REVIEW: Sensors for Proteolytic Activity Visualization and Their Application in Animal Models of Human Diseases
A. A. Bogdanov, Jr.1,2,3,a*, I. D. Solovyev2,4, and A. P. Savitsky2,4
1University of Massachusetts Medical School, Department of Radiology, Laboratory of Molecular Imaging Probes, MA 01655, Worcester, USA2A. N. Bach Institute of Biochemistry, Federal Research Center “Fundamentals of Biotechnology”, Russian Academy of Sciences, Laboratory of Molecular Imaging, 119071 Moscow, Russia
3Lomonosov Moscow State University, Faculty of Bioengineering and Bioinformatics, 119991 Moscow, Russia
4A. N. Bach Institute of Biochemistry, Fundamentals of Biotechnology Federal Research Center, Russian Academy of Sciences, Laboratory of Physical Biochemistry, 119071 Moscow, Russia
* To whom correspondence should be addressed.
Received October 6, 2018; Revised October 6, 2018; Accepted October 6, 2018
Various sensors designed for optical and photo(opto)acoustic imaging in living systems are becoming essential components of basic and applied biomedical research. Some of them including those developed for determining enzyme activity in vivo are becoming commercially available. These sensors can be used for various fluorescent signal detection methods: from whole body tomography to endoscopy with miniature cameras. Sensor molecules including enzyme-cleavable macromolecules carrying multiple quenched near-infrared fluorophores are able to deliver their payload in vivo and have long circulation time in bloodstream enabling detection of enzyme activity for extended periods of time at low doses of these sensors. In the future, more effective “activated” probes are expected to become available with optimized sensitivity to enzymatic activity, spectral characteristics suitable for intraoperative imaging of surgical field, biocompatibility and lack of immunogenicity and toxicity. New in vivo optical imaging methods such as the fluorescence lifetime and photo(opto)acoustic imaging will contribute to early diagnosis of human diseases. The use of sensors for in vivo optical imaging will include more extensive preclinical applications of experimental therapies. At the same time, the ongoing development and improvement of optical signal detectors as well as the availability of biologically inert and highly specific fluorescent probes will further contribute to the introduction of fluorescence imaging into the clinic.
KEY WORDS: sensor molecules, optical and photo(opto)acoustic imaging, fluorescent signal detectionDOI: 10.1134/S0006297919140013
Abbreviations: ABP, activity-based probe; AFI, autofluorescence imaging; CPP, cell-penetrating peptide; FITC, fluorescein isothiocyanate; FMT, fluorescence molecular tomography; ICG, indocyanine green; MB, methylene blue; MFS, macromolecular fluorescent sensor; MMP, matrix metalloproteinase; MPEG-gPLL, methoxypolyethylene glycol-graft-poly(L-lysine) copolymer; NIR, near-infrared; PSA, prostate-specific antigen; qNIRF-ABP, quenched near-infrared fluorescent activity-based probe; uPA, urokinase-type plasminogen activator.
Many affected tissues actively secrete proteolytic proenzymes that are
then activated outside the cells resulting in the local increase in the
proteolytic activity, for example, during cancer progression or
inflammation. Some clinical visualization (imaging) methods used in
living systems, i.e., magnetic resonance imaging (MRI) and positron
emission tomography (PET), have been successfully applied for
non-invasive detection of enzymatic activity in tissues [1-4]. Recently, fluorescent
sensors of enzymatic activity have been tested in various types of
therapy monitoring due to the fact that, unlike radiation, optical
imaging is non-ionizing and safe even if imaging procedures are
performed repeatedly; optical images can be obtained even with small
doses of sensors [5, 6].
Fluorescence, as a particular case of luminescence, is a property of
various organic and inorganic molecules and materials. Electromagnetic
radiation in the visible or ultraviolet ranges is absorbed by compounds
resulting in their transition to the excited state, after which their
molecules emit photons of lower energies. In general, excitation light
always has a shorter wavelength (higher energy) than emitted light.
Since excitation radiation and fluorescence can be distinguished based
on spectral (frequency) and temporal characteristics, the presence of
highly sensitive sensor molecules might allow identification of
picomolar concentrations of enzymes in biological samples. This is why
technologies for visualization and measurement of enzymatic activity
using fluorescent substrates have been continuously evolving during the
last several decades and the developed methods became commonly adopted
largely due to their high sensitivity. As a rule, the used molecules
are fluorogenic, i.e., able to form fluorescent reaction products as a
result of enzymatic catalysis. Such fluorogenic substrates have been
used not only for analyzing the kinetics of enzymatic processes in
solutions, but also for acquiring images with a micron resolution and
high sensitivity. However, more and more specialized probes and sensors
have been recently designed for in vivo visualization, i.e., for
monitoring of both normal and pathophysiological processes, as well as
tracking of individual cells. Such sensors are needed to solve complex
problems that require visualization of gene expression and its
regulation, activation and differentiation of stem cells, and
intracellular signaling. The main limitation of fluorescence imaging in
living systems is impossibility of signal detection in deep tissues.
The detection limit is determined by the depth to which the excitation
light penetrates and at which fluorescence is emitted. Almost all
biological tissues absorb and scatter light in the visible and
ultraviolet ranges. However, long-wavelength excitation light [from
far-red to near-infrared (NIR), 700-1500 nm] can penetrate living
matter to a depth of several centimeters [7].
Fluorescent dyes that emit photons with lower energy (red and NIR),
which are rarely absorbed by molecules in tissues, can be detected at a
depth of several millimeters to a centimeter. A secondary, but
extremely important factor is light scattering in the tissue that
reduces the intensity of the observed signal and complicates its
spatial localization. However, early advances in visualization using
animal models of human disease have sparked an interest in fluorescent
imaging as an approach to diagnose human diseases, in particular, those
caused by inflammation or involving inflammation component in their
pathogenesis (e.g., atherosclerosis, osteoarthritis). Intraoperative
imaging of the surgical field using fluorescent molecules capable of
identifying cancerous lesions is particularly important [8-10]. For example, fluorescent
probes accumulating in metastases or sentinel lymph nodes will enable
surgeons to perform selective dissections and ensure complete removal
of metastases by using fluorescent “navigation” during
laparoscopy and other surgical procedures without the use of
radioactive labels [11, 12].
In addition, fluorescence imaging allows the so-called multiplexing of
various molecules, since fluorescent methods can detect simultaneously
several (in some cases, up to 8-10) target molecules using fluorescent
sensors with different spectral characteristics [13].
PROTEOLYTIC ACTIVITY IMAGING AND ITS USE IN THE THERAPY AND
DIAGNOSTICS OF DISEASES
The importance of estimating the levels of enzymatic activity in the diagnostic medicine has been affirmed by clinical experience of many years. Currently, several routine blood tests are based on enzymatic activity measurements, e.g., identification of serine protease prostate-specific antigen (PSA) in diagnostics of prostate cancer. Given a relatively low cost, versatility, and high efficiency of clinical analyzes of enzymatic activity, the need for performing anatomically precise mapping and simultaneous quantitative analysis of enzymatic activity in patients often seems questionable. However, in many diseases (cancer, atherosclerosis, infections with obscure etiology), clinicians often follow initial diagnosis with invasive procedures for more detailed diagnostics (for example, biopsy and exploratory surgery) or treatment (radiation treatment of cancer, surgical excisions, and thrombectomy/embolectomy). These procedures can be performed with greater efficiency and safety when the exact location of the lesion in the affected tissue has been detected in advance. In addition, the choice of treatment may depend on the degree of disease spread (for example, tumor metastasis) by taking into account available anatomical data.
Strategies aimed at the visualization and detection of enzyme activity carry considerable potential for diagnostics and prediction of a wide range of diseases. There is ample evidence that dysregulation of enzyme activity plays a role in the etiology and/or progression of cancer, atherosclerosis, stroke and heart disease, diabetes, arthritis, multiple sclerosis, Alzheimer’s disease [14], human immunodeficiency virus (HIV) infection, and other infections. For example, various proteolytic cascades involving urokinase-type plasminogen activator (uPA) [15], matrix metalloproteinases (MMPs) [16] and cysteine proteases [17] participate in the growth of cancerous tumors. It is believed that these enzymes contribute to the formation of defects in the extracellular matrix, thereby allowing cancer cells and activated stromal cells to invade surrounding tissues [18]. MMPs are expressed by cells in various types of cancer and their expression levels correlate with the tumor progression [19], invasiveness [20, 21], metastasis [22] and degree of vascularization [23]. Elevated levels of expression and activity of cysteine proteases cathepsins and uPA were found in some tumors [24-27]; elevated levels of cathepsins B, D, and L were observed in mammary adenocarcinoma, thus serving as a prognostic factor [28-31]. Inflammatory processes in cardiovascular disorders, chronic obstructive pulmonary disease, and cystic fibrosis are mediated by the activation of the corresponding signaling pathways in neutrophils and accompanied with the release of elastase and myeloperoxidase as a result of secretion of azurophilic granules and NETosis. Synthesis and secretion of hypochlorite anion, a product of myeloperoxidase activity, leads to the activation of matrilysin (MMP-7) and may contribute to the loss of atherosclerotic plaque integrity resulting from the fibrous surface layer degradation [32, 33]. In cancer therapy, proteasome inhibitors have been used in the treatment of multiple myeloma [34]. Enzyme inhibitors have been also used to inhibit activated AMP protein kinase [35], MMP-9 and MMP-14 [36, 37], cathepsin S [38] and cathepsins B, L, S and T [39].
FLUORESCENT MOLECULES IN CLINICAL PRACTICE
Autofluorescence imaging (AFI) is commonly used in clinical diagnostics as a component of endoscopic examination. AFI is based on fluorescence emitted by endogenous tissue biomolecules, such as collagen, flavins and other cofactors, porphyrins, and some related compounds. In some cases, emission wavelength of these compounds noticeably changes with changes in the tissue metabolic state, which can be used, for example, as an indicator of tumor malignancy. Currently, novel methods are being developed based on the systemic delivery of fluorescent dyes and fluorescently labeled macromolecules that are then distributed throughout the body. The success of this approach depends on the ability of fluorescent probe signal to provide accurate information about physiological processes and diagnostic signatures of diseases. In turn, this largely depends on the probe specificity for a given process and its ability to generate a signal sufficiently strong for detection. Autofluorescence of biological tissues is another factor affecting the quality of in vivo imaging. Various components of biological macromolecules and tissues (hemoglobin, tryptophan, NADH, pyridoxine, collagen, elastin, flavins, porphyrins, nucleotides, etc.) absorb photons in the visible range; some of these molecules are fluorescent.
An “ideal” in vivo fluorescent sensor (either endogenous or exogenous) should have the following properties: (i) absorbance in the range of 700-900 or 1000-1700 nm; (ii) high quantum yield; (iii) narrow excitation band with the maximally possible bathochromic shift from the photon absorbance region; (iv) high photochemical stability; (v) low toxicity and low phototoxicity; (vi) biocompatibility and ability to be degraded and removed from the body; (vii) availability of chemically activated derivatives [40].
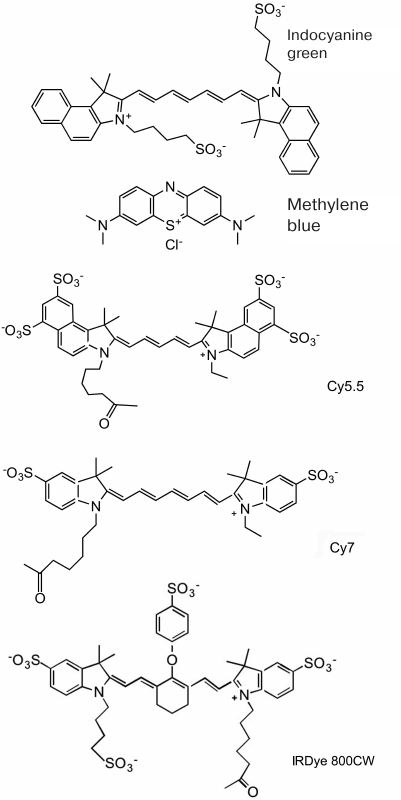
Out of all NIR fluorescent dyes, only indocyanine green dye (ICG) and methylene blue (MB) are currently approved for clinical application (Fig. 1). The first one is used as a nonspecific fluorescent contrast tracer for intraoperative angiography, visualization of ducts, and visualization of sentinel lymph nodes in cancer [41-43]. The main use of MB is methemoglobinemia therapy (i.e., MB has been used as a tracer for imaging only in clinical trials and for the same purposes as ICG). In the clinic, 5-aminolevulinic acid is used to visualize tumors as a fluorophore precursor. It does not fluoresce by itself but is involved in the biosynthesis of fluorescent protoporphyrin IX that accumulates in cancer cells due to the low activity of ferrochelatase, thus contributing to the tumor (glioma, bladder carcinoma) detection and therapy. Protoporphyrin IX fluorescence (625-710 nm) is used to map tumor location during neurosurgical procedures. It was reported that fluorescence imaging ensures more complete resection of brain tumors and improves the clinical outcome [44, 45].
Fig. 1. Chemical structure of fluorophores frequently used in the design of sensors for in vivo optical imaging of enzymatic activity. Approved for clinical use: indocyanine green; methylene blue. Cy7 and IRDye 800CW are examples of cyanine fluorophores used for research in animals.
FLUORESCENT SENSORS AS SUBSTRATES FOR PROTEOLYTIC ACTIVITY
VISUALIZATION IN LIVING SYSTEMS
While proteomic data and measuring the content of specific mRNAs provide semi-quantitative information on the expression levels of various enzymes in biological tissues, many enzymes are activated post-translationally. Therefore, a direct approach to determine enzymatic activity in intact tissues and whole organisms is often required. Fluorescent sensors are suitable for this task due to their ability to undergo rapid transformation to a fluorescent state on a sub-second scale under the influence of enzymatic catalysis.
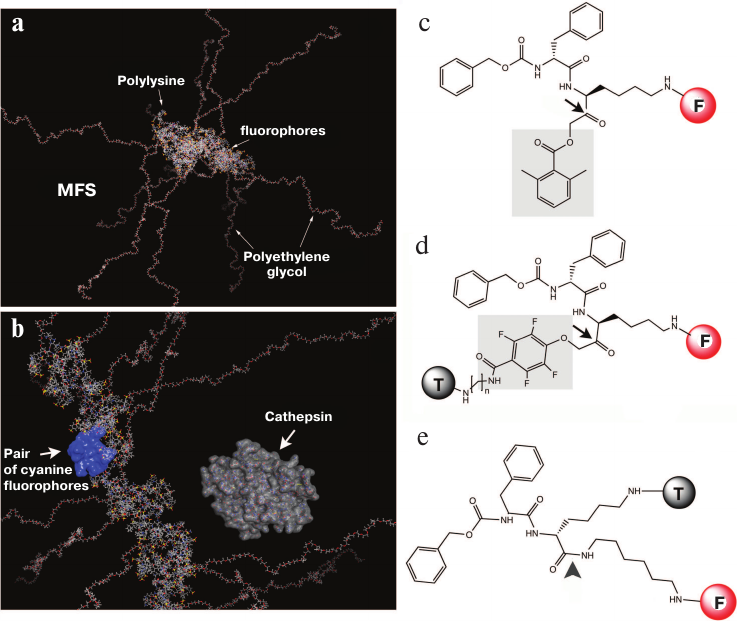
There are three main strategies for obtaining a signal based on which conclusions can be drawn about the presence or absence of enzymatic activity in the tissue. The first one is detection of a signal generated via degradation of optically “inactive” molecules and macromolecules by enzymes with the formation of optically active reaction products. This approach is often used for detecting proteases using macromolecular fluorescent sensors (MFSs) with covalently bound fluorophores [5] (Fig. 2, a and b).
Fig. 2. Models and structures of the macromolecular fluorescent sensor (MFS) based on MPEG-gPLL (a, b) and low-molecular-weight protease sensors (c-e). Main elements of the MPEG-gPLL fragment structure (20 monomers; molecular model obtained with the Molecular Operating Environment, MOE) are shown. The fragment is conjugated with the IRDye 800CW fluorophores; for clarity, MPEG simplified elongated conformation is shown (a); MPEG-gPLL model with the surface map of a pair of interacting IRDye 800CW dyes (H-dimer) highlighted in blue. For size comparison, a molecule of cathepsin B that cleaves various derivatives of modified MPEG-gPLL is shown (b). Low-molecular-weight sensors: c) ABP; d) qNIRF-ABP; e) low-molecular-weight substrate of lysosomal enzymes. Electrophilic groups are shown in gray; arrows indicate the sites of nucleophilic attack by the sulfhydryl group of the cysteine protease active center (adapted from [46-49]).
The second strategy is based on the molecule synthesis from low-molecular-weight substrates (precursors) that can be retained within a certain volume due to the reactivity of the reaction products [50]. One enzyme molecule is able to activate locally a variety of substrates associated with the fluorophore. If fluorescent reaction products are retained within the region of enzymatic reaction for a time period required for their detection without total signal reduction due to the self-quenching effect, then the fluorescence signal is amplified. This approach can be used to detect the activity of myeloperoxidase [51].
The third approach uses substrate analogs (activity-based probes, ABPs) that specifically react with the active sites of enzyme molecules. These reactions are irreversible and inhibit enzyme activity (e.g., there are ABPs that specifically and irreversibly inhibit cysteine proteases) (Fig. 2, c and d) [46]. Because of low molecular weight, such sensors easily penetrate tissue barriers; as a rule, they are extremely reactive due to the presence of electrophilic groups and susceptible to hydrolysis. As a result, these sensors produce a high background signal and have rather low selectivity in living systems (some of these sensors were modified to include quenching groups to reduce background fluorescence [47]). Sensors synthesized without introduction of reactive electrophilic groups form positively charged lysosomotropic fluorophores after cleavage with cathepsins (Fig. 2e) [49].
Sensor molecules intended for detecting proteolytic activity in living systems were originally developed based on enzyme-cleaved macromolecules (i.e., quasi-substrates) consisting of the polypeptide backbone modified with the fluorescent dye fluorescein isothiocyanate (FITC). FITC reacts with lysine ε-amino groups in polypeptides at high pH values to form the corresponding thiocarbamates [52]. The first detailed description of in vitro cell imaging using a macromolecule carrying fluorescein residues [53] demonstrated that in the albumin molecule labeled with several FITC molecules, FITC fluorescence is quenched resulting in the decrease in fluorescence intensity and lifetime. Endocytosis of FITC-labeled albumin by cultured cells caused an increase in the fluorescence intensity and a sharp 6-fold (from 0.5 to 3 ns) extension of the fluorescence lifetime. Control experiments using fluorescein-conjugated poly(L-lysine) and poly(D-lysine) demonstrated that the increase in the fluorescence lifetime and intensity for FITC-BSA was due to intracellular proteolysis, since no similar increase was observed with the inert D-isomer that is not cleaved by cellular proteases [53]. Later, FITC-labeled macromolecules were used to monitor cell migration in a three-dimensional matrix [54]. These studies have shown that the fluorescence intensity of macromolecules covalently modified with fluorophores depends nonlinearly on the fluorophore (F) concentration and decreases with the increase in the density of bound fluorophore after a certain local concentration [F] is reached. This effect can be explained by the energy transfer from the F molecule in its excited state to another F molecule, i.e., by dynamic Förster or Dexter self-quenching mechanisms. The probability of self-quenching depends on the distance between the fluorophores: W(r) = (1 + (r/R0)6)–1, where R0 is the Förster radius corresponding to 50% probability of energy transfer W(r) = 0.5. In the case of covalent fluorophore coupling to the polypeptide, the distance between individual fluorophore molecules is with a high probability less than 4 nm, i.e., within the Förster range at which energy transfer is highly probable (1.5-6 nm). Nevertheless, despite the fact that Förster dynamic self-quenching theory is widely used for interpreting the resonance energy transfer, it is not always suitable for describing self-quenching effects in macromolecules containing more than two closely spaced covalently linked fluorophores. If the Förster distance uniquely determined the effectiveness of self-quenching, then self-quenching would be observed for any pair of fluorophores located at a distance less than 6 nm from each other. However, there is a known example of commercially available dye Alexa Fluor 488. Although spectrally identical to fluorescein, this dye is less susceptible to self-quenching in its conjugates with proteins (Fig. 3).
Fig. 3. Effect of fluorophore molecule structure on its self-quenching in conjugates with proteins. Fluorescein (b) conjugates with antibodies (c) are much more susceptible to self-quenching than conjugates of spectrally identical but structurally different Alexa Fluor 488 (a) with antibodies. The data indicate the contribution of static mechanisms based on the interactions between the molecules to the dye self-quenching in covalent conjugates with polypeptides.
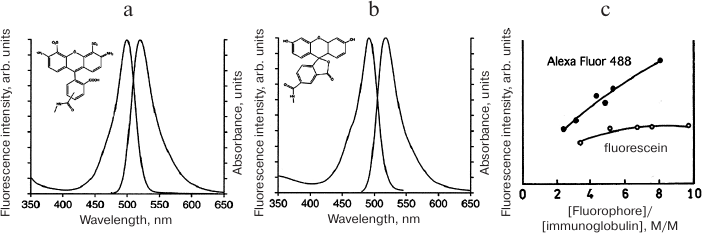
This example proves that the static quenching occurs due to the formation of non-fluorescent dimers of the F-F type in the ground state accompanied by photon absorption and return to the ground state without emitting a photon. Unlike dynamic quenching that is promoted by temperature increase, static quenching decreases with the increase in temperature, since the association constant of fluorophores KS = [F-F]/([F] × [F]) depends on temperature. In the simplest case, the fluorophore concentration [F0] is [F0] = [F] + [F-F], where [F] is the concentration of fluorescent monomers and [F-F] is the concentration of non-fluorescent dimers. According to the Stern–Volmer law, the dependence of the ratio between the fluorescence intensity in the absence of self-quenching F0 and the observed fluorescence in the presence of self-quenching F can be expressed as F0/F = 1 + KS × [F]. In general, self-quenching is caused by the combined effect of dynamic and static quenching and F0/F = 1 + Kobs × [F], where the observed Kobs = (KD + KS) + KD × KS × [F], where KD is the Stern–Volmer quenching constant. In the absence of dynamic self-quenching, the average fluorescence lifetime in a system consisting of F and F-F is determined by the lifetime of F, i.e., non-quenched single fluorophore associated with the macromolecule. However, in the presence of dynamic interactions between F and F-F dimers, the average lifetime can be much shorter. These theoretical considerations apply to the fluorescent dyes with the fluorescence maxima in the NIR range. For example, cyanines are highly susceptible to static quenching via the non-radiative resonant energy transfer. They form the so-called H-aggregates at an extremely close Förster distance from each other, which results in the excitation radiation transfer not only to the chemically different acceptor molecules but also between the same cyanine molecules. Complete aggregation of cyanines is usually prevented by special modifications with the formation of covalently linked sulfo-groups that increase the total charge of the molecules.
Early studies in cell cultures using fluorescently labeled proteins have been very important for subsequent development of probes for proteolysis imaging. An increase in the fluorescence intensity resulting from the cleavage of a macromolecular substrate is due to the conformational and chemical changes in the cleavage products. Most polypeptides have secondary and tertiary structures defined by hydrogen bonds and hydrophobic interactions of amino acid residues. If reactive groups in the polypeptide chain (first of all, ε-amino groups of lysines) are spatially located close to each other, then covalent binding of F to these groups is likely to result in low overall fluorescence of the molecule. The mobility of polypeptide chains in native protein globules is low compared to their synthetic poly- and oligo(amino acid) analogues. For example, poly(lysine) molecule exists in a disordered conformation at pH above 8.5. The conformational flexibility of linear and branched synthetic poly(amino acids) can potentially lead to an increased number of interacting fluorophores due to (i) a higher total number of available reactive groups and (ii) mobility that facilitates formation of the transitional or stable dimers (or their aggregates). The probability of fluorescent dye quenching in this case is high. Therefore, the use of NIR-fluorescent F molecules for modification of plasma proteins with a purpose of probe design has both advantages and disadvantages. The advantages include the availability of individually purified or recombinant plasma proteins, as well as their homogeneity and low immunogenicity in the native form. The disadvantages are a limited number of reactive groups, usually short circulation times in the bloodstream, an uptake of these molecules by the reticuloendothelial system cells, and a possibility of immune response induction by fluorophores linked to proteins. Linear synthetic poly(amino acids) have short circulation times in the bloodstream; their negative or positive charge can result in the high uptake levels in the kidneys and potentially induce undesirable platelet aggregation. However, the immunogenicity of poly(amino acids) whose side groups are highly modified with various ligands is usually lower that of proteins.
The design of macromolecular fluorescent sensors (MFSs) capable of detecting the catalytic activity of proteases in living systems is also based on the effect of dissociation of non-fluorescent cyanine F covalently linked directly to the enzyme-hydrolyzed polymer (Fig. 2a). Cyanine aggregates dissociate after substrate enzymatic hydrolysis that disrupts the sensor integrity (i.e., by cleavage of the linker or of the macromolecular carrier to which the fluorophores are chemically attached) (Fig. 4). As a result, fluorescence occurs instead of non-radiative energy transfer.
Fig. 4. Fluorescence self-quenching and detection of proteolytic activity using MFSs. a) Proteolytic cleavage of MFS at the peptide bond (arrow) leads to MFS fragmentation and separation of the Cy5.5-Cy5.5 pair which abolishes self-quenching; b) absorption spectra of the original MFS and the products of its trypsinolysis (the products contain significantly more fluorophore monomers); c) excitation and emission spectra of MFS before and after trypsinolysis; d) visualization of the effect of trypsinolysis (excitation, 610-650 nm; emission, >700 nm); on the left – a sample of MFS with no trypsin added; on the right – MFS in the presence of trypsin; e) blended bright field and NIR microscopy images of the tumor sample (thin section). Central panel shows fluorescent images in the NIR range; V, vessel lumen; N, necrotic tissue. Adapted from [48, 55, 56].
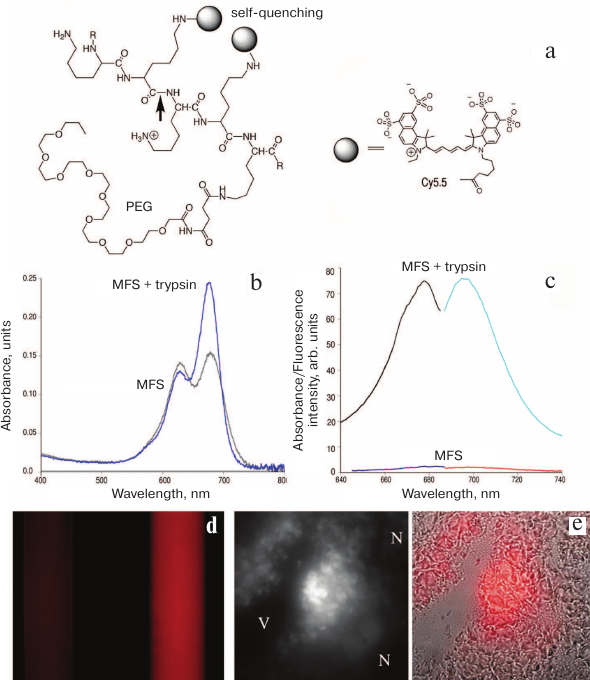
Currently, activated macromolecular NIR sensor substrates for proteolysis imaging in living animals are commercially available. These MFSs are synthesized using methoxy polyethylene glycol (MPEG)-grafted poly(L-lysine) (PLL) copolymers (MPEG-gPLL; for example, Prosense® from Perkin-Elmer) containing peptide bonds that are subject to hydrolysis by various cysteine and serine proteases (Fig. 4). Partial covalent modification of lysine ε-amino groups with activated polyethylene glycol derivatives (PEGylation, i.e., covalent binding of polyethylene glycol to other molecules or surfaces) is necessary in order to circumvent the problems associated with a very rapid removal of these MFSs from the bloodstream and immunogenicity of PLL. In addition, MPEG is necessary to increase the hydrodynamic radius and, consequently, the circulation time of MFSs in vivo. The resulting so-called sterically protected grafted copolymer (Figs. 2a and 4a) can be used for both diagnostic visualization and drug delivery (see [55] and table).
Macromolecular enzyme-activated sensors and their use for optical
imaging in animals
Notes: FI, fluorescence intensity; FMT, fluorescence-mediated
tomography.
One of the advantages of PEGylation is that due to a very high level of hydration (on average, three coordinated water molecules per one PEG mono-ethylene oxide monomer) and a very high segment flexibility, MPEG molecules create the envelope of the so-called soft matter around the PLL central molecule. As a result, PLL and fluorophores covalently associated with it are protected from the rapid removal from the bloodstream. Another important property of MPEG is its ability to prevent or slow down activation of the complement system and, as a result, to suppress immune response in the presence of biomacromolecules. In the course of MFS optimization, we found that an average MPEG mass of 5000 Da is optimal for achieving MFS monoexponential elimination from the bloodstream and preventing formation of antibodies against PLL. The resulting MPEGs based on MPEG-gPLL and used in initial in vivo experiments had an approximate molecular weight of 350-450 kDa and carried ~50-55 available for modification free amino groups per molecule [88]. However, not all available amino groups can be efficiently modified by the excess of the activated cyanine dye ester in aqueous media due to steric limitations. Depending on the fluorophore structure, it is usually possible to bind approximately 30-45 molecules of large cyanine fluorophores per MFS molecule. The result is an MFS in which most of the fluorophores are in the non-fluorescent form. The background fluorescence of the MFS is usually low, and the efficiency of its activation depends on several factors: (i) ability of dyes to be quenched, i.e., the strength of dye dimerization; (ii) fluorescence quantum yield after sensor fragmentation; and (iii) ease of formation of acylated PLL fragments during proteolysis. These parameters differ among various NIR fluorophores, although not very much. Spectral properties of MFS-bound F molecules depend on their density on PLL. If the MFS molecule is modified with less than 12 fluorophores, their absorption spectra reflect a small percentage of close interaction between the individual dye molecules (the absorbance peak with the hypsochromic shift is very small) (Fig. 5c). If the density is high (more than 35 F per MFS molecule), there is a pronounced peak in the absorption spectrum characteristic of the H-dimers that becomes less pronounced after MFS proteolytic cleavage. Fluorescent properties change dramatically after proteolysis leading to a strong increase in fluorescence intensity (Fig. 4, c and d). Out of tested F molecules, the heptamethine IRDye 800CW bound to the MFS displayed a multiple increase in fluorescence intensity after proteolysis (fragmentation). MFS cleavage by a model protease depends on whether the density of cyanine dye on PLL is high or low and whether MPEG chains are attached to the PLL backbone by ester (i.e., metastable) linkers or not. Metastable linkers are hydrolyzed by non-specific esterases; they also quickly break in alkaline or acidic media. MFSs containing such linkers are readily degraded into small fragments, which results in a large increase in the fluorescence intensity and lifetime. In general, intensity changes are higher for cyanine dyes, which emit photons in the far-red range of spectrum, e.g., Cy5.5 (Fig. 1). Fluorescence intensity increase is concomitant to a fluorescence lifetime increase (up to 5 times). Fluorescence lifetime of far-red fluorophores is longer than the lifetimes of NIR fluorophores such as Cy7 and IRDye 800CW (Fig. 1). However, in vivo visualization experiments show that heptamethine dyes have advantages besides the excitation and fluorescence spectral ranges. It is assumed that MPEG protective chains might interfere with the enzymatic cleavage of bonds in the MFSs. This scenario is unlikely for lysosomal hydrolases with relatively small hydrodynamic diameters; however, large matrix proteases secreted and activated in the extracellular space, for example, in the extracellular matrix, display low hydrolysis reaction rates. In any case, the extent of MFS fragmentation depends on whether the protease and the sensor colocalize within the same tissue volume for a sufficiently long period of time. While model serine proteases exhibit high catalytic activity, real catalytic constants of in vivo reactions can be significantly lower, which undoubtedly affects the sensitivity of MFSs. However, because of the prolonged MFS circulation in the bloodstream and cleavage in the cells of experimental tumors, it was possible to successfully visualize xenografted tumors in mice due to a high level of amplification of the NIR fluorescent signal in the tissues (Fig. 5).
Fig. 5. Accumulation and cleavage of MFS in human tumor xenografts in athymic mice. a) Xenograft images in the visible light and in the NIR range obtained 24 h after intravenous administration of MFS (MPEG-gPLL carrying self-quenching Cy5.5 molecules). b) Intravital fluorescent two-channel microscopy using two-photon excitation of EGFP fluorescence and confocal microscopy in the NIR fluorescence channel. Images were obtained in mice with the implanted glioma tumor expressing EGFP after injecting MFS at a depth of ~100 μm from the skin surface; the depth distance between the images is 15 μm. c) Flow cytometry of tumor cells. Arrow indicates the fraction of cells with high NIR fluorescence intensity of the Cy5.5 dye. d) Fluorescence microscopy of tumor cells: 1) EGFP fluorescence; 2) NIR fluorescence; 3) merged image. Cells lacking EGFP (arrow) displayed the brightеcт NIR fluorescence. Adapted from [56, 89].
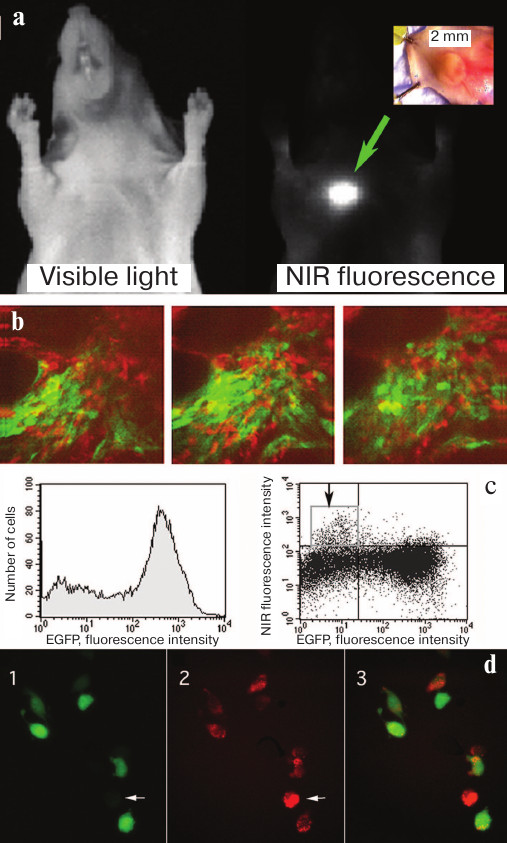
Visualization in animals allowed us to detect femtomolar amounts of fluorophores (0.8 pmol) with the preservation of sufficiently high signal/background ratio of 4.6 (visualization time, 5 min). Imaging of small fluorophore amounts in animals revealed that Cy5.5 can be detected in the concentrations up to 5 μM (250 pmol fluorophore) at a depth of 5 mm from the surface (exposure time, 10 s; signal/background ratio, 9.4). This was possible because of several factors including MFS accumulation due to a high permeability of vascular walls, high levels of cathepsin expression in tumor stroma and cancer cells, and location of MFS cleavage products in lysosomes (i.e., inside the cells). Further analysis using the two-channel transdermal microscopy of tumors expressing GFP showed that it is stromal cells that take up and cleave MFS (Fig. 5, b-d).
The use of PLL with naturally “encoded” specificity to cathepsins has proven to be possible in numerous in vivo applications. There are also many hydrolases, including cathepsins, that cleave PLL with either free or acylated amino groups in MFSs. These sensors have been originally developed without taking into account the high selectivity and specificity of this type of hydrolases. The specificity was provided by using iodoacetylated MFS as a matrix for binding synthetic peptides with specific sequences and carrying free cysteines and free amino groups [88]. The amino groups were modified with various cyanine fluorophores (e.g., Cy5.5). The resulting MFSs exhibited a 350-fold increase in the fluorescence intensity after reaction with hydrolases that cleaved the linkers. The addition of linkers can also contribute to solving the problem of a potential lack of accessibility of the PLL backbone in the MFS. However, even such intricate design does not prevent MFS hydrolysis by several extracellular proteases (including cathepsin B secreted by cells [90]) leading to cleavage of both the PLL backbone and the linkers. One possible solution is the use of poly(D-lysine) that is not hydrolyzed by natural proteases. This approach was used to synthesize a stable non-hydrolyzed long-circulating MFS (Angiosense®) that has been widely used to monitor blood volume in organs using optical imaging [91].
In some cases, MFSs were derived from macromolecules that are different in their composition and structure from the MPEG-gPLL. In particular, polyamidoamine (PAMAM) covalently bonded to Cy5.5-labeled peptides was used as a macromolecular substrate for MMP-7 for detecting intestinal neoplasia. Introduction of this optically inactive (due to self-quenching of Cy5.5) MFS in experimental tumors expressing active MMP-7 resulted in a 2.2-fold increase in the fluorescence intensity already 3 h after its intravenous administration [83]. Ex vivo visualization made it possible to observe a 6-fold signal increase and allowed to identify tumors with a diameter of 10 mm. Unlike MPEG-gPLL, MFSs based on PAMAM dendrimers are not completely cleaved by proteases. A broad substrate specificity of most proteases complicates visualization of specific classes of these enzymes in living systems. If the goal of imaging is to use proteases to amplify the signal rather than to visualize specific proteolytic activity, various transport molecules on the cell membrane surface can be used to deliver fluorescent sensor molecules, for example, avidin–rhodamine X conjugate (Av–3ROX). This high-molecular-weight mannosylated conjugate has an affinity for lectin that is present on the surface of some metastasizing cells (e.g., ovarian, colon, stomach, and pancreatic cancer cells). When bound to lectin, the conjugate is internalized and cleaved by lysosomal proteases into fluorescent fragments after dissociation of avidin tetramers into monomers. This approach is based both on a broad specificity of lysosomal enzymes and the ability of cell surface lectins to specifically bind mannans in avidin [92]. Intravital imaging in the models of ovarian cancer metastasis using Av–3ROX allowed to observe submillimeter tumors with a sensitivity of 92% and specificity of 98% with a low background signal. In the later studies, a non-immunogenic avidin substitute was used for linking to serum albumin galactosamine (GmSA) with similar results [93].
Therefore, enzyme substrate sensors have important advantages: 1) they do not inhibit enzymatic activity (i.e., do not cause irreversible inactivation of the enzymes); 2) they are highly soluble in aqueous media; 3) they are non-toxic at concentrations much higher than the substrate dose required for in vivo imaging; 4) they have a long half-life in circulation, which allows the substrate to accumulate in the tissues as a result of locally increased vascular permeability (for example, in experimentally obtained tumors).
PHARMACOKINETICS OF MFSs DESIGNED FOR VISUALIZATION OF ENZYMATIC
ACTIVITY IN LIVING SYSTEMS
MFSs derived from MPEG and poly(amino acid) long-circulating graft copolymers (e.g., MPEG-gPLL) carry at least one element (amino acid or amino acid sequence) recognized by proteases. As mentioned above, these MFSs are capable of delivering F followed by the enzyme-mediated activation. These are large molecules that are usually uptaken in small quantities in the organs of the reticuloendothelial system (liver and spleen). The only exception is MFSs synthesized from copolymers with a low degree of PEGylation or a very low degree of modification of the central element of the molecule (e.g., with 10% amino groups). In this case, MFSs are usually removed from the bloodstream quickly (half-lifetime, <1 h). It is known that MFS biodistribution in animals of various species (rats, rabbits, and primates) approximately corresponds to the half-lifetime of MPEG-gPLL in which all residual amino groups are modified, for example, by chelating moieties. The half-lifetime of PEGylated enzyme-sensitive macromolecular sensors in the bloodstream in different species varies from 24 (rat) to 32 h (rabbit). Prolonged circulation in the bloodstream gives a slow increase in the MFS extravasation in the area of increased vascular permeability. In tumors and sites of inflammation, the vascular walls usually have a high permeability, but are not freely permeable even for molecules with a size of 5-10 nm (similar to MFSs). If we assume that the process of MFS extravasation is determined by the boundary conditions of mass transfer through a semipermeable vessel wall, then it can be described by the Kedem–Katchalsky equation [94]. Then the transfer rate of a substance (in this case, macromolecular MFS) dissolved in the blood through the blood vessel wall (Js) can be expressed as:
Js = ωRT (Cv − Ce) + Jv (1 − σ)C, (1)
where C = 1/2(Cv + Ce) is the average between MFS concentrations in the bloodstream (Cv) and the extravascular space (Ce); Jv is the fluid volume flow; σ is the Staverman's reflection coefficient (in general, 0 < σ < 1); ω is the solute permeability coefficient, RT is a derivative of temperature and the universal gas constant. The first term of the equation describes the diffusion transfer, and the second term describes the convection velocity. If we take that temperature is constant, then: ωRT ~ PS, where PS is the product of the surface filtration area and vessel permeability (permeability surface product).
The majority of techniques of fluorescence imaging in animals are based on measuring fluorescence intensity in a certain organ (area of interest) over time. In a general case, the total fluorescence intensity (Io) measured in an organ is:
Io(t) = Ie(t) + Iv(t), (2)
where Ie is extravascular fluorescence; Iv is fluorescence intensity in the bloodstream. However, for a macromolecular self-quenching MFS that circulates in the bloodstream in an intact state for a long time, Iv is close to 0; therefore, Eq. (2) can be simplified:
Io(t) ≅ Ie(t).
Changes in the fluorescence intensity outside the blood vessels (i.e., total fluorescence in the target organ (Ie) in the absence of self-quenching fluorescent MFS cleavage products released by the enzyme) will be proportional to the increase in the concentration of free F (L) released by the cleavage of MFS or another macromolecule carrier. We can assume that during the time of measurement, an increase in the fluorescence intensity is directly proportional to the rate of the enzyme-catalyzed release of F from MFS. In this case, the initial rate of fluorescence intensity change can be described as:
dIo/dt = dIe/dt = A(dL/dt)
and the rate of MFS cleavage into fluorescent fragments by the enzyme can be expressed as:
dL/dt = a⋅c⋅Dt, (3)
where Dt is MFS concentration in the target organ at any time; c is dimensionless concentration of F in the MFS macromolecule; and a is proportionality coefficient. Thus, in the presence of a non-zero blood flow (Jv) through the target organ, the intensity of extravascular fluorescence measured in the target organ can be expressed in general terms as:
Io ≅ Ie = A⋅Dt⋅c, (4)
where Dt can be defined using the initial known dose of intravenously administered MFS, Do:
Dt = Do⋅B(1 – exp(–B⋅t)),
where B = (PS⋅Ie)/(Ve⋅Kav); PS is a derivative of the surface area and permeability of blood vessels through which transfer of MFS occurs; Jv is the blood flow velocity; Ve is the extravascular volume; and Kav is the available fraction of the extravascular volume (extravascular fraction). Some of the above parameters can be estimated experimentally using microscopy and fluorescent imaging of the whole body. It should be noted that, in general case, the kinetics of release (or formation) of fluorescent products is non-linear and Eq. (3) is written in a simplified form. Fluorescent products of MFS cleavage by proteases can be retained in the extravascular space. Later, in the absence of fluorescence concentration quenching, the dependence of Io on Dt can be described as a first approximation by Eq. (4). However, in the ideal case, the rate of changes in Io should reflect the order of enzymatic reaction which is generally unknown for most lysosomal hydrolases or other enzymes that can simultaneously participate in the MFS hydrolysis in the extracellular matrix.
FLUORESCENCE IMAGING IN THE FLUORESCENCE LIFETIME MODE USING
MFSs
Until very recently, preclinical approaches to optical imaging have been primarily based on fluorescence excitation and measurements of its intensity using continuous (continuous wave, CW) light sources. However, in living systems, measurement of fluorescence intensity has some obvious drawbacks. The main disadvantage is that the signal depends on numerous parameters, most of which are difficult to control. Fluorescence intensity depends on both the concentration of F and the lifetime of the molecule excited state after photon absorption. In general case, the contribution of F concentration and lifetime cannot be estimated separately, since both the local concentration of the molecule and the lifetime of the F signal influence fluorescence intensity independently. The efficiency of fluorescence quenching in activated sensor molecules is usually below 100%, which can lead to an increased background signal that hinders the measurement of fluorescence generated by the enzymatic activity. Moreover, the most serious problem in in vivo imaging with enzyme substrates as sensor molecules is often related to the effect of sensor activation in the organs of the reticuloendothelial system (for example, non-specific activation due to degradation in the liver) which leads to a high non-specific background signal. In order to distinguish between the background signal and the measured signal (in our case, the one generated by the enzymatic activity), methods based on fluorescence lifetime can be used. Depending on the type of equipment, either the frequency domain or the time domain of the fluorescent molecule lifetime in the excited state can be determined in vivo. The advantage of lifetime-based method is a unique pattern of fluorescence decay of any particular population of F molecules that is proportional to the quantum yield of the process and generally does not depend on the F concentration at constant temperature. Other benefits of lifetime visualization include (i) independence of measurements on the intensity of excitation radiation, (ii) ability in some cases to distinguish between signals corresponding to the diffusion photons of the excitation radiation and actual fluorescence, and (iii) possibility of multiplexing, i.e., simultaneous visualization or measurement of signals from several fluorophores even in the absence of significant differences between their fluorescence spectra.
We have previously proposed and developed an experimental approach to visualizing fluorescence lifetime parameters in the time domain in vivo using analysis of multi-exponential signal attenuation curves. This method allows estimation of relative amounts of fluorescent components with long and short fluorescence lifetimes by analyzing their contribution to the total signal. Using this approach, we were able to identify the presence in experimental animals of fluorescent hydrolysis products with long fluorescence lifetime (Fig. 6, a-c).
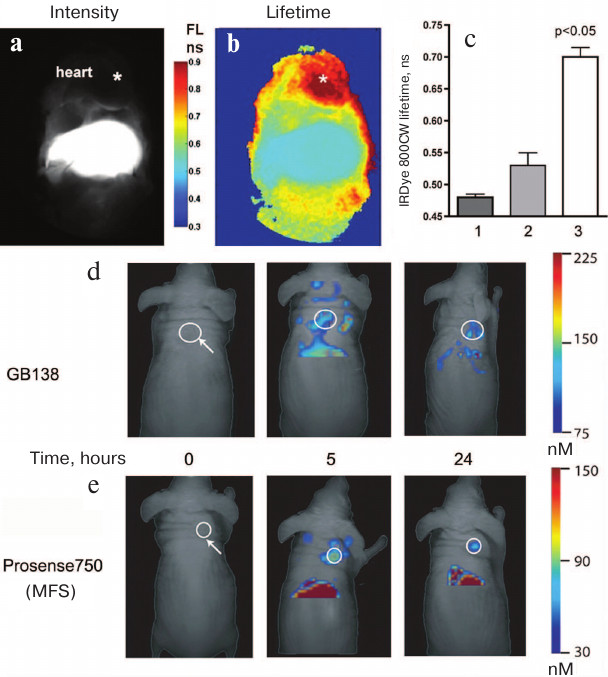
We have shown that measuring the differences in the lifetime allows in vivo visualization of internal organs that cannot be studied individually in the fluorescence intensity measuring mode. Thus, we were able to visualize experimental myocardial infarction in mice due to the elimination of a strong background signal from the liver, since it had a shorter average lifetime than the signal in the area of myocardial infarction [65] (Fig. 6, b and c). The very fact of the presence of proteolytic cleavage products with different fluorescent lifetimes for the protease-specific MFS is somewhat paradoxical, since it is believed that NIR fluorophores in MPEG-gPLL-based sensors are quenched solely due to the static effect and that their lifetime should not experience changes [95].
Fig. 6. In vivo imaging using the fluorescence lifetime method and FMT. a) The image obtained using traditional CW excitation in the NIR range shows a high level of fluorescence in the liver that obscures fluorescence in the heart region. IRDye 800CW fluorescence was measured after MFS injection and compared with the changes in fluorescence intensity in experimental myocardial infarction in situ. b) Fluorescence lifetime imaging shows that longer fluorescence lifetime in the heart region is a consequence of the MFS cleavage typical for the ischemic region in the myocardium. c) An increase in fluorescence lifetime in the chest of mice with myocardial infarction (3) was significantly higher than in the liver of control mice (1) or liver of mice in the myocardial infarction experimental group (2); p < 0.05. FMT images showing accumulation of (d) ABP (GB138) and (e) MFS (Prosense 750) in subcutaneous human adenocarcinomas (white circles) xenografted into mice. Adapted from [96, 97].
LOW-MOLECULAR-WEIGHT SENSORS FOR PROTEOLYTIC ACTIVITY
VISUALIZATION
The products of MFS cleavage in the extracellular space (e.g., those generated by MMPs, uPA, and thrombin) are fluorescent fragments that can quickly diffuse and be removed from the tissues thereby reducing the sensitivity of enzymatic activity detection. Therefore, as an alternative to MFSs, sensor molecules have been proposed based on a pair of Cy5.5-labeled peptides consisting of three elements: 1) positively charged oligo(arginine) peptide capable of penetration into the cell (cell-penetrating peptide, CPP); 2) protease-sensitive linker; and 3) negatively charged fragment masking the CPP charge. The principle of operation of the so-called activated CPPs (ACPPs) lies in the fact that ACPPs can accumulate in the extracellular space without penetrating into the cells until the linker is cleaved by an extracellular MMP, which results in the CPP separation from the neutralizing peptide, thereby facilitating CPP interaction with the cell surface. The sensor cleaved by the enzyme will localize to tumor cells, while the original sensor will be eventually removed from the tissue, thereby creating a contrast between tissues with high and low proteolytic activities. It was shown that in cultured tumor cells and carcinoma biopsy specimens containing activated MMPs (MMP2 and 9), there is a 2- to 3-fold increase in NIR fluorescence over the background. Subsequent studies have shown that ACPPs can be used for imaging of several different types of cancer (including breast cancer) and that ACPP accumulation is the highest at the margin of the tumor stroma [98].
Alternative approaches using ABPs (fluorogenic irreversible enzyme inhibitors) imply the use of reactive groups for covalent binding of quenched F in the enzyme active site [99, 100]. These probes emit fluorescent signal only after covalently modifying a specific protease target. Thus, the use of quenched NIR fluorescent ABPs (qNIRF-ABPs, Fig. 2d) targeting lysosomal cysteine proteases allowed to detect in vivo tumors in xenografted nude mice (the signal from the tumors was approximately 9 times higher than the signal from the surrounding tissues) and to observe a decrease in the enzyme activity after therapy with an appropriate enzyme inhibitor [101]. The time periods required to measure NIR signal are different for ABPs and MFSs, which makes direct comparison of these two sensors problematic. However, the amplification effect does not always lead to an overall increase in the optical signal [96]. When used for fluorescence molecular tomography (FMT), low-molecular-weight cathepsin-specific qNIRF-ABPs produced high fluorescence in tumors and low background signal in the liver and spleen. Comparison of the total FMT signal in subcutaneous (ectopic) breast tumors showed that, in the case of qNIRF-ABP, the probe concentration in the tumor was higher. However, the MFS Prosense® provided better contrast between the tumors and lung tissue in the experimental animals and a much brighter total signal at later times as compared to qNIRF-ABP (see Fig. 6, d and e) [96]. The observed differences between qNIRF-ABPs and MFSs are apparently due to the differences in their biodistribution and activation. qNIRF-ABPs rapidly diffuse in the extravascular space and are rapidly excreted. In contrast, MFSs diffuse into the extravascular space much slower and are slowly uptaken by the cells. MFS fluorescent fragments are retained in the cells and emit for longer periods of time than qNIRF-ABPs, since the latter are subjects to rapid cleavage, exocytosis, and capture into lysosomes after covalent or non-covalent interactions with enzymes. Experimental comparison of properties of ABPs and fluorogenic substrates has contributed to sensor optimization and allowed an improved design of low-molecular-weight sensors of cysteine cathepsins [49] (Fig. 2e).
Various sensors for optical and photo(opto)acoustic imaging techniques in living systems have become important components in basic and applied biomedical research. Many of them, including those developed directly for determining enzymatic activity in vivo via enzyme-mediated activation, are now commercially available. These sensors can be used for various fluorescent signal detection methods – from the whole-body tomography to endoscopy using miniature cameras. Sensor molecules are able to deliver a useful payload in a form of fluorophores and have long in vivo circulation times thus allowing detection of the corresponding enzymatic activities for extended periods of time at low sensor concentrations. In the future, more efficient activated probes are expected to emerge with an optimized sensitivity to enzymatic activity, spectral characteristics suitable for intraoperative imaging of the surgical field, biocompatibility, and lack of immunogenicity and toxicity. Newer preclinical optical imaging methods, such as fluorescence lifetime imaging and photo(opto)acoustic imaging, will contribute to more advanced methods for early diagnostics of human diseases. The use of sensors for in vivo optical imaging will include more extensive preclinical applications of the experimental therapy. At the same time, the ongoing development and improvement of optical signal detectors, as well as availability of biologically inert and highly specific fluorescent probes, will further contribute to the introduction of fluorescence imaging into clinical practice.
Funding
This work was supported in part by the Ministry of Science and Higher Education of the Russian Federation (project 14.W03.31.0023).
REFERENCES
1.Moats, R. A., Fraser, S. E., and Meade, T. J.
(1997) A “smart” magnetic resonance imaging agent that
reports on specific enzymatic activity, Angewandte Chemie Int.
Ed., 36, 726-731.
2.Gambhir, S. S., Herschman, H. R., Cherry, S. R.,
Barrio, J. R., Satyamurthy, N., Toyokuni, T., Phelps, M. E., Larson, S.
M., Balatoni, J., Finn, R., Sadelain, M., Tjuvajev, J., and Blasberg,
R. (2000) Imaging transgene expression with radionuclide imaging
technologies, Neoplasia, 2, 118-138.
3.Bennett, J. J., Tjuvajev, J., Johnson, P.,
Doubrovin, M., Akhurst, T., Malholtra, S., Hackman, T., Balatoni, J.,
Finn, R., Larson, S. M., Federoff, H., Blasberg, R., and Fong, Y.
(2001) Positron emission tomography imaging for herpes virus infection:
implications for oncolytic viral treatments of cancer, Nature
Medicine, 7, 859-863.
4.Bogdanov, A. J., Matuszewski, L., Bremer, C.,
Petrovsky, A., and Weissleder, R. (2002) Oligomerization of
paramagnetic substrates results in signal amplification and can be used
for MR imaging of molecualr targets, Mol. Imaging, 1,
16-23.
5.Law, B., and Tung, C. H. (2009) Proteolysis: a
biological process adapted in drug delivery, therapy, and imaging,
Bioconj. Chem., 20, 1683-1695.
6.Edgington, L., Verdoes, M., and Bogyo, M. (2011)
Functional imaging of proteases: recent advances in the design and
application of substrate-based and activity-based probes, Curr.
Opin. Chem. Biol., 15, 798-805.
7.Pogue, B. W., Davis, S. C., Song, X., Brooksby, B.
A., Dehghani, H., and Paulsen, K. D. (2006) Image analysis methods for
diffuse optical tomography, J. Biomed. Opt., 11,
33001.
8.Troyan, S. L., Kianzad, V., Gibbs-Strauss, S. L.,
Gioux, S., Matsui, A., Oketokoun, R., Ngo, L., Khamene, A., Azar, F.,
and Frangioni, J. V. (2009) The FLARE intraoperative near-infrared
fluorescence imaging system: a first-in-human clinical trial in breast
cancer sentinel lymph node mapping, Ann. Surg. Oncol.,
16, 2943-2952.
9.Hutteman, M., van der Vorst, J. R., Mieog, J. S.,
Bonsing, B. A., Hartgrink, H. H., Kuppen, P. J., Lowik, C. W.,
Frangioni, J. V., van de Velde, C. J., and Vahrmeijer, A. L. (2011)
Near-infrared fluorescence imaging in patients undergoing
pancreaticoduodenectomy, Eur. Surg. Res., 47, 90-97.
10.Boogerd, L. S., Handgraaf, H. J., Lam, H. D.,
Huurman, V. A., Farina-Sarasqueta, A., Frangioni, J. V., van de Velde,
C. J., Braat, A. E., and Vahrmeijer, A. L. (2017) Laparoscopic
detection and resection of occult liver tumors of multiple cancer types
using real-time near-infrared fluorescence guidance, Surg.
Endosc., 31, 952-961.
11.Mondal, S. B., Gao, S., Zhu, N., Liang, R.,
Gruev, V., and Achilefu, S. (2014) Real-time fluorescence image-guided
oncologic surgery, Adv. Cancer Res., 124, 171-211.
12.Van der Vorst, J. R., Schaafsma, B. E., Verbeek,
F. P., Keereweer, S., Jansen, J. C., van der Velden, L. A., Langeveld,
A. P., Hutteman, M., Lowik, C. W., van de Velde, C. J., Frangioni, J.
V., and Vahrmeijer, A. L. (2013) Near-infrared fluorescence sentinel
lymph node mapping of the oral cavity in head and neck cancer patients,
Oral Oncol., 49, 15-19.
13.Crane, L. M., Themelis, G., Pleijhuis, R. G.,
Harlaar, N. J., Sarantopoulos, A., Arts, H. J., van der Zee, A. G.,
Ntziachristos, V., and van Dam, G. M. (2011) Intraoperative
multispectral fluorescence imaging for the detection of the sentinel
lymph node in cervical cancer: a novel concept, Mol. Imaging
Biol., 13, 1043-1049.
14.Cataldo, A. M., and Nixon, R. A. (1990)
Enzymatically active lysosomal proteases are associated with amyloid
deposits in Alzheimer brain, Proc. Natl. Acad. Sci. USA,
87, 3861-3865.
15.Edwards, D. R., and Murphy, G. (1998) Cancer.
Proteases – invasion and more, Nature, 394,
527-528.
16.Fang, J., Shing, Y., Wiederschain, D., Yan, L.,
Butterfield, C., Jackson, G., Harper, J., Tamvakopoulos, G., and Moses,
M. A. (2000) Matrix metalloproteinase-2 is required for the switch to
the angiogenic phenotype in a tumor model, Proc. Natl. Acad. Sci.
USA, 97, 3884-3889.
17.Murray, G. I., Duncan, M. E., O'Neil, P., Melvin,
W. T., and Fothergill, J. E. (1996) Matrix metalloproteinase-1 is
associated with poor prognosis in colorectal cancer, Nat. Med.,
2, 461-462.
18.Chambers, A. F., and Matrisian, L. M. (1997)
Changing views of the role of matrix metalloproteinases in metastasis,
J. Natl. Cancer Inst., 89, 1260-1270.
19.Folkman, J. (1999) Angiogenic zip code, Nat.
Biotechnol., 17, 749.
20.Davidson, B., Goldberg, I., Kopolovic, J.,
Lerner-Geva, L., Gotlieb, W. H., Ben-Baruch, G., and Reich, R. (1999)
MMP-2 and TIMP-2 expression correlates with poor prognosis in cervical
carcinoma – a clinicopathologic study using immunohistochemistry
and mRNA in situ hybridization, Gynecol. Oncol., 73,
372-382.
21.Kanayama, H., Yokota, K., Kurokawa, Y., Murakami,
Y., Nishitani, M., and Kagawa, S. (1998) Prognostic values of matrix
metalloproteinase-2 and tissue inhibitor of metalloproteinase-2
expression in bladder cancer, Cancer, 82, 1359-1366.
22.Sakakibara, M., Koizumi, S., Saikawa, Y., Wada,
H., Ichihara, T., Sato, H., Horita, S., Mugishima, H., Kaneko, Y., and
Koike, K. (1999) Membrane-type matrix metalloproteinase-1 expression
and activation of gelatinase A as prognostic markers in advanced
pediatric neuroblastoma, Cancer, 85, 231-239.
23.Shalinsky, D. R., Brekken, J., Zou, H.,
McDermott, C. D., Forsyth, P., Edwards, D., Margosiak, S., Bender, S.,
Truitt, G., Wood, A., Varki, N. M., and Appelt, K. (1999) Broad
antitumor and antiangiogenic activities of AG3340, a potent and
selective MMP inhibitor undergoing advanced oncology clinical trials,
Ann. NY Acad. Sci., 878, 236-270.
24.Smith, H. W., and Marshall, C. J. (2010)
Regulation of cell signalling by uPAR, Nat. Rev. Mol. Cell
Biol., 11, 23-36.
25.Jedeszko, C., and Sloane, B. F. (2004) Cysteine
cathepsins in human cancer, Biol. Chem., 385,
1017-1027.
26.Mohamed, M. M., and Sloane, B. F. (2006) Cysteine
cathepsins: multifunctional enzymes in cancer, Nat. Rev. Cancer,
6, 764-775.
27.Keppler, D., Sameni, M., Moin, K., Mikkelsen, T.,
Diglio, C. A., and Sloane, B. F. (1996) Tumor progression and
angiogenesis: cathepsin B & Co, Biochem. Cell Biol.,
74, 799-810.
28.Foekens, J. A., Kos, J., Peters, H. A., Krasovec,
M., Look, M. P., Cimerman, N., Meijer-van Gelder, M. E.,
Henzen-Logmans, S. C., van Putten, W. L., and Klijn, J. G. (1998)
Prognostic significance of cathepsins B and L in primary human breast
cancer, J. Clin. Oncol., 16, 1013-1021.
29.Harbeck, N., Alt, U., Berger, U., Kruger, A.,
Thomssen, C., Janicke, F., Hofler, H., Kates, R. E., and Schmitt, M.
(2001) Prognostic impact of proteolytic factors (urokinase-type
plasminogen activator, plasminogen activator inhibitor 1, and
cathepsins B, D, and L) in primary breast cancer reflects effects of
adjuvant systemic therapy, Clin. Cancer Res., 7,
2757-2764.
30.Jagodic, M., Vrhovec, I., Borstnar, S., and
Cufer, T. (2005) Prognostic and predictive value of cathepsins D and L
in operable breast cancer patients, Neoplasma, 52,
1-9.
31.Garcia, M., Platet, N., Liaudet, E., Laurent, V.,
Derocq, D., Brouillet, J. P., and Rochefort, H. (1996) Biological and
clinical significance of cathepsin D in breast cancer metastasis,
Stem Cells, 14, 642-650.
32.Macphee, C. H., Nelson, J. J., and Zalewski, A.
(2005) Lipoprotein-associated phospholipase A2 as a target of therapy,
Curr. Opin. Lipidol., 16, 442-446.
33.Fu, X., Kassim, S. Y., Parks, W. C., and
Heinecke, J. W. (2001) Hypochlorous acid oxygenates the cysteine switch
domain of pro-matrilysin (MMP-7). A mechanism for matrix
metalloproteinase activation and atherosclerotic plaque rupture by
myeloperoxidase, J. Biol. Chem., 276, 41279-41287.
34.Orlowski, R. Z. (2004) Bortezomib and its role in
the management of patients with multiple myeloma, Expert Rev.
Anticancer Ther., 4, 171-179.
35.Evans, J. M., Donnelly, L. A., Emslie-Smith, A.
M., Alessi, D. R., and Morris, A. D. (2005) Metformin and reduced risk
of cancer in diabetic patients, Brit. Med. J., 330,
1304-1305.
36.Devy, L., Rabbani, S. A., Stochl, M., Ruskowski,
M., Mackie, I., Naa, L., Toews, M., van Gool, R., Chen, J., Ley, A.,
Ladner, R. C., Dransfield, D. T., and Henderikx, P. (2007) PEGylated
DX-1000: pharmacokinetics and antineoplastic activity of a specific
plasmin inhibitor, Neoplasia, 9, 927-937.
37.Devy, L., Huang, L., Naa, L., Yanamandra, N.,
Pieters, H., Frans, N., Chang, E., Tao, Q., Vanhove, M., Lejeune, A.,
van Gool, R., Sexton, D. J., Kuang, G., Rank, D., Hogan, S., Pazmany,
C., Ma, Y. L., Schoonbroodt, S., Nixon, A. E., Ladner, R. C., Hoet, R.,
Henderikx, P., Tenhoor, C., Rabbani, S. A., Valentino, M. L., Wood, C.
R., and Dransfield, D. T. (2009) Selective inhibition of matrix
metalloproteinase-14 blocks tumor growth, invasion, and angiogenesis,
Cancer Res., 69, 1517-1526.
38.Burden, R. E., Gormley, J. A., Jaquin, T. J.,
Small, D. M., Quinn, D. J., Hegarty, S. M., Ward, C., Walker, B.,
Johnston, J. A., Olwill, S. A., and Scott, C. J. (2009)
Antibody-mediated inhibition of cathepsin S blocks colorectal tumor
invasion and angiogenesis, Clin. Cancer Res., 15,
6042-6051.
39.Elie, B. T., Gocheva, V., Shree, T., Dalrymple,
S. A., Holsinger, L. J., and Joyce, J. A. (2010) Identification and
pre-clinical testing of a reversible cathepsin protease inhibitor
reveals anti-tumor efficacy in a pancreatic cancer model,
Biochimie, 92, 1618-1624.
40.Funovics, M., Weissleder, R., and Tung, C. H.
(2003) Protease sensors for bioimaging, Anal. Bioanal. Chem.,
377, 956-963.
41.Leblond, F., Davis, S. C., Valdes, P. A., and
Pogue, B. W. (2010) Pre-clinical whole-body fluorescence imaging:
review of instruments, methods and applications, J. Photochem.
Photobiol. B, 98, 77-94.
42.Marshall, M. V., Rasmussen, J. C., Tan, I.-C.,
Aldrich, M. B., Adams, K. E., Wang, X., Fife, C. E., Maus, E. A.,
Smith, L. A., and Sevick-Muraca, E. M. (2010) Near-infrared
fluorescence imaging in humans with indocyanine green: a review and
update, Open Surg. Oncol. J., 2, 12-25.
43.Sevick-Muraca, E. M. (2012) Translation of
near-infrared fluorescence imaging technologies: emerging clinical
applications, Annu. Rev. Med., 63, 217-231.
44.Stummer, W., Pichlmeier, U., Meinel, T.,
Wiestler, O. D., Zanella, F., and Reulen, H. J. (2006)
Fluorescence-guided surgery with 5-aminolevulinic acid for resection of
malignant glioma: a randomised controlled multicentre phase III trial,
Lancet Oncol., 7, 392-401.
45.Feigl, G. C., Ritz, R., Moraes, M., Klein, J.,
Ramina, K., Gharabaghi, A., Krischek, B., Danz, S., Bornemann, A.,
Liebsch, M., and Tatagiba, M. S. (2010) Resection of malignant brain
tumors in eloquent cortical areas: a new multimodal approach combining
5-aminolevulinic acid and intraoperative monitoring, J.
Neurosurg., 113, 352-357.
46.Lee, J., and Bogyo, M. (2010) Development of
near-infrared fluorophore (NIRF)-labeled activity-based probes for
in vivo imaging of legumain, ACS Chem. Biol., 5,
233-243.
47.Verdoes, M., Oresic Bender, K., Segal, E., van
der Linden, W. A., Syed, S., Withana, N. P., Sanman, L. E., and Bogyo,
M. (2013) Improved quenched fluorescent probe for imaging of cysteine
cathepsin activity, J. Am. Chem. Soc., 135,
14726-14730.
48.Bogdanov, A. A., and Mazzanti, M. L. (2013)
Fluorescent macromolecular sensors of enzymatic activity for in
vivo imaging, Prog. Mol. Biol. Transl., 113,
349-387.
49.Yim, J. J., Tholen, M., Klaassen, A., Sorger, J.,
and Bogyo, M. (2018) Optimization of a protease activated probe for
optical surgical navigation, Mol. Pharm., 15,
750-758.
50.Querol, M., and Bogdanov, A., Jr. (2006)
Amplification strategies in MR imaging: activation and accumulation of
sensing contrast agents (SCAs), J. Magn. Reson. Imaging,
24, 971-982.
51.Wadghiri, Y. Z., Hoang, D. M., Leporati, A.,
Gounis, M. J., Rodriguez-Rodriguez, A., Mazzanti, M. L., Weaver, J. P.,
Wakhloo, A. K., Caravan, P., and Bogdanov, A. A., Jr. (2018)
High-resolution imaging of myeloperoxidase activity sensors in human
cerebrovascular disease, Sci. Rep., 8, 7687.
52.Maeda, H., Ishida, N., Kawauchi, H., and
Tsujimura, K. (1969) Reaction of fluorescein-isothiocyanate with
proteins and amino acids. I. Covalent and non-covalent binding of
fluorescein-isothiocyanate and fluorescein to proteins, J.
Biochem., 65, 777-783.
53.French, T., So, P. T., Weaver, D. J., Jr.,
Coelho-Sampaio, T., Gratton, E., Voss, E. W., Jr., and Carrero, J.
(1997) Two-photon fluorescence lifetime imaging microscopy of
macrophage-mediated antigen processing, J. Microsc., 185,
339-353.
54.Horino, K., Kindezelskii, A. L., Elner, V. M.,
Hughes, B. A., and Petty, H. R. (2001) Tumor cell invasion of model
3-dimensional matrices: demonstration of migratory pathways, collagen
disruption, and intercellular cooperation, FASEB J., 15,
932-939.
55.Bogdanov, A. A., Mazzanti, M., Castillo, G., and
Bolotin, E. (2012) Protected graft copolymer (PGC) in imaging and
therapy: a platform for the delivery of covalently and non-covalently
bound drugs, Theranostics, 2, 553-576.
56.Weissleder, R., Tung, C. H., Mahmood, U., and
Bogdanov, A., Jr. (1999) In vivo imaging of tumors with
protease-activated near-infrared fluorescent probes, Nat.
Biotechnol., 17, 375-378.
57.Mahmood, U., Tung, C. H., Bogdanov, A., Jr., and
Weissleder, R. (1999) Near-infrared optical imaging of protease
activity for tumor detection, Radiology, 213,
866-870.
58.Ntziachristos, V., Tung, C. H., Bremer, C., and
Weissleder, R. (2002) Fluorescence molecular tomography resolves
protease activity in vivo, Nat. Med., 8,
757-760.
59.Bremer, C., Tung, C. H., Bogdanov, A., Jr., and
Weissleder, R. (2002) Imaging of differential protease expression in
breast cancers for detection of aggressive tumor phenotypes,
Radiology, 222, 814-818.
60.Wunderbaldinger, P., Turetschek, K., and Bremer,
C. (2003) Near-infrared fluorescence imaging of lymph nodes using a new
enzyme sensing activatable macromolecular optical probe, Eur.
Radiol., 13, 2206-2211.
61.Wunder, A., Tung, C. H., Muller-Ladner, U.,
Weissleder, R., and Mahmood, U. (2004) In vivo imaging of
protease activity in arthritis: a novel approach for monitoring
treatment response, Arthritis Rheum., 50, 2459-2465.
62.Lai, W. F., Chang, C. H., Tang, Y., Bronson, R.,
and Tung, C. H. (2004) Early diagnosis of osteoarthritis using
cathepsin B sensitive near-infrared fluorescent probes,
Osteoarthritis Cartilage, 12, 239-244.
63.Chen, J., Tung, C. H., Mahmood, U.,
Ntziachristos, V., Gyurko, R., Fishman, M. C., Huang, P. L., and
Weissleder, R. (2002) In vivo imaging of proteolytic activity in
atherosclerosis, Circulation, 105, 2766-2771.
64.Marten, K., Bremer, C., Khazaie, K., Sameni, M.,
Sloane, B., Tung, C. H., and Weissleder, R. (2002) Detection of
dysplastic intestinal adenomas using enzyme-sensing molecular beacons
in mice, Gastroenterology, 122, 406-414.
65.Goergen, C., Chen, H., Bogdanov, A. J., Sosnovik,
D., and Kumar, A. (2012) In vivo fluorescence lifetime detection
of an activatable probe in infarcted myocardium, J. Bomed.
Optics, 17, 056001.
66.Grimm, J., Kirsch, D. G., Windsor, S. D., Kim, C.
F., Santiago, P. M., Ntziachristos, V., Jacks, T., and Weissleder, R.
(2005) Use of gene expression profiling to direct in vivo
molecular imaging of lung cancer, Proc. Natl. Acad. Sci. USA,
102, 14404-14409.
67.Nahrendorf, M., Sosnovik, D. E., Waterman, P.,
Swirski, F. K., Pande, A. N., Aikawa, E., Figueiredo, J. L., Pittet, M.
J., and Weissleder, R. (2007) Dual channel optical tomographic imaging
of leukocyte recruitment and protease activity in the healing
myocardial infarct, Circ. Res., 100, 1218-1225.
68.Alencar, H., Funovics, M. A., Figueiredo, J.,
Sawaya, H., Weissleder, R., and Mahmood, U. (2007) Colonic
adenocarcinomas: near-infrared microcatheter imaging of smart probes
for early detection – study in mice, Radiology,
244, 232-238.
69.Ignat, M., Aprahamian, M., Lindner, V., Altmeyer,
A., Perretta, S., Dallemagne, B., Mutter, D., and Marescaux, J. (2009)
Feasibility and reliability of pancreatic cancer staging using
fiberoptic confocal fluorescence microscopy in rats,
Gastroenterology, 137, 1584-1592 e1581.
70.Ding, S., Blue, R. E., Moorefield, E., Yuan, H.,
and Lund, P. K. (2017) Ex vivo and in vivo noninvasive
imaging of epidermal growth factor receptor inhibition on colon
tumorigenesis using activatable near-infrared fluorescent probes,
Mol. Imaging, 16, 1536012117729044.
71.Haller, J., Hyde, D., Deliolanis, N., de Kleine,
R., Niedre, M., and Ntziachristos, V. (2008) Visualization of pulmonary
inflammation using noninvasive fluorescence molecular imaging, J.
Appl. Physiol., 104, 795-802.
72.Sheth, R. A., Upadhyay, R., Stangenberg, L.,
Sheth, R., Weissleder, R., and Mahmood, U. (2009) Improved detection of
ovarian cancer metastases by intraoperative quantitative fluorescence
protease imaging in a pre-clinical model, Gynecol. Oncol.,
112, 616-622.
73.Habibollahi, P., Figueiredo, J. L., Heidari, P.,
Dulak, A. M., Imamura, Y., Bass, A. J., Ogino, S., Chan, A. T., and
Mahmood, U. (2012) Optical imaging with a cathepsin B activated probe
for the enhanced detection of esophageal adenocarcinoma by dual channel
fluorescent upper GI endoscopy, Theranostics, 2,
227-234.
74.Nahrendorf, M., Waterman, P., Thurber, G.,
Groves, K., Rajopadhye, M., Panizzi, P., Marinelli, B., Aikawa, E.,
Pittet, M. J., Swirski, F. K., and Weissleder, R. (2009) Hybrid in
vivo FMT-CT imaging of protease activity in atherosclerosis with
customized nanosensors, Arterioscler. Thromb. Vasc. Biol.,
29, 1444-1451.
75.Blau, R., Epshtein, Y., Pisarevsky, E., Tiram,
G., Israeli Dangoor, S., Yeini, E., Krivitsky, A., Eldar-Boock, A.,
Ben-Shushan, D., Gibori, H., Scomparin, A., Green, O., Ben-Nun, Y.,
Merquiol, E., Doron, H., Blum, G., Erez, N., Grossman, R., Ram, Z.,
Shabat, D., and Satchi-Fainaro, R. (2018) Image-guided surgery using
near-infrared Turn-ON fluorescent nanoprobes for precise detection of
tumor margins, Theranostics, 8, 3437-3460.
76.Tung, C. H., Mahmood, U., Bredow, S., and
Weissleder, R. (2000) In vivo imaging of proteolytic enzyme
activity using a novel molecular reporter, Cancer Res.,
60, 4953-4958.
77.Jaffer, F. A., Kim, D. E., Quinti, L., Tung, C.
H., Aikawa, E., Pande, A. N., Kohler, R. H., Shi, G. P., Libby, P., and
Weissleder, R. (2007) Optical visualization of cathepsin K activity in
atherosclerosis with a novel, protease-activatable fluorescence sensor,
Circulation, 115, 2292-2298.
78.Jaffer, F. A., Tung, C. H., Gerszten, R. E., and
Weissleder, R. (2002) In vivo imaging of thrombin activity in
experimental thrombi with thrombin-sensitive near-infrared molecular
probe, Arterioscler. Thromb. Vasc. Biol., 22,
1929-1935.
79.Bremer, C., Tung, C. H., and Weissleder, R.
(2001) In vivo molecular target assessment of matrix
metalloproteinase inhibition, Nat. Med., 7, 743-748.
80.Lamfers, M. L., Gianni, D., Tung, C. H., Idema,
S., Schagen, F. H., Carette, J. E., Quax, P. H., Van Beusechem, V. W.,
Vandertop, W. P., Dirven, C. M., Chiocca, E. A., and Gerritsen, W. R.
(2005) Tissue inhibitor of metalloproteinase-3 expression from an
oncolytic adenovirus inhibits matrix metalloproteinase activity in
vivo without affecting antitumor efficacy in malignant glioma,
Cancer Res., 65, 9398-9405.
81.Chen, J., Tung, C. H., Allport, J. R., Chen, S.,
Weissleder, R., and Huang, P. L. (2005) Near-infrared fluorescent
imaging of matrix metalloproteinase activity after myocardial
infarction, Circulation, 111, 1800-1805.
82.Deguchi, J. O., Aikawa, M., Tung, C. H., Aikawa,
E., Kim, D. E., Ntziachristos, V., Weissleder, R., and Libby, P. (2006)
Inflammation in atherosclerosis: visualizing matrix metalloproteinase
action in macrophages in vivo, Circulation, 114,
55-62.
83.Scherer, R. L., VanSaun, M. N., McIntyre, J. O.,
and Matrisian, L. M. (2008) Optical imaging of matrix
metalloproteinase-7 activity in vivo using a proteolytic
nanobeacon, Mol. Imaging, 7, 118-131.
84.Klohs, J., Baeva, N., Steinbrink, J., Bourayou,
R., Boettcher, C., Royl, G., Megow, D., Dirnagl, U., Priller, J., and
Wunder, A. (2009) In vivo near-infrared fluorescence imaging of
matrix metalloproteinase activity after cerebral ischemia, J. Cereb.
Blood Flow Metab., 29, 1284-1292.
85.Messerli, S. M., Prabhakar, S., Tang, Y., Shah,
K., Cortes, M. L., Murthy, V., Weissleder, R., Breakefield, X. O., and
Tung, C. H. (2004) A novel method for imaging apoptosis using a
caspase-1 near-infrared fluorescent probe, Neoplasia, 6,
95-105.
86.Hsiao, J. K., Law, B., Weissleder, R., and Tung,
C. H. (2006) In vivo imaging of tumor associated urokinase-type
plasminogen activator activity, J. Biomed. Opt., 11,
34013.
87.Ho, G., Morin, J., Delaney, J., Cuneo, G., Yared,
W., Rajopadhye, M., Peterson, J. D., and Kossodo, S. (2013) Detection
and quantification of enzymatically active prostate-specific antigen
in vivo, J. Biomed. Opt., 18, 101319.
88.Tung, C. H., Bredow, S., Mahmood, U., and
Weissleder, R. (1999) Preparation of a cathepsin D sensitive
near-infrared fluorescence probe for imaging, Bioconj. Chem.,
10, 892-896.
89.Bogdanov, A. A., Lin, C. P., Simonova, M.,
Matuszewski, L., and Weissleder, R. (2002) Cellular activation of the
self-quenched fluorescent reporter probe in tumor microenvironment,
Neoplasia, 4, 228-236.
90.Sloane, B. F., Yan, S., Podgorski, I., Linebaugh,
B. E., Cher, M. L., Mai, J., Cavallo-Medved, D., Sameni, M., Dosescu,
J., and Moin, K. (2005) Cathepsin B and tumor proteolysis: contribution
of the tumor microenvironment, Semin. Cancer Biol., 15,
149-157.
91.Zhang, R., Brennan, M. L., Fu, X., Aviles, R. J.,
Pearce, G. L., Penn, M. S., Topol, E. J., Sprecher, D. L., and Hazen,
S. L. (2001) Association between myeloperoxidase levels and risk of
coronary artery disease, JAMA, 286, 2136-2142.
92.Hama, Y., Urano, Y., Koyama, Y., Kamiya, M.,
Bernardo, M., Paik, R. S., Shin, I. S., Paik, C. H., Choyke, P. L., and
Kobayashi, H. (2007) A target cell-specific activatable fluorescence
probe for in vivo molecular imaging of cancer based on a
self-quenched avidin-rhodamine conjugate, Cancer Res.,
67, 2791-2799.
93.Hama, Y., Urano, Y., Koyama, Y., Gunn, A. J.,
Choyke, P. L., and Kobayashi, H. (2007) A self-quenched
galactosamine-serum albumin-rhodamineX conjugate: a “smart”
fluorescent molecular imaging probe synthesized with clinically
applicable material for detecting peritoneal ovarian cancer metastases,
Clin. Cancer Res., 13, 6335-6343.
94.Kedem, O., and Katchalsky, A. (1958)
Thermodynamic analysis of the permeability of biological membranes to
non-electrolytes, Biochim. Biophys. Acta, 27,
229-246.
95.Kumar, A. T. N., Rice, W. L., Lopez, J. C.,
Gupta, S., Goergen, C. J., and Bogdanov, A. A., Jr. (2016)
Substrate-based near-infrared imaging sensors enable fluorescence
lifetime contrast via built-in dynamic fluorescence quenching elements,
ACS Sensors, 1, 427-436.
96.Blum, G., Weimer, R. M., Edgington, L. E., Adams,
W., and Bogyo, M. (2009) Comparative assessment of substrates and
activity based probes as tools for non-invasive optical imaging of
cysteine protease activity, PLoS One, 4, e6374.
97.Goergen, C. J., Chen, H. H., Bogdanov, A.,
Sosnovik, D. E., and Kumar, A. T. (2012) In vivo fluorescence
lifetime detection of an activatable probe in infarcted myocardium,
J. Biomed. Opt., 17, 056001.
98.Olson, E. S., Aguilera, T. A., Jiang, T., Ellies,
L. G., Nguyen, Q. T., Wong, E. H., Gross, L. A., and Tsien, R. Y.
(2009) In vivo characterization of activatable cell penetrating
peptides for targeting protease activity in cancer, Integr.
Biol. (Camb.), 1, 382-393.
99.Kato, D., Boatright, K. M., Berger, A. B., Nazif,
T., Blum, G., Ryan, C., Chehade, K. A., Salvesen, G. S., and Bogyo, M.
(2005) Activity-based probes that target diverse cysteine protease
families, Nat. Chem. Biol., 1, 33-38.
100.Blum, G., Mullins, S. R., Keren, K., Fonovic,
M., Jedeszko, C., Rice, M. J., Sloane, B. F., and Bogyo, M. (2005)
Dynamic imaging of protease activity with fluorescently quenched
activity-based probes, Nat. Chem. Biol., 1, 203-209.
101.Blum, G., von Degenfeld, G., Merchant, M. J.,
Blau, H. M., and Bogyo, M. (2007) Noninvasive optical imaging of
cysteine protease activity using fluorescently quenched activity-based
probes, Nat. Chem. Biol., 3, 668-677.