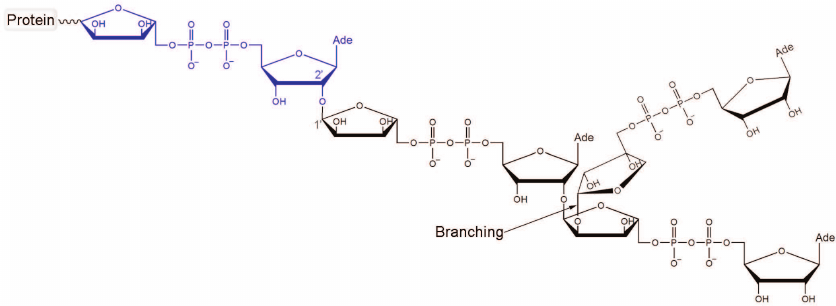
Fig. 1. Chemical structure of PAR. The first ADP-ribose unit attached to the acceptor protein is shown in blue. Polymer branching takes place when the “nicotinamide” ribose of PAR gets involved in the nucleophilic substitution reaction. The ratio between the ADP-ribose units attached via the elongation and branching reactions is 41 : 1 [10].
The binding of PARP-1 to DNA breaks results in the modification of proteins involved in DNA metabolism, as well as in the enzyme automodification [11, 12]. Poly(ADP-ribosyl)ation leads to the reorganization of chromatin structure and mobilization of DNA repair proteins to eliminate the damage [13-16]. In particular, automodified PARP-1 forms a complex with the excision repair protein XRCC1 which, in turn, interacts with DNA polymerase β and DNA ligase III [17, 18]. Since PARP-1 is a key DNA repair enzyme in tumor cells, great attention has been given to the search for its inhibitors exhibiting the anti-proliferative effect by themselves or acting in a combination with DNA-damaging agents [19-22]. Three synthetic inhibitors of PARP-1 have been recently approved for the treatment of breast and ovarian cancer: olaparib, rucaparib, and niraparib [23-25]. The cellular functions of PARP-1, as well as its suppression, have been analyzed in detail in many reviews [26-31].
Less is known about the molecular mechanism of the ADP-ribosylation reaction. The active site of PARP-1 catalytic domain includes the donor (NAD+) binding site and the acceptor (PAR) binding site [32]. The NAD+ molecule supposedly forms hydrogen bonds with Gly863 and a hydrophobic contact with Tyr907 (similar to the nicotinamide fragment mimics for which the structures of the enzyme–inhibitor complex have been determined [33, 34]). Some assumptions on the acceptor substrate binding can be made based on the PARP-1 crystal structure with an inactive structural analogue, whose adenosine diphosphate fragment forms a hydrophobic contact with Met890 and hydrogen bonds with Lys903 and Glu988 [35]. The carboxyl group of Glu988 is located near the scissile N-glycosidic bond of NAD+ and presumably acts as a general base that activates the nucleophilic group of the acceptor substrate and/or participates in the transition state stabilization [33, 35, 36].
So far, there is no reliable information on the relative orientation of PARP-1 substrates required for the reaction, as well as on the structure of transition states and intermediates. Therefore, an immediate challenge is to model the enzyme–substrate complexes on the basis of the available crystallographic data. A detailed study of PARP-1 molecular interactions with the substrates is not only of fundamental interest but may also create a basis for the rational design of effective competitive inhibitors. Successful solution of this task is now possible due to the following factors: the availability of the multidomain structure of the apo form (PDB ID 4dqy) [37, 38], the structure of the catalytic domain with the bound acceptor substrate analogue (1a26) [35], and recently released structure of the PARP-1 catalytic domain with the bound donor substrate analogue (6bhv) [39].
MATERIALS AND METHODS
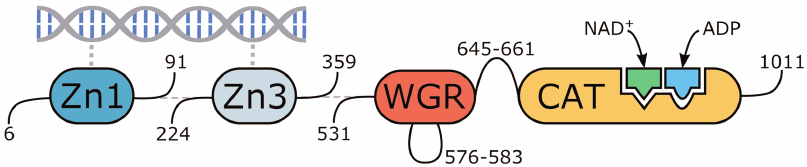
The molecular model of human PARP-1 was constructed based on the 4dqy crystal structure (chains A, B, C, M, and N). The coordinates of the missing loop 576-583 in the WGR domain were transferred from the 2cr9 structure. The coordinates of the loop 645-661 between the WGR and catalytic domains were predicted with the Modeller 9.20 program (Fig. 2) [40]. The coordinates of the NAD+ analogue were transferred from the 6bhv structure, after which its benzamide fragment was converted to nicotinamide by replacement of the corresponding carbon atom with nitrogen. The coordinates of the Arg878 side chain, which interacts with the NAD+ adenine group and displays significant conformational mobility, were also taken from 6bhv. The coordinates of ADP as a structural analogue of the acceptor substrate were transferred from the 1a26 structure. Matt 1.00 was used for the superimposition of structures [41].
Fig. 2. Multidomain organization of the obtained human PARP-1 model with bound DNA, NAD+, and ADP molecules.
Next, the structure was optimized and studied by molecular dynamics (MD) simulation using AmberTools 15 and Amber 14 [42, 43] installed on the Moscow State University supercomputer [44]. Hydrogen atoms were added considering the ionization properties of residues; in particular, the imidazole ring of the active site residue His862 was modeled with protonated Νδ1 atom. The structure was surrounded by a layer (12 Å) of TIP3P water, and sodium ions were added to neutralize the negative net charge. At the first stage of energy minimization of the obtained system (2500 steps of the steepest descent algorithm + 2500 steps of the conjugate gradient algorithm), the coordinates of the protein, DNA, and substrates were kept fixed by the positional restraints of 2 kcal/(mol⋅Å2) on heavy atoms. The second minimization stage (5000 steepest descent steps + 5000 conjugate gradient steps) was carried out without restraints. The system was then heated up from 0 to 300 K with positional restraints of 1 kcal/(mol⋅Å2) on the protein, DNA, and substrate atoms (250 ps, constant volume) and equilibrated at 300 K (500 ps, constant pressure). The acquisition of the equilibrium conformation of the substrates was confirmed by analyzing the root-mean-square deviation of their atoms from the initial positions. Subsequently, using the prepared structure, a 5000-ps trajectory of the equilibrium MD simulation was calculated and analyzed. The integration step was 0.002 ps, taking into account the use of the SHAKE algorithm. The non-bonded cut-off distance was 10 Å. The temperature and pressure were controlled using the Langevin and Berendsen algorithms. The ff14SB force field [45] was used to describe the protein and DNA with molecular mechanics, and parameters from the Amber Parameter Database [46-48] were used to describe NAD+ and ADP molecules.
A structure with the relative orientation of the substrates close to the reactive configuration was selected from the equilibrium simulation frames and subjected to energy minimization (5000 steepest descent steps + 5000 conjugate gradient steps). The obtained structure was used in covalent docking calculations with the Lead Finder 1.1.15 program [49, 50]: the ADP molecule was built up to the PAR fragments composed of two ADP-ribose units. The missing groups of atoms were attached to the C5′ atom (elongation, branching) and C1′ atom (branching). In addition, conventional docking of 3-aminobenzamide and 7-methylguanine inhibitors into the active site was performed with Lead Finder (substrate molecules were preliminary removed from the PARP-1 structure). VMD 1.9.2 was used for the visualization of the structures [51].
RESULTS AND DISCUSSION
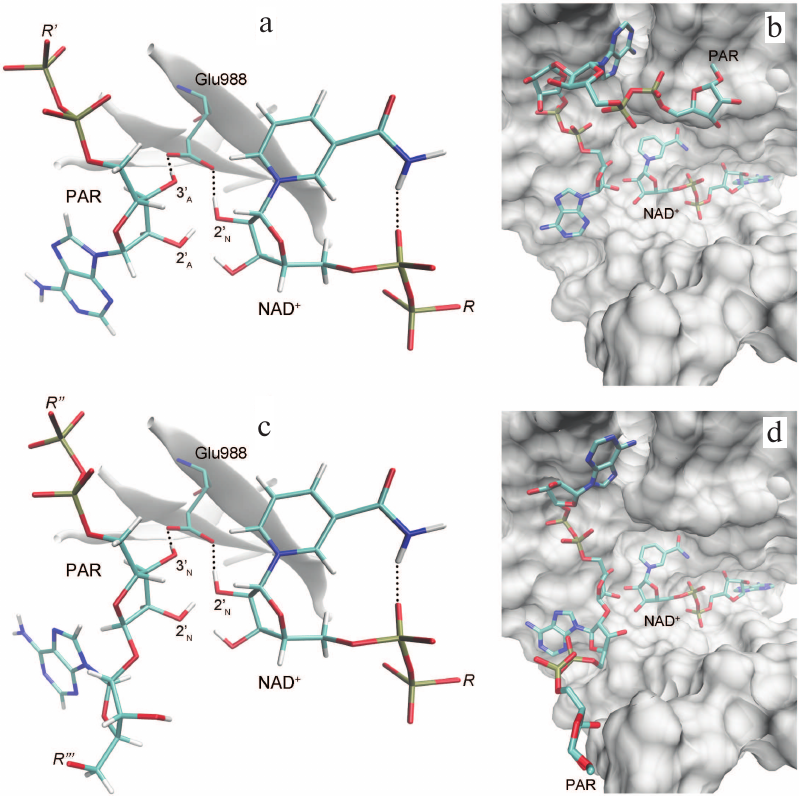
Based on the available set of PARP-1 crystal structures, we constructed for the first time the MD model of the enzyme–substrate complex with NAD+ and PAR terminal fragment in the form of ADP molecule [Fig. S1 in Supplement to this paper on the web site of the journal (http://protein.bio.msu.ru/biokhimiya) and Springer site (Link.springer.com)]. The obtained solvated system included, besides the substrates, 703 amino acid residues, 52 nucleotides, 2 Zn2+ ions, 49 Na+ ions, and 73,281 water molecules. Analysis of the PARP-1 equilibrium simulation trajectory has revealed the following important intermolecular interactions. NAD+ nicotinamide group forms two hydrogen bonds with Gly863 (table and Fig. S2 in the Supplement) and π-stacking with Tyr907 side chain, which is consistent with the results of homology modeling obtained using the structures of other superfamily members (diphtheria toxin and exotoxin A) [34, 52]. The adenine ribose of NAD+ interacts via a hydrogen bond with the Nε2 atom of His862 imidazole ring. The adenine substituent of the PAR terminal ribose forms persistent hydrophobic contact with the Met890 side chain, and the pyrophosphate group forms a hydrogen bond with the Lys903 amino group which, in turn, stabilizes the position of Glu988 residue crucial for catalysis. The carboxyl group of Glu988 forms hydrogen bonds with the 2′N-hydroxyl group of NAD+ and the 3′A-hydroxyl group of the PAR fragment, providing the required relative orientation of the donor and acceptor substrates (Fig. 3a; subscripts N and A denote nicotinamide and adenine ribose, respectively).
Fig. 3. Relative positions of NAD+ and PAR in the models of PARP-1 enzyme–substrate complexes obtained by MD and docking methods: a, b) elongation; c, d) branching. It can be seen that the pyrophosphate group attached to the attacking PAR ribose occupies very similar positions.
Key interactions in the PARP-1 active site revealed by the equilibrium
MD simulation. Mean distances are presented with standard
deviations
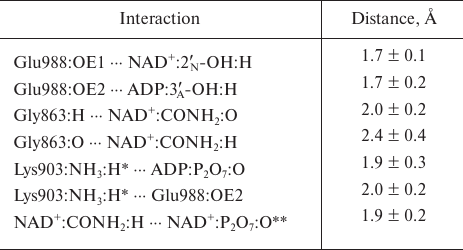
* For each frame from the trajectory, the minimum of the distances to
the hydrogen atoms of Lys903 amino group was taken into account because
of the ability of this group to undergo rotation.
** Intramolecular hydrogen bond of NAD+.
Here we should mention that some authors consider that Glu988 also forms a hydrogen bond with the 2′-hydroxyl group of PAR and acts as a proton acceptor upon the nucleophilic attack by the SN2 mechanism [35, 36, 53, 54]. However, during the simulation, we failed to observe the formation of the above-mentioned hydrogen bond or the reactive in-line configuration of the ADP:O2′A, NAD+:C1′N, and NAD+:N1N atoms, typical for the SN2 mechanism. The mean distance O2′A ⋅⋅⋅ C1′N was 4.2 Å; the angle O2′A ⋅⋅⋅ C1′N ⋅⋅⋅ N1N was 135° (Fig. 4), whereas the distance required for the nucleophilic attack is ~3 Å, and the angle should not deviate significantly from 180° [55]. This suggests that the PARP-1-catalyzed reaction of ADP-ribosylation may have an alternative SN1-like mechanism (Fig. 5). Apparently, an oxocarbenium ion intermediate is formed first and stabilized due to the negative charge of the Glu988 carboxyl group. The reactive center proceeds to the planar configuration, which facilitates subsequent attack by the 2′-hydroxyl group of the acceptor. In view of the fact that PAR is a negatively charged biopolymer, its binding in the PARP-1 active site as an acceptor substrate may also contribute to the formation of the oxocarbenium ion. Interestingly, a similar mechanism was established for other ADP-ribosyltransferase superfamily members such as PARP-10 [56], diphtheria toxin [57, 58], exotoxin A [59, 60], and iota toxin [61, 62].
Fig. 4. Angle vs. distance distribution for the PARP-1 reactive center atoms obtained by the equilibrium MD simulation. Each point corresponds to a frame from the trajectory; the structure used for docking (t = 3234 ps) is marked in green.
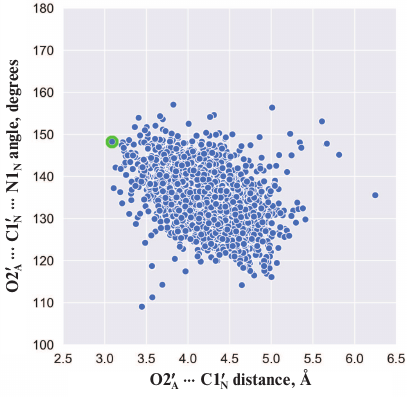
Fig. 5. Proposed SN1-like mechanism of ADP-ribosylation catalyzed by PARP-1. The negatively charged carboxyl group of Glu988 stabilizes the intermediate (oxocarbenium ion).
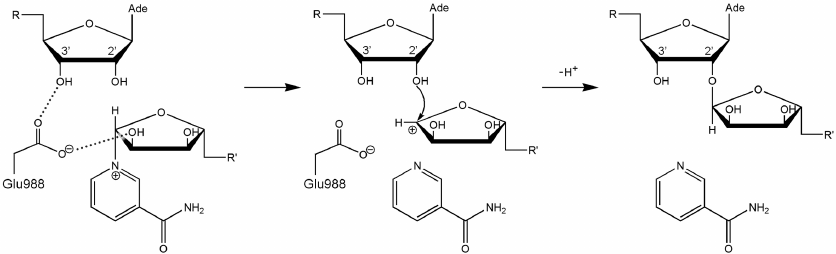
Considering possible formation of the oxocarbenium ion in the PARP-1 active site, we can also propose a mechanism for the initiation of PAR synthesis, when the ADP-ribose acceptor is a glutamate or aspartate residue on the protein surface subjected to modification. In this case, one of the oxygen atoms of the modified carboxyl group may occupy the position of the PAR 2′-hydroxyl group near the scissile glycosidic bond. The negative charge on the carboxyl group of the acceptor protein promotes NAD+ cleavage with the oxocarbenium ion formation, followed by the nucleophilic attack. This explains the fact that the E988Q and E988A mutations significantly reduce the ability of PARP-1 to catalyze the elongation reaction but have little effect on the initiation stage [36]. Glu988 provides the reactive orientation of the substrates (NAD+ and PAR) and stabilizes the intermediate during the PAR chain growth. However, this residue plays less important role at the initiation stage, when its stabilizing function is performed by the carboxyl group of the acceptor protein.
The obtained MD structure of PARP-1 characterizes the relative orientation of NAD+ and attacking PAR ribose in the case of the model ADP molecule, but does not provide information on the position of the growing polymer chain. Construction of more complex PAR fragments consisting of two ADP-ribose units was carried out by covalent docking. For this purpose, we selected a frame from the MD simulation trajectory in which the relative positions of the substrates were close to the reactive configuration (Fig. 4), and the NAD+ nicotinamide ribose was in the 3′-exo conformation. In this conformation, the C2′N, C1′N, O4′N, and C4′N atoms are all in the same plane (as in the oxocarbenium ion), which should facilitate formation of the reaction intermediate. Then, the missing groups of atoms were attached to the ADP molecule, producing coordinates of the acceptor substrate (PAR) for the elongation and branching reactions. In elongation, a new unit is transferred to the terminal adenine ribose of PAR; the reactive orientation of the substrates is shown in Fig. 3, a and b. Polymer branching occurs periodically, when a new unit is transferred to the “nicotinamide” ribose (Fig. 1) [10, 63, 64]. To undergo this reaction, PAR is considered to be rotated 180°, which allows the 2′N-OH group to occupy the reactive position of 2′A-OH (Fig. S3 in the Supplement). The position of the pyrophosphate group attached to the attacking ribose, meanwhile, does not undergo significant changes [35]. To model the PAR position corresponding to the branching reaction, ADP adenine group was replaced by the ADP-ribose moiety with inversion of the C1′ atom configuration (Fig. 3, c and d). One can compare the orientations of the growing polymer chain for the elongation and branching reactions shown in Fig. 3, b and d.
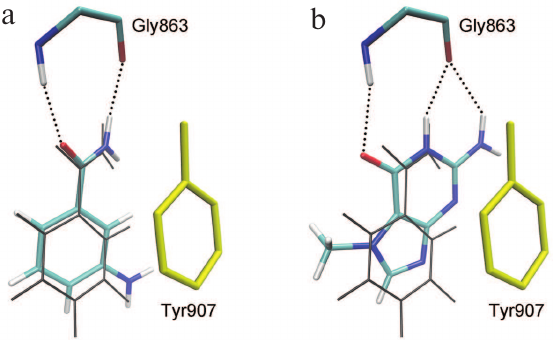
The relevance of the obtained model of the enzyme–substrate complex and the importance of the established interactions in the active site of the MD structure were confirmed by docking of two known PARP-1 inhibitors proposed to be competitive. The first compound, 3-aminobenzamide, is a well-studied structural analogue of NAD+ nicotinamide group [65-67]. The second compound, 7-methylguanine, is a new PARP inhibitor with promising pharmacokinetics and toxicity profiles. This nucleic acid metabolite was found to accelerate apoptotic death of cancer cells in combination with cisplatin and doxorubicin in vitro and to exert no significant adverse effects on the organism in preliminary in vivo tests [68-70]. Molecular docking has shown that 3-aminobenzamide and 7-methylguanine occupy the binding site of the NAD+ nicotinamide group, forming substrate-specific interactions with the Gly863 and Tyr907 residues (Fig. 6). In the case of 7-methylguanine, the functional amide group is built in the fused-ring system, but it does not prevent formation of hydrogen bonds with Gly863 (Fig. 6b).
Fig. 6. Docking poses of 3-aminobenzamide (a) and 7-methylguanine (b) in the PARP-1 active site. The phenyl group of Tyr907 (shown in yellow) forms π-stacking interactions with the inhibitors; coordinates of NAD+ nicotinamide group in the PARP-1 MD structure are shown in gray.
This research has resulted in the development of the enzyme–substrate complex model of human PARP-1. Analysis of the MD trajectory of the complex allowed us to describe the relative orientation of the donor substrate (NAD+ molecule) and the acceptor substrate (PAR fragment), as well as their interactions with the active site residues, where Gly863, Lys903, and Glu988 play a crucial role. This led us to conclude that the PARP-1-catalyzed synthesis of PAR may have the SN1-like mechanism involving formation of oxocarbenium ions. Docking methods showed that the binding site of the NAD+ nicotinamide group is a target for 3-aminobenzamide and 7-methylguanine inhibitors. The obtained model of the enzyme–substrate complex could be used in further design of the next-generation PARP inhibitors. In particular, it might serve as a basis for hybrid quantum mechanics/molecular mechanics modeling of the PARP-1 intermediate structure for screening of molecules complementary to the corresponding conformational state of the active site.
Funding. This study was supported by the Russian Foundation for Basic Research (projects 18-315-00389 mol_a and 17-08-01614 A).
Acknowledgements. The research was carried out using the equipment of the shared research facilities of HPC computing resources at the Lomonosov Moscow State University.
Conflict of interest. The authors declare no conflict of interest.
Compliance with ethical standards. This article does not contain description of studies involving animals or human participants performed by any of the authors.
REFERENCES
1.Cohen, M. S., and Chang, P. (2018) Insights into
the biogenesis, function, and regulation of ADP-ribosylation, Nat.
Chem. Biol., 14, 236-243.
2.Taniguchi, T. (1987) Reaction mechanism for
automodification of poly(ADP-ribose) synthetase, Biochem. Biophys.
Res. Commun., 147, 1008-1012.
3.Lin, H. (2007) Nicotinamide adenine dinucleotide:
beyond a redox coenzyme, Org. Biomol. Chem., 5,
2541-2554.
4.Naegeli, H., Loetscher, P., and Althaus, F. R.
(1989) Poly ADP-ribosylation of proteins. Processivity of a
post-translational modification, J. Biol. Chem., 264,
14382-14385.
5.Menard, L., Thibault, L., and Poirier, G. G. (1990)
Reconstitution of an in vitro poly(ADP-ribose) turnover system,
Biochim. Biophys. Acta, 1049, 45-58.
6.Tao, Z., Gao, P., and Liu, H. W. (2009)
Identification of the ADP-ribosylation sites in the PARP-1
automodification domain: analysis and implications, J. Am. Chem.
Soc., 131, 14258-14260.
7.Altmeyer, M., Messner, S., Hassa, P. O., Fey, M.,
and Hottiger, M. O. (2009) Molecular mechanism of
poly(ADP-ribosyl)ation by PARP1 and identification of lysine residues
as ADP-ribose acceptor sites, Nucleic Acids Res., 37,
3723-3738.
8.Drenichev, M. S., and Mikhailov, S. N. (2015)
Poly(ADP-ribose) – a unique natural polymer structural features,
biological role and approaches to the chemical synthesis,
Nucleosides Nucleotides Nucleic Acids, 34, 258-276.
9.Miwa, M., Ishihara, M., Takishima, S., Takasuka,
N., Maeda, M., Yamaizumi, Z., Sugimura, T., Yokoyama, S., and Miyazawa,
T. (1981) The branching and linear portions of poly(adenosine
diphosphate ribose) have the same alpha(1→2) ribose-ribose
linkage, J. Biol. Chem., 256, 2916-2921.
10.Keith, G., Desgres, J., and de Murcia, G. (1990)
Use of two-dimensional thin-layer chromatography for the components
study of poly(adenosine diphosphate ribose), Anal. Biochem.,
191, 309-313.
11.Mendoza-Alvarez, H., and Alvarez-Gonzalez, R.
(1993) Poly(ADP-ribose) polymerase is a catalytic dimer and the
automodification reaction is intermolecular, J. Biol. Chem.,
268, 22575-22580.
12.Mendoza-Alvarez, H., and Alvarez-Gonzalez, R.
(1999) Biochemical characterization of mono(ADP-ribosyl)ated
poly(ADP-ribose) polymerase, Biochemistry, 38,
3948-3953.
13.Hassler, M., and Ladurner, A. G. (2012) Towards a
structural understanding of PARP1 activation and related signaling
ADP-ribosyl-transferases, Curr. Opin. Struct. Biol., 22,
721-729.
14.Schreiber, V., Dantzer, F., Ame, J. C., and de
Murcia, G. (2006) Poly(ADP-ribose): novel functions for an old
molecule, Nat. Rev. Mol. Cell Biol., 7, 517-528.
15.Hassa, P. O., Haenni, S. S., Elser, M., and
Hottiger, M. O. (2006) Nuclear ADP-ribosylation reactions in mammalian
cells: where are we today and where are we going? Microbiol. Mol.
Biol. Rev., 70, 789-829.
16.Jagtap, P., and Szabo, C. (2005) Poly(ADP-ribose)
polymerase and the therapeutic effects of its inhibitors, Nat. Rev.
Drug Discov., 4, 421-440.
17.Brem, R., and Hall, J. (2005) XRCC1 is required
for DNA single-strand break repair in human cells, Nucleic Acids
Res., 33, 2512-2520.
18.Masson, M., Niedergang, C., Schreiber, V.,
Muller, S., Menissier-de Murcia, J., and de Murcia, G. (1998) XRCC1 is
specifically associated with poly(ADP-ribose) polymerase and negatively
regulates its activity following DNA damage, Mol. Cell. Biol.,
18, 3563-3571.
19.Jain, P. G., and Patel, B. D. (2019) Medicinal
chemistry approaches of poly ADP-ribose polymerase 1 (PARP1) inhibitors
as anticancer agents – a recent update, Eur. J. Med.
Chem., 165, 198-215.
20.Martin, S. A., Lord, C. J., and Ashworth, A.
(2008) DNA repair deficiency as a therapeutic target in cancer,
Curr. Opin. Genet. Dev., 18, 80-86.
21.Cepeda, V., Fuertes, M. A., Castilla, J., Alonso,
C., Quevedo, C., Soto, M., and Perez, J. M. (2006) Poly(ADP-ribose)
polymerase-1 (PARP-1) inhibitors in cancer chemotherapy, Recent Pat.
Anticancer Drug Discov., 1, 39-53.
22.Nilov, D. K., Yashina, K. I., Gushchina, I. V.,
Zakharenko, A. L., Sukhanova, M. V., Lavrik, O. I., and Svedas, V. K.
(2018) 2,5-Diketopiperazines: a new class of poly(ADP-ribose)polymerase
inhibitors, Biochemistry (Moscow), 83, 152-158.
23.Frampton, J. E. (2015) Olaparib: a review of its
use as maintenance therapy in patients with ovarian cancer,
BioDrugs, 29, 143-150.
24.Mittica, G., Ghisoni, E., Giannone, G., Genta,
S., Aglietta, M., Sapino, A., and Valabrega, G. (2018) PARP inhibitors
in ovarian cancer, Recent Pat. Anticancer Drug Discov.,
13, 392-410.
25.Zimmer, A. S., Gillard, M., Lipkowitz, S., and
Lee, J. M. (2018) Update on PARP inhibitors in breast cancer, Curr.
Treat. Options Oncol., 19, 21.
26.Ray Chaudhuri, A., and Nussenzweig, A. (2017) The
multifaceted roles of PARP1 in DNA repair and chromatin remodeling,
Nat. Rev. Mol. Cell Biol., 18, 610-621.
27.Ryu, K. W., Kim, D. S., and Kraus, W. L. (2015)
New facets in the regulation of gene expression by ADP-ribosylation and
poly(ADP-ribose) polymerases, Chem. Rev., 115,
2453-2481.
28.Curtin, N. J., and Szabo, C. (2013) Therapeutic
applications of PARP inhibitors: anticancer therapy and beyond, Mol.
Aspects Med., 34, 1217-1256.
29.Ferraris, D. V. (2010) Evolution of
poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors. From concept to
clinic, J. Med. Chem., 53, 4561-4584.
30.Virag, L., and Szabo, C. (2002) The therapeutic
potential of poly(ADP-ribose) polymerase inhibitors, Pharmacol.
Rev., 54, 375-429.
31.Malyuchenko, N. V., Kotova, E. Y., Kulaeva, O.
I., Kirpichnikov, M. P., and Studitskiy, V. M. (2015) PARP1 inhibitors:
antitumor drug design, Acta Naturae, 7, 27-37.
32.Barkauskaite, E., Jankevicius, G., and Ahel, I.
(2015) Structures and mechanisms of enzymes employed in the synthesis
and degradation of PARP-dependent protein ADP-ribosylation, Mol.
Cell, 58, 935-946.
33.Ruf, A., Mennissier de Murcia, J., de Murcia, G.,
and Schulz, G. E. (1996) Structure of the catalytic fragment of
poly(ADP-ribose) polymerase from chicken, Proc. Natl. Acad. Sci.
USA, 93, 7481-7485.
34.Ruf, A., de Murcia, G., and Schulz, G. E. (1998)
Inhibitor and NAD+ binding to poly(ADP-ribose) polymerase as
derived from crystal structures and homology modeling,
Biochemistry, 37, 3893-3900.
35.Ruf, A., Rolli, V., de Murcia, G., and Schulz, G.
E. (1998) The mechanism of the elongation and branching reaction of
poly(ADP-ribose) polymerase as derived from crystal structures and
mutagenesis, J. Mol. Biol., 278, 57-65.
36.Marsischky, G. T., Wilson, B. A., and Collier, R.
J. (1995) Role of glutamic acid 988 of human poly-ADP-ribose polymerase
in polymer formation. Evidence for active site similarities to the
ADP-ribosylating toxins, J. Biol. Chem., 270,
3247-3254.
37.Langelier, M. F., Planck, J. L., Roy, S., and
Pascal, J. M. (2012) Structural basis for DNA damage-dependent
poly(ADP-ribosyl)ation by human PARP-1, Science, 336,
728-732.
38.Langelier, M. F., Eisemann, T., Riccio, A. A.,
and Pascal, J. M. (2018) PARP family enzymes: regulation and catalysis
of the poly(ADP-ribose) posttranslational modification, Curr. Opin.
Struct. Biol., 53, 187-198.
39.Langelier, M. F., Zandarashvili, L., Aguiar, P.
M., Black, B. E., and Pascal, J. M. (2018) NAD+ analog
reveals PARP-1 substrate-blocking mechanism and allosteric
communication from catalytic center to DNA-binding domains, Nat.
Commun., 9, 844.
40.Sali, A., and Blundell, T. L. (1993) Comparative
protein modelling by satisfaction of spatial restraints, J. Mol.
Biol., 234, 779-815.
41.Menke, M., Berger, B., and Cowen, L. (2008) Matt:
local flexibility aids protein multiple structure alignment, PLoS
Comput. Biol., 4, e10.
42.Case, D. A., Berryman, J. T., Betz, R. M.,
Cerutti, D. S., Cheatham, T. E., 3rd, et al. (2015) AMBER 2015,
University of California, San Francisco.
43.Salomon-Ferrer, R., Case, D. A., and Walker, R.
C. (2013) An overview of the Amber biomolecular simulation package,
WIREs Comput. Mol. Sci., 3, 198-210.
44.Voevodin, V. V., Zhumatiy, S. A., Sobolev, S. I.,
Antonov, A. S., Bryzgalov, P. A., Nikitenko, D. A., Stefanov, K. S.,
and Voevodin, V. V. (2012) Practice of “Lomonosov”
supercomputer, Open Systems J. (Moscow), 7, 36-39.
45.Maier, J. A., Martinez, C., Kasavajhala, K.,
Wickstrom, L., Hauser, K. E., and Simmerling, C. (2015) ff14SB:
improving the accuracy of protein side chain and backbone parameters
from ff99SB, J. Chem. Theory Comput., 11, 3696-3713.
46.Walker, R. C., de Souza, M. M., Mercer, I. P.,
Gould, I. R., and Klug, D. R. (2002) Large and fast relaxations inside
a protein: calculation and measurement of reorganization
energies in alcohol dehydrogenase, J. Phys. Chem. B, 106,
11658-11665.
47.Pavelites, J. J., Gao, J., Bash, P. A., and
MacKerell, A. D., Jr. (1997) A molecular mechanics force field for
NAD+, NADH, and the pyrophosphate groups of nucleotides,
J. Comput. Chem., 18, 221-239.
48.Meagher, K. L., Redman, L. T., and Carlson, H. A.
(2003) Development of polyphosphate parameters for use with the AMBER
force field, J. Comput. Chem., 24, 1016-1025.
49.Stroganov, O. V., Novikov, F. N., Stroylov, V.
S., Kulkov, V., and Chilov, G. G. (2008) Lead finder: an approach to
improve accuracy of protein–ligand docking, binding energy
estimation, and virtual screening, J. Chem. Inf. Model.,
48, 2371-2385.
50.Zakharenko, A. L., Sukhanova, M. V., Khodyreva,
S. N., Novikov, F. N., Stroylov, V. S., Nilov, D. K., Chilov, G. G.,
Svedas, V. K., and Lavrik, O. I. (2011) Improved procedure of the
search for poly(ADP-ribose) polymerase-1 potential inhibitors with the
use of the molecular docking approach. Mol. Biol. (Moscow),
45, 517-521.
51.Humphrey, W., Dalke, A., and Schulten, K. (1996)
VMD: visual molecular dynamics, J. Mol. Graph., 14,
33-38.
52.Ivanisenko, N. V., Zhechev, D. A., and
Ivanisenko, V. A. (2017) Structural modeling of NAD+ binding
modes to PARP-1, Russ. J. Genet. Appl. Res., 7,
574-579.
53.Bellocchi, D., Costantino, G., Pellicciari, R.,
Re, N., Marrone, A., and Coletti, C. (2006)
Poly(ADP-ribose)polymerase-catalyzed hydrolysis of NAD+:
QM/MM simulation of the enzyme reaction, ChemMedChem, 1,
533-539.
54.Alemasova, E. E., and Lavrik, O. I. (2019)
Poly(ADP-ribosyl)ation by PARP1: reaction mechanism and regulatory
proteins, Nucleic Acids Res., 47, 3811-3827.
55.Yang, S.-Y., Fleurat-Lessard, P., Hristov, I.,
and Ziegler, T. (2004) Free energy profiles for the identity
SN2 reactions Cl– + CH3Cl and
NH3 + H3BNH3: a constraint
ab initio molecular dynamics study, J. Phys. Chem. A,
108, 9461-9468.
56.Kleine, H., Poreba, E., Lesniewicz, K., Hassa, P.
O., Hottiger, M. O., Litchfield, D. W., Shilton, B. H., and Luscher, B.
(2008) Substrate-assisted catalysis by PARP10 limits its activity to
mono-ADP-ribosylation, Mol. Cell, 32, 57-69.
57.Bell, C. E., and Eisenberg, D. (1996) Crystal
structure of diphtheria toxin bound to nicotinamide adenine
dinucleotide, Biochemistry, 35, 1137-1149.
58.Parikh, S. L., and Schramm, V. L. (2004)
Transition state structure for ADP-ribosylation of eukaryotic
elongation factor 2 catalyzed by diphtheria toxin, Biochemistry,
43, 1204-1212.
59.Jorgensen, R., Merrill, A. R., Yates, S. P.,
Marquez, V. E., Schwan, A. L., Boesen, T., and Andersen, G. R. (2005)
Exotoxin A–eEF2 complex structure indicates ADP ribosylation by
ribosome mimicry, Nature, 436, 979-984.
60.Jorgensen, R., Wang, Y., Visschedyk, D., and
Merrill, A. R. (2008) The nature and character of the transition state
for the ADP-ribosyltransferase reaction, EMBO Rep., 9,
802-809.
61.Tsuge, H., Nagahama, M., Oda, M., Iwamoto, S.,
Utsunomiya, H., Marquez, V. E., Katunuma, N., Nishizawa, M., and
Sakurai, J. (2008) Structural basis of actin recognition and arginine
ADP-ribosylation by Clostridium perfringens iota-toxin, Proc.
Natl. Acad. Sci. USA, 105, 7399-7404.
62.Tsurumura, T., Tsumori, Y., Qiu, H., Oda, M.,
Sakurai, J., Nagahama, M., and Tsuge, H. (2013) Arginine
ADP-ribosylation mechanism based on structural snapshots of iota-toxin
and actin complex, Proc. Natl. Acad. Sci. USA, 110,
4267-4272.
63.Rolli, V., O'Farrell, M., Menissier-de Murcia,
J., and de Murcia, G. (1997) Random mutagenesis of the poly(ADP-ribose)
polymerase catalytic domain reveals amino acids involved in polymer
branching, Biochemistry, 36, 12147-12154.
64.Kistemaker, H. A., Overkleeft, H. S., van der
Marel, G. A., and Filippov, D. V. (2015) Branching of poly(ADP-ribose):
synthesis of the core motif, Org. Lett., 17,
4328-4331.
65.Banasik, M., and Ueda, K. (1994) Inhibitors and
activators of ADP-ribosylation reactions, Mol. Cell. Biochem.,
138, 185-197.
66.Nguewa, P. A., Fuertes, M. A., Cepeda, V.,
Alonso, C., Quevedo, C., Soto, M., and Perez, J. M. (2006)
Poly(ADP-ribose) polymerase-1 inhibitor 3-aminobenzamide enhances
apoptosis induction by platinum complexes in cisplatin-resistant tumor
cells, Med. Chem., 2, 47-53.
67.Zheng, Y. D., Xu, X. Q., Peng, F., Yu, J. Z., and
Wu, H. (2011) The poly(ADP-ribose) polymerase-1 inhibitor
3-aminobenzamide suppresses cell growth and migration, enhancing
suppressive effects of cisplatin in osteosarcoma cells, Oncol.
Rep., 25, 1399-1405.
68.Nilov, D. K., Tararov, V. I., Kulikov, A. V.,
Zakharenko, A. L., Gushchina, I. V., Mikhailov, S. N., Lavrik, O. I.,
and Svedas, V. K. (2016) Inhibition of poly(ADP-ribose) polymerase by
nucleic acid metabolite 7-methylguanine, Acta Naturae, 8,
108-115.
69.Nilov, D., Kirsanov, K., Antoshina, E.,
Maluchenko, N., Feofanov, A., Kurgina, T., Zakharenko, A., Khodyreva,
S., Gerasimova, N., Studitsky, V., Lavrik, O., and Svedas, V. (2018)
7-Methylguanine: a natural DNA repair inhibitor and a promising
anticancer compound, FEBS Open Bio, 8, P.09-198-W.
70.Maluchenko, N., Nilov, D., Feofanov, A., Lys, A.,
Kutuzov, M., Gerasimova, N., and Studitsky, V. (2019) 7-Methylguanine
traps PARP-1 on nucleosomes: spFRET microscopy study, Microsc.
Microanal., 25 (S2), 1282-1283.
Supplementary Figures S1-S3 (PDF)