REVIEW: Development of Methodology for Analyzing RNA Structure and Its Application in Molecular Biology and Virology
S. K. Vasilenkoa,b
State Research Center of Virology and Biotechnology Vector, 630559 Koltsovo, Novosibirsk Region, Russia
Received June 20, 2019; Revised July 27, 2019; Accepted August 5, 2019
The review covers three independent blocks of research. The first one is discovery, isolation, and investigation of snake venom RNases and their use in studying RNA macrostructure. It has been established that snake venom RNases are not specific to the primary RNA structure but rather to the RNA helical conformation (double, single, or hybrid helix). Snake venom RNases hydrolyze RNA to short oligomers with the 5′-terminal phosphate. Analysis of the kinetics and products of tRNA hydrolysis exemplifies the use of snake venom RNases for deciphering RNA macrostructure. The second block is devoted to the principle formulated by the author for analyzing the primary structure of nucleic acids and describes the method of direct RNA sequencing that has been developed with author’s participation. The third block describes the results of genotyping and etiologic control of epidemic influenza A viruses circulating in the Soviet Union in 1968 to 1992. The method for comparative analysis of genome sequences of viral isolates has made it possible to detect and characterize epidemic influenza virus strains that had emerged in the circulation as a result of reactivation of inactivated vaccines.
KEY WORDS: snake venoms, ribonuclease, RNA, macrostructure, primary structure, molecular virologyDOI: 10.1134/S0006297919110014
Abbreviations: HA, hemagglutinin; PDE, phosphodiesterase; RNP, ribonucleoprotein.
I am deeply grateful for the opportunity to review the results of my
scientific work from more than half a century in the anniversary issue
of the Biochemistry (Moscow) journal. First of all, I owe my
biological education and preparation for independent scientific
research to my alma mater, the Department of Biochemistry of the
Biological Faculty, Moscow State University. I entered MSU in 1954,
immediately after graduation from a school in Kostroma. As a
second-year student, I joined the Department of Animal Biochemistry.
During that time, the Professors and Adjuncts working and teaching at
this Department were those who had received their education before the
Russian Revolution of 1917 and who followed the traditions founded by
fathers of Russian biology. The most brilliant representative of the
“old guard” was Professor Sergey Evgenievich Severin, an
outstanding scientist, brilliant teacher, and founder of the Department
of Animal Biochemistry. I have been extremely lucky to be taught at the
Department headed by S. E. Severin. I remember our first meeting with
the Department members and teachers and the introductory lecture by S.
E. Severin, in which he had outlined the prospects of our learning. As
far as I understood from this lecture, he believed that it was
impossible to prepare a qualified specialist within such a short time.
The goal of the university education and, in particular, the task of
the Department, was to give the students fundamental knowledge and
experimental skills that would help them to conduct their own
scientific activities. Although it seemed that S. E. Severin did not
engage much with juniors and sophomores; he was well-informed about all
events taking place at the Department. Mainly, we were looked after by
the Department scientists who attentively observed and strictly guided
our laboratory activities. Once, I happened to become the object of
this strictness. I do not remember which experiment exactly I was
performing, but it took a week and my results were very good. However,
when reporting the experiment to my supervisor, it became clear that I
had neglected to control the temperature. Although it was only a few
degrees higher than recommended and did not affect the results, I had
to repeat the entire experiment from the beginning. At that moment, I
believed it to be an unnecessary cavil, but with time, I’ve
appreciated the wisdom of this decision. I’ve remembered this
incident for my whole life and unnoticeably for myself, it has changed
my outlook to the quality of the experiments. Outstanding experimental
skills of researchers graduated from the Department of Biochemistry
have been more than once mentioned by Russian and foreign
specialists.
S. E. Severin mostly worked with senior students. He regularly participated in the discussion on their projects and diploma works, which he criticized more frequently (sometimes rather severely) than praised. Often, he turned to the problems unrelated to the subject discussed at the seminar. I remember the conversation initiated by him about the role of ideology in the formation of a naturalist. Another time, there was a lecture on the role of ethics in the communication between humans. Many of us even did not know such words. It should be remembered that it was 1958; the communist ideology was in full power, and no books were available on these subjects. Later, I understood that S. E. Severin was preparing us for independent work; he put in the accents and behavior markers. These talks by S. E. Severin were priceless for me. His principles have helped me in difficult times of my life to find solutions for both scientific and everyday problems. During my senior year, S. E. Severin proposed to supervise my diploma work. Of course, I happily agreed. The goal of this work was to study the influence of parathyroid gland on oxidative phosphorylation based on indirect data. The hypothesis was confirmed, and the diploma defense was successful. I believe I have a right to consider myself a disciple of S. E. Severin, because I had been greatly influenced by him and carried the memory of him through many years.
After graduating from Moscow State University, I was accepted to the Novosibirsk Institute of Organic Chemistry, Siberian Branch of the Academy of Sciences of the USSR, to the laboratory of D. G. Knorre. The direction of our future studies was discussed at the laboratory meeting, and it was concluded that the best choice would be studying the primary structure of ribonucleic acids. I was tasked (almost by chance) with developing the methodology for the nucleotide sequence analysis in polyribonucleotides. At that time, researchers could determine only the nucleotide composition of nucleic acids, whereas deciphering the nucleotide sequence was only a dream. I had also to provide the necessary “tool set” that, first of all, included ribonucleases (RNases) and phosphodiesterases (PDEs) with different specificity. D. G. Knorre advised me to pay a special attention to PDEs from snake venoms as enzyme cleaving monoribonucleotides from the 3′-ends of polynucleotides. He believed that kinetic analysis of PDE-catalyzed RNA hydrolysis would provide useful information on the nucleotide sequence in the RNA chain. Soon after that, I traveled to Tashkent to collect snake venoms and brought specimens from five snake species. Such was the start of the application of snake venom nucleases for investigating RNA structure.
We had studied snake venom RNases from 1963 to 1982 with some interruptions. The majority of studies have been performed in my laboratory at the Novosibirsk Institute of Bioorganic Chemistry and later at the Novosibirsk Institute of Molecular Biology (Glavmicrobioprom). All tested venoms of Central Asian snakes were found to contain RNases. Analysis of enzymatic properties of these RNases revealed their unique specificity to the substrate helical conformation. We were the first to identify enzymes with such specificity. Snake venom RNases still remain unique enzymes for more than half a century, since no similar RNases have been found. An article summarizing the accumulated information has never been written; therefore, I am taking the opportunity to fill the gap in the present review. The results of our studies on the kinetics of hydrolysis of natural RNAs with snake venom RNases were extremely interesting and sometimes paradoxical. Here, I present these data in detail, since they have served as a basis for the use of RNases in deciphering the structures of RNAs and their complexes with proteins.
The results of our studies have been reported at scientific conferences, published in central scientific journals, and acknowledged in the scientific circles, and have considerably influenced the development of concepts on the structural organization of RNAs and RNPs (ribonucleoproteins). By now, they have become a part of the biological science history. Therefore, I decided to omit a rather large body of studies related to the use of snake venom RNases in elucidation of the macrostructure of specific tRNA complexes with aminoacyl-tRNA synthetases and elongation factor EF-Tu, as well as the macrostructure of bacterial ribosomes. If the readers desire to make themselves familiar with this information, they might turn to the scientific publications of that period [1-5]. Here, I will present only the seminal data that had been of particular importance.
DETECTION, CHARACTERIZATION, AND PRACTICAL USE OF RIBONUCLEASES
FROM SNAKE VENOMS
Detection and general characteristics of snake venom RNases. Comparative analysis of phosphodiesterase activity in snake venoms revealed that for some venoms, the cleavage rate of bis-(para-nitrophenyl)phosphate did not correlate with the cleavage rate of natural RNAs. Based on this finding, it was suggested that snake venoms, in addition to PDEs, contain endonucleases that could not be tested by reaction with a synthetic substrate. Chromatographic analysis of venoms from five snake species revealed in all of them the endoribonuclease activity that could be quantitatively fractionated from the PDE activity. Venoms from gyurza (Vipera lebetina), cobra (Naja naja oxiana), and Pallas’ pit viper (Gloydius halys) were used in further studies, because, on one hand, they contained the greatest amounts of the enzyme, and, on the other hand, this choice allowed us to compare the properties of nucleases of snakes from different taxa. The procedure developed by us for the isolation of RNases and PDEs included successive fractionation of venoms by gel-filtration on Sephadex G-75 and chromatography on SE-cellulose (Fig. 1) [6-9].
Fig. 1. Chromatography profiles of snake venoms: a) pit viper; b) gyurza; c) cobra; 1) gel-chromatography on Sephadex G-75 (3 × 120 cm); 2) re-chromatography of RNase fractions on SE-cellulose (1 × 60 cm). Solid line, absorption at 280 nm; dashed line, PDE activity; dash-dotted line, RNase activity.
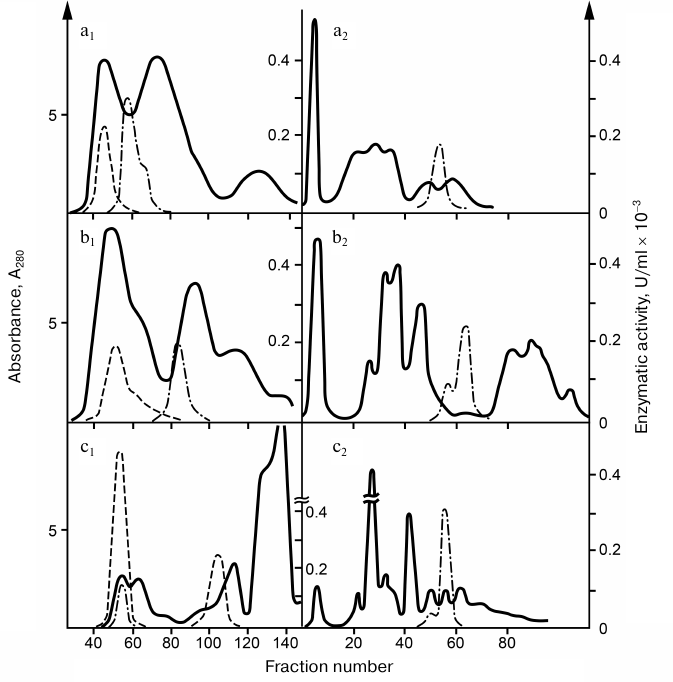
RNases preparations were purified (100-400 times) and completely free from interfering admixtures. The RNase activity of cobra venom was 15-20 and 5-10 times higher than the RNase activities in the gyurza and pit viper venoms, respectively. The pH optimum of RNases was within 7.6-8.0. The molecular mass of the enzyme was 17 kDa in gyurza and cobra and 35 kDa in pit viper as determined by SDS-electrophoresis. The active form of RNase from the cobra and gyurza venoms consisted of two identical subunits, partially dissociating under conditions of gel-chromatography.
Snake venom RNases (RNases V1) [8, 9] were characterized as metal-dependent RNA hydrolases activated by magnesium ions and inhibited by zinc and copper ions, mercapto compounds, and chelating agents [7]. The final products of polyribonucleotide hydrolysis by these enzymes were short oligonucleotides with phosphate residue at the 5′-end [10, 11]. RNases V1 were nonspecific to the RNA sequence. The DNase activity of RNases V1 was approximately 600 times lower than the RNase activity in the enzyme preparation and could be associated with DNase admixtures. Other than molecular masses, RNases V1 from different venoms were virtually identical in their enzymatic properties.
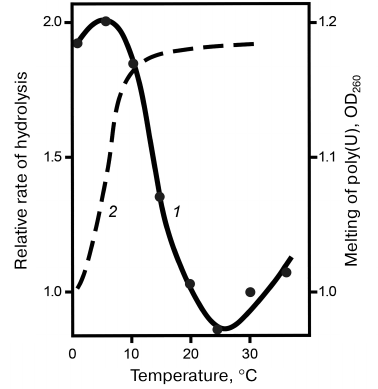
Specificity of snake venom RNases to the polyribonucleotide macrostructure. Specificity of RNases to homopolyribonucleotides. Despite the absence of specificity to the nature of nitrogen bases, RNases V1 were found to hydrolyze synthetic homopolyribonucleotides at considerably different rates. Thus, the reaction rate decreased in the following order: poly(A) > poly(C) > poly(I) > poly(U), with poly(G) virtually resistant to hydrolysis. The difference in the specificity of RNases V1 to different homopolymers was explained by the influence of polyribonucleotide secondary structure, the conformation and stability of which depended on the nitrogen bases. This explanation was confirmed by comparing the hydrolysis rates of free poly(U) and poly(G) with the hydrolysis rates of same nucleotides in the content of complementary double helix. Figure 2 shows that poly(U) in the complex with poly(A) was hydrolyzed approximately 10 times faster, whereas poly(G) in the complex with poly(C) was hydrolyzed at same rate as poly(C). This raised the question on the importance of substrate helical conformation for its hydrolysis with RNase V1. This question was answered by the studies on the temperature dependence of poly(U) hydrolysis. Figure 3 shows that within a certain temperature interval, the hydrolysis rate inversely depended on the reaction temperature, which is unusual within the framework of traditional enzymatic reaction thermodynamics and can be explained only if we assume that hydrolysis of the phosphodiester bond at the site of its direct contact with the enzyme is preceded by the poly(U) region transition to the helical conformation [12, 13].
Fig. 2. Kinetic curves of (a) poly(U) and (b) poly(G) hydrolysis in the double helix (1) and free state (2) by RNase from cobra venom.
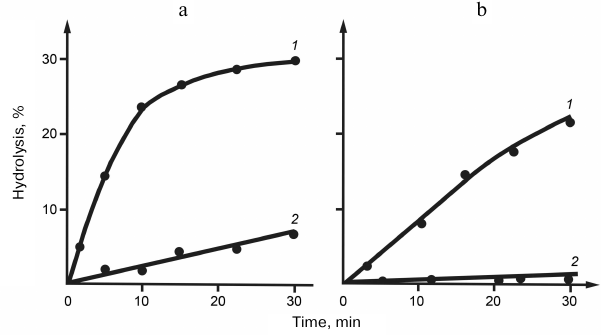
Fig. 3. Temperature dependence of the relative rate of poly(U) hydrolysis (1); poly(U) melting curve (2).
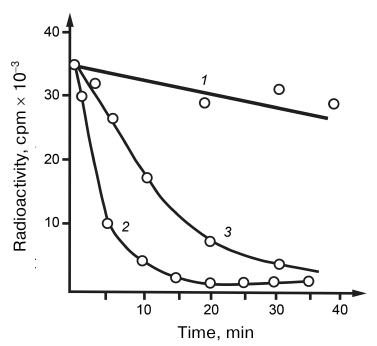
The substrate features of hybrid RNA–DNA helices have been studied for the hydrolysis of 32P-labeled transcripts. As shown in Fig. 4, RNA in the hybrid helix was degraded more rapidly than RNA in a free state. The ability of RNase V1 from cobra venom to hydrolyze RNA in the hybrid helix evidenced once again that the enzyme is specific to the polyribonucleotide helical conformation independently of the mechanism of its formation (double, hybrid, or single-stranded helix). Having in mind such specificity, it may be assumed that the RNA loop regions unable to acquire helical conformation because of fixed positions of their ends should be resistant to hydrolysis. We were the first who described the specificity of RNases to the helical RNA structure. RNases V1 differ from their nearest analogs, cellular processing RNases working co-transcriptionally in the poorly studied intermediate state of the synthesized RNA chain macrostructure, in the fact that they require the presence of helical conformation only in the attacked RNA region. Therefore, snake venom RNase can be considered a degenerate processing RNase. It may be assumed that enzymes with such specificity do not correspond and even contradict the general structure of metabolism and can exist only in antimetabolic systems.
Fig. 4. Hydrolysis of DNA–RNA hybrid helices by (1) RNase A and (2) RNase from cobra venom; 3) hydrolysis curve of free RNA by RNase from cobra venom.
Specificity of RNases to natural polyribonucleotides. Unlike homopolyribonucleotides with their monotonous secondary structure, natural RNAs have complex three-dimensional structure with alternating helical and loop regions. Elucidation of the impact of RNA macrostructure on the specificity of RNase from cobra venom was important for analyzing the macrostructures of both RNA itself and RNP complexes. The studies on the products of initial stages of hydrolysis of the MS-2 phage RNA labeled at the 3′-end by enzymatic ligation with 32P revealed high RNase selectivity to the specific regions of RNA chain. Thus, the hydrolysis sites remained virtually unchanged at different extent of hydrolysis, which indicated retention of the RNA macrostructure at the early stages of hydrolysis. Comparison of RNA breaks induced by RNases V1 from cobra, gyurza, and pit viper venoms confirmed that these nucleases have identical specificity. This allowed us to view RNases as functionally important components of snake venoms, although these enzymes by themselves are not considered toxins.
The biological role of snake venom RNases remains unclear. First, all substrates of these enzymes are located inside the cells. Therefore, the enzymes have to cross the cell membrane without disturbing its integrity, which seems rather problematic. Among all speculative hypotheses, the most plausible idea is that RNases can be aimed at inhibiting the immune response of an organism to snake venom. It is possible that RNase interacts with an immune cell receptor and then gets internalized with its help into the cytoplasm, where it attaches to the nuclear membrane and degrades mRNAs released from the nucleus, thus preventing cell involvement in the immunity formation.
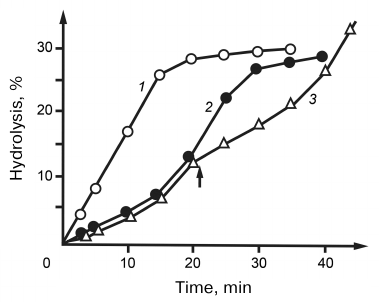
Kinetic features of tRNA hydrolysis by RNases from snake venoms. The above-described high selectivity of RNase V1 to helical RNA structures has opened new approaches for topographic analysis of helical elements in RNA molecules. When we were starting our studies, researchers could use only RNases specific to non-helical loop RNA regions that usually represent less than a half of RNA polynucleotide chain. Numerous attempts to identify RNA sites involved in the contact with protein ligands revealed only that such sites are not located in loop regions. Another feature is that the RNA breaks in the loop region leads to a more rapid destabilization of RNA macrostructure and loss of the RNA native conformation compared to the breaks in the helical regions. The reason for this difference is that a break in the loop region results in the conversion of a less mobile loop region into highly mobile end fragments, whereas if the break happens in the helical fragment, the conformation of the RNA chain can be maintained.
The studies on the hydrolysis of natural RNAs by RNases V1 revealed for the first time that this reaction occurred with auto-acceleration. This phenomenon was most noticeable for the hydrolysis of tRNAs – the reaction rate increased 3-4 times after the break of several phosphodiester bonds (Fig. 5). We suggested that such unusual hydrolysis kinetics could be caused by the influence of tRNA tertiary structure and assumed that native tRNAs are different from hydrolysis intermediates in their substrate features. This assumption was confirmed by the studies on the effect of fresh additions of tRNA during its hydrolysis. As shown in Fig. 5, introduction of additional tRNA to the medium at the stage of reaction acceleration led to the immediate decrease in the hydrolysis rate. On one hand, this dependence evidenced an increased affinity of RNase V1 to the native tRNA; on the other hand, it indicated that stabilization of the enzyme–substrate complex was accompanied by a decrease in the hydrolysis rate. We explained this increased stability of the tRNA–RNase complex by a decrease in the entropy factor due to more rigid conformation of native tRNA as compared to the products of its hydrolysis. This explanation seems quite reasonable if we assume that the enzyme molecule contained a substrate-binding site different from the enzyme catalytic site. It was suggested that such sites were present in RNase III from Escherichia coli [14]. The limited mobility of RNase relatively to tRNA in its primary complex could hinder transition to the hydrolysis-competent complex, which was manifested by the observed decrease in the initial rate of tRNA hydrolysis. If we assume that the internal mobility of the substrate molecule and stability of the enzyme–substrate complex were in inverse correlation, then its most stable variants should form, first of all, during enzyme interaction with the most fixed elements of the tRNA tertiary structure. In its turn, this should limit the number of phosphodiester bonds subjected to the primary attack by the enzyme. This hypothesis was confirmed by studying the topography of primary RNase-induced breaks in nine different tRNAs: phenylalanine, valine, asparagine and tryptophan tRNAs from yeast; phenylalanine, valine, methionine, glutamine, and tryptophan tRNAs from E. coli; and tryptophan tRNA from bovine liver [1-4, 15, 16]. When the breaks produced by tRNA hydrolysis were superimposed to the corresponding macrostructure models, all breaks without exception were found in the helical regions. Unlike unambiguously addressed breaks in the central part of the molecule, the 3′-end of tRNA was attacked at five closely located phosphodiester bonds, which was apparently due to the increased flexibility of this region. Radioactivity measurements showed than during the early stages of hydrolysis, ~80% of all breaks were introduced in the acceptor stem. In addition to the acceptor stem, breaks in the anticodon stem at positions 28-30 and 41-43 were common for all tRNAs. Other helical regions were rather resistant to hydrolysis. Breaks in the D-stem at position 13 were found only in tRNAPhe. In tRNATrp , all three RNases caused breaks in the acceptor stem only at positions 65 and 69. Based on these data, we suggested a mechanism for the enzyme–substrate complex formation: the primary binding site of the enzyme binds to the least mobile central fragment of tRNA; the catalytic site can interact only with bonds nearest to the primary binding site. The more mobile are these sites, the more variants of the hydrolysis-competent complex can form [17]. The existence of this mechanism is supported by the fact that the RNase-attacked bonds are arranged within a radius of 20-25 Å around the central concave part of the tRNA molecule. It should be noted that some results of our studies retain their significance nowadays. First of all, this relates to the RNase from snake venom which does not have analogs and remains the only enzyme that can be used for testing helical regions in RNA molecules without disturbing their spatial structure.
Fig. 5. Kinetic curves of hydrolysis of poly(A) (1) and yeast tRNA (2). 3) Kinetic curves of hydrolysis by RNase from cobra venom. The arrow indicates addition of a fresh portion of tRNA to the reaction mixture.
DEVELOPMENT OF METHODS FOR THE ANALYSIS OF RNA PRIMARY
STRUCTURE
In 1963, at the meeting on determining the nucleotide sequence of tRNA, I proposed a new approach for decoding the nucleotide sequence of oligonucleotides. The essence of the proposal was as follows: the products of incomplete oligonucleotide hydrolysis by the snake venom PDE that sequentially cleaves 5′-nucleotides from the 3′-end of substrate molecule, are separated by their molecular masses by ion-exchange chromatography under conditions when oligonucleotides exit from column according to their length with a step of one nucleotide. Subsequent 3′-terminal analysis of intermediates (3′-end nucleotide transforms into nucleoside at alkaline hydrolysis) should ensure the deciphering of the oligonucleotide nucleotide sequence [18]. My proposal was approved by the participants of the meeting, who also supported our decision to collaborate on this topic with the Institute of Chemistry of Natural Compounds (later, Institute of Bioorganic Chemistry, USSR Academy of Sciences). At this time, we did not have individual RNAs. To obtain model oligonucleotides, we had to hydrolyze total RNA by RNase A and fractionate the reaction products by chromatography in 7 M urea. The total pentanucleotide fraction was re-chromatographed in acidic solution. For our further studies, we selected a fraction of the 2G2A1C composition. Although isolation and purification of this pentaribonucleotide took about 2 months, only a few days were spent to determine its primary structure [19]. Some months earlier, a study identical to our work in its design was published by the laboratory of Holly. In this laboratory, several individual tRNAs had been already isolated by countercurrent distribution, and due to the new method for oligonucleotide sequencing, Holly and colleagues were able to quickly establish the primary structure of alanine tRNA. This was the first deciphered sequence of natural RNA. For this work, Robert W. Holly was awarded the Nobel Prize in Physiology or Medicine in 1968. In the works that appeared soon after, chromatography was replaced by two-dimensional polyacrylamide gel electrophoresis, which considerably simplified the procedure, and the sequencing method has been then generally accepted. The principle formulated by us and Holly group has become the basis for the well-known chemical method of DNA sequencing by Maxam and Gilbert.
Later, my group had to change the subject of our studies and switch to the investigation of the features of snake venom RNases from and isolation of individual tRNAs. The need for the direct RNA sequencing emerged again after we had started molecular studies of RNA viruses. RNA sequencing methods existing at that time had a common drawback: RNA hydrolysis required for the sequence analysis was enzymatic and took place under conditions when both secondary and tertiary RNA structures strongly hindered uniform fragmentation of the RNA chain that could be achieved only by chemical hydrolysis under denaturing conditions. On the other hand, chemical fragmentation of RNA makes it difficult to arrange the hydrolysis products in the order corresponding to the RNA nucleotide sequence. While searching for the method for direct RNA sequencing, we came to the conclusion that it had to satisfy two conditions: uniform fragmentation of RNA chain combined with a reliable method for the analysis of its degradation products. Originally, we had considered two independent approaches for RNA sequencing. According to the first option, 32P-labeled phosphate is introduced by polynucleotide kinase into the RNA 5′-end. RNA is then partially hydrolyzed by 5′-endoribonuclease combined with snake venom PDE, so that the distribution of breaks along the chain is as uniform as possible. The resulting hydrolysate is treated with T4 RNA ligase that links non-helical intramolecular RNA fragments into rings. In these ring-like structures, the 5′-terminal 32P-phosphate forms the phosphodiester bond and became resistant to the phosphatase. After phosphatase-catalyzed removal of 5′-Pi phosphate residues from the RNA fragments not involved in the cyclization, the hydrolysate is fractionated by electrophoresis in polyacrylamide gel. Positions of radioactive bands in the electrophoregram should correspond to the positions of nucleotides in the RNA chain, and the nature of the labeled nucleotide could be identified after elution of the correspondent RNA fragment from the gel and its alkaline hydrolysis. This is possible due to the transition of radioactive phosphate onto the 3′-terminal nucleotide of the initial fragment during cyclization of the RNA fragment and its subsequent alkaline hydrolysis. This proposed method appeared reliable because it represented a combination of repeatedly tested procedures. The two issues that remained unclear were the limiting length of fragments for the cyclization with ligase and the effect of the residual secondary structure of RNA fragments in the hydrolysate on cyclization. The second approach was technically easier than the first one but had more uncertainties. It was proposed to perform mild chemical hydrolysis producing approximately 2-4 breaks per RNA molecule and then, using polynucleotide phosphokinase, to attach 32Pi to 5′-hydroxyls released during hydrolysis. After this, the radioactive reaction mixture is separated by polyacrylamide gel electrophoresis; radioactive fractions are cut from the gel, subjected to alkaline hydrolysis, and separated by electrophoresis. The developed radioactive spots could be identified by their electrophoretic mobility, and the nucleotide order in the RNA chain should correspond to the order of radioactive bands in the polyacrylamide gel. Before its experimental verification, this new approach for RNA sequencing seemed purely hypothetical to me; I was not quite sure that it was possible. Soon after this, I was invited to spend six months at the Strasbourg Institute of Molecular and Cellular Biology (France). There, I made acquaintance with the English biochemist John Stanley, who was finishing his internship and appeared to be relatively free. I shared my ideas with him and proposed to test them in practice. After some thought and hesitation, he agreed and in two days, he presented a detailed plan scheduled by the day, collected all necessary equipment and reagents, discussed with me all unclear points, and began to implement our plans. It should be noted that Strasbourg Institute had been already engaged in structural analysis of nucleic acids for 10 years, so all the equipment and reagents we needed for our work were available, which greatly facilitated our task. During that time, I was very busy with my main research project and somewhat departed from our plans. Imagine my surprise when exactly two weeks after the work had been started, radiant John appeared with a developed X-ray film covered with black spots. We immediately started to decipher the sequence of our tRNA (it was tRNAPhe from Bacillus stearothermophilus) and then compared it with the already known sequence. Incredibly, but the sequences completely coincided. For this, only 5 µg of tRNA was used, almost a million times less than we expected in 1962! Our success became immediately known at the Institute. During the following two days, John had to explained in detail the results of our experiment to everyone. Soon, he wrote an article and flew to London to arrange its publication in the Nature journal. There, he was promised that the article would be published in the nearest future. Indeed, it appeared in the July issue of Nature in 1978, four months after we had received the first results [20]. The method had been used and fully confirmed in several laboratories. It could be applied for analyzing the sequences of RNAs with the tertiary structure that blocks transcription. Such regions were found, for example, in RNAs of viruses and viroids. Our method appeared shortly before the development of the first sequence analyzers. Automated methods have rapidly devalued almost all manual approaches, including our method, because it was soon found how to convert RNA with a complex tertiary structure into cDNA. However, our method still has some advantages over automated analysis. One of the advantages has been mentioned above; besides, our method allows to fix and identify minor bases in the RNA chain. It is possible that it will be applied in the future in the diagnostics of severe diseases associated with disorders in the minor base synthesis.
Our method presents one more unsolved riddle: although RNA hydrolysis was carried out in formamide under guaranteed denaturing conditions, i.e., when the reactivity of all phosphodiester bonds should be the same, the breaks in the RNA chain was considerably different from random, which completely contradicted the laws of statistical distribution. It appeared that after the first break, the RNA chain becomes resistant to further hydrolysis. I can only suggest my explanation. Formally, RNA hydrolysis takes place in water-free formamide. Free water that accidentally happens to be present in the reaction mixture should quickly evaporate at 100°C. However, we observed that RNA chain breaks by conventional hydrolysis with the participation of water. If it were not the case, formation of 5′-hydroxyl at the site of the chain break would remain inexplicable. Most likely, the reaction occurs with the participation of hydration water bound to either RNA or formamide. It was shown in control experiments that RNA hydrolysis in formamide proceeded more intensively than in deionized water; therefore, formamide should act as a catalyst of hydrolysis and for this, it has to interact specifically with the phosphodiester bond. It is known that the complex stability is in inverse correlation with its mobility. In our case, the thermal mobility of phosphodiester bonds is directly related to the polynucleotide chain mobility, and a break in the middle of the polynucleotide chain can become impossible. Therefore, initial hydrolysis stages include point hydrolysis predominantly of the original RNA molecules. This hypothesis is supported by the following observation: 13-nucleotide RNA fragments had only one hydrolysis site on each end of the RNA molecule. This means that 13-nucleotide fragments cannot be hydrolyzed deeper under the used conditions. Most likely, the breaks of phosphodiester bonds nearest to the fragment ends appeared during the first acts of hydrolysis of native RNA molecule.
MOLECULAR AND ETIOLOGICAL ANALYSIS OF CURRENT DIVERSITY OF HUMAN
INFLUENZA VIRUS
Genotyping of influenza A virus epidemic strains circulating on the USSR territory in 1968-1989 and studies of vaccine strain entry into the epidemic circulation. In 1985, the department I headed turned to the analysis of genomes of epidemiologically relevant viruses. From 1986 to 1990 in the laboratory of my disciple N. A. Petrov, more than 100,000 nucleotides from several tens of hemagglutinin (HA) genes and ~100 other genes of influenza A viruses, as well as of some other viruses, had been sequenced. This became possible due to a new method for direct sequencing of viral RNAs based on the combination of chemical sequencing with enzymatic extension of radioactive primer on the viral genome template. This technique can be used for the analysis of crude total nucleic acid material isolated from the concentrated clarified allantoic fluid of chicken embryos infected with the influenza virus. Using comparative analysis of gene sequences of circulating viral variants, we planned to answer the questions on the vital activity and epidemiology of influenza A viruses, in particular, to elucidate whether the reversal of influenza A virus vaccine strains could occur during their natural circulation. Moreover, we wanted to study the specificity and regularities of the antigenic drift of the dominating influenza A(H3N2) subtype viruses.
It is known that during the interepidemic period, the variety of genome sequences and antigenic characteristics of the circulating pathogens increases. Thus, in the middle 1980s, influenza viruses of the A(H3N2) antigenic subtype were represented by at least three antigenic subgroups. The differences in the genomes of all these strains completely satisfied the laws of natural variability of influenza A viruses related to the antigenic drift. However, along with the viruses dominating in the epidemic process, there were cases of so-called antigenic anachronisms atypical for the natural variability. These atypical anachronisms have been found in the USSR and neighboring countries. We have analyzed and compared genomes of some influenza viruses of the antigenic subtype A(Н1N1), which allowed us to assume their direct relation with the vaccine strains created on the basis of the A/PR/8-34(H1N1) virus.
Influenza viruses of the A/Mongolia/128/86(H1N1) subtype (five strains) were isolated from children in Mongolia in 1985-1986. Viruses of the A/Moscow/771/88(H1N1) subtype (three strains) were isolated from adults in February 1988. The nucleotide sequences of viral genomes were established through the revertase-catalyzed extension of 5′-labeled oligonucleotide primers on total vital RNA as a template followed by chemical sequencing of the reaction products. Genome sequencing of the A/Moscow/771/88(H1N1) influenza virus revealed its relationship with the reassortant vaccine strain A/Kiev/59/79(H1N1) that had been widely used in 1981-1985 as an inactivated vaccine. The HA gene of the A/Moscow/771/88 virus contained all four unique silent substitutions that distinguished the HA gene of the A/Kiev/59/79/P strain from other isolates of the A(H1N1) subtype. Moreover, two additional mutations, C737T and A912G were detected. Partial sequencing of the NP, PB1, PB2, PA, M, and NS genes of the A/Moscow/771/88 virus showed their full identity to the corresponding genes of the vaccine strain. The only difference was the T58A substitution in the M gene that was identical to the substitution in the laboratory variant A/PR/8/34 that was parental to the variant used in the USSR. The other antigenic anachronism, A/Mongolia/128/86(H1N1), was related to the other vaccine strain, A/Leningrad/54/1, that had been used as a component of inactivated vaccine in 1979-1981. The НA gene of the A/Mongolia/128/86 virus contained all unique substitutions inherent in the НA gene of the vaccine strain and different from it only by a silent mutation at position 536. Analysis of the PB2, PB1, NP, NS, and M genes sequences showed that all of them, as well as the vaccine strain, had originated from the laboratory variant of the A/PR/8/34(H1N1) virus. The T58A substitution in the M gene and in the virus A/Moscow/771/88 indicated their common origin. Moreover, we have also found evidence of the relationship between some (H3N2) subtype viruses isolated by us in 1982 and live attenuated vaccine A/1533/17(H3N2) used in the USSR in 1974-1982. Therefore, detailed analysis of genome sequences of the majority of atypical viral isolates has established their direct relationship with previously used whole-virion vaccines, both inactivated reassortant and attenuated [21-26]. Theoretically, individual genes of vaccine strains in the epidemic circulation could occur as a result of their “uptake” by virulent viruses through reassortment; however, we have not observed such reassortment in the studied cases – all genes of the viral anachronisms were related only to the genes of the corresponding vaccine strain. Realistic assessment of the current situation with influenza vaccines prompts us to fear that purely technical improvements in the inactivation of viral preparations would not guarantee prevention of biosphere contamination with vaccine strains. Therefore, it seems essential to review the general strategy of influenza prevention with vaccines and to abolish the widespread use of whole-virion vaccines [26].
Dynamics of mutations in the antigenic A(H3N2) subtype viruses during their circulation from 1968 to 1977. Influenza viruses of the antigenic A(H3N2) subtype reappeared in the human population in 1968 after a long-term interruption and caused a pandemic of influenza. Since then, they have maintained epidemic activity while being subjected to the antigenic drift. During the first decade, their dominant variants had changed rather rapidly (every 1-3 years), which resulted in the irreversible alteration in the existing antigenic variety. Such a long-term (above 20 years) persistence of the epidemic caused by the influenza A(H3N2) viruses has presented a rare opportunity for studying fast microevolutionary processes in natural populations. It is commonly known that the molecular basis of antigenic drift is accumulation of mutations in the HA gene accompanied by changes of the dominant variant. The major reason for the fixation of new mutations is a continuous increase in the collective immunity of the host population. Continuous circulation of the virus between the hosts in a partially immune population ensures selection and retention of the drift mutations.
In order to elucidate specific features of the current antigenic subgroups of the influenza A(H3N2) viruses and properties of the antigenic drift that had occurred within 20 years, we determined the sequence of the antigenic HA protein from the most typical representative of this subgroup on the USSR territory. We also determined HA sequences from some international etalons and viruses of past years: A/Hong Kong/1/68, A/Victoria/36/72, A/Leningrad/538/4, A/Leningrad/337/76, A/Leningrad/385/80, A/Philippines/2/82, A/USSR/31/85, A/USSR/3/85, A/USSR/3/86, A/USSR/281/86, A/Riga/9977/86, A/Victoria/7/87, A/Sichuan/2/87, A/USSR/21/88 [26-33].
Analysis of amino acid substitutions in HAs of the influenza A(H3N2) viruses existing at that time revealed that more than a half of them were repeated mutations at the positions that had already mutated in the antigenic drift since 1968. Moreover, most of these repeated mutations were reversions that restored the protein structure of the earlier drift variant or the A/Hong Kong/1/68 prototype.
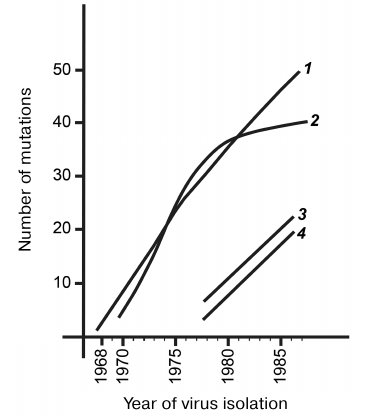
Regardless of the sequence diversity and antigenic features of different influenza A(H3N2) virus subgroups, all observed differences were consistent with the antigenic drift resulting from the development of epidemic process during the interaction between the hosts and virus populations. Mutations in the viral genes can be clearly subdivided into two fundamentally different groups. One group is silent mutations that (due to the genetic code degeneracy) do not alter protein structure and exert no direct influence on the antigenic drift. Figure 6 shows the dynamics of accumulation of such silent nucleotide substitutions in the HA gene of the influenza A(H3N2) viruses during 20 years of continuous antigenic drift (curve 1). The region encoding the most variable heavy subunit of the HA protein was analyzed in more than 50 genes. The maximum spread for each curve point did not exceed three substitutions, which represented an increase in the number of mutations per year. The shape of curve 1 indicates random character of nucleotide substitutions, their neutrality for the virus functioning, and a constant rate of their accumulation. The neutralizing function of silent substitutions is also indicated by their uniform distribution along the entire gene length. Due to the constant rate of mutagenesis and random appearance of both direct and reverse silent mutations, the entire evolutionary history of the viral population during the studied period is imprinted in gene sequences. The entire evolutionary history of any isolate can be interpreted by reconstructing its phylogenetic relationships with other related isolates.
Fig. 6. Increase in the number of differences in the HA gene and corresponding protein from the prototypical strain during antigenic drift: 1) silent nucleotide substitutions in the HA gene of the influenza A(H3N2) virus prototype A/Hong Kong/1/68(H3N2); 2) amino acid differences from the prototype A/Hong Kong/1/68(P3T2); 3) silent nucleotide substitutions in the HA gene of the influenza A(H1N1) viruses relatively to the A/USSR/20/77(H1N1) prototype; 4) differences of amino acids in the HA1 protein of the influenza A(H3N2) viruses relatively to the gene of the A/USSR/20/77(H1N1) prototype.
Unlike silent mutations, mutations resulting in amino acid substitutions directly affect viral phenotype and undergo selection.
The curve 2 in Fig. 6 shows the number of differences in the HA1 region from the prototype A/Hong Kong/1/68 and is based on more than 50 structures; the spread in the number of mutations for each year does not exceed two substitutions. During the first decade of the drift, amino acid substitutions had accumulated linearly; however, during the second decade, this process had significantly slowed down and eventually almost stopped. This was observed for almost entire current population of A/(H3N2) viruses, despite the fact that they belong to very different antigenic subgroups. Linear accumulation of amino acid substitutions during the initial drift phase indicates their randomness and neutrality for the HA molecule that performs numerous functions in the influenza virus life cycle. When the immune pressure from the host increases, the presence of multiple new HA variants offers temporary selective advantage. However, the very nature of substitutions and the number of positions in the molecule permitting such quasi-free changes are very limited. It may be assumed that the host’s immune pressure gradually distorts the native structure of the prototypical protein, and as a result, accumulation of substitutions prevents the functioning of this protein. Therefore, selection becomes bidirectional and stabilizes the number of differences at the level of 40-50. At the same time, substitutions continued to be fixed, but they occur mainly within the same limited regions of antigenic determinants and often lead to the reversion/restoration of the earlier versions of protein structure. Transition of A/(H3N2) viruses to the stabilizing drift phase indicates that they have practically exhausted their potential for variability that has allowed them to exist in the epidemic regime for more than 20 years, at least in the human population. Limited drift indicates that the epidemic phase of the human influenza A virus is also limited. Inevitable antigenic drift leads into a kind of evolutionary “dead end”. Human influenza viruses have to renew their gene pool from time to time either at the expense of HA genes from animal reservoirs characterized by negligible antigenic drift or by maintaining intact viral genomes in the human population in the absence of circulation and the drift inevitably associated with it.
In addition to the more or less completed studies described above, I have been also involved in small-scale studies, some of which, I believe, deserve to be mentioned in the review. First of all, it was genomic sequencing of viruses other than orthomyxoviruses (mainly, picorno- and poxviruses). Thus, we have sequenced genomes of encephalomyocarditis and foot and mouth disease viruses from the picornavirus family (Picornaviridae) and genes (~10,000 nt) from vaccinia and smallpox viruses from the poxvirus family (Poxviridae). In 1982, our Center started collaboration with the Institute of Foot and Mouth Disease of the Ministry of Agriculture of the USSR in the city of Vladimir. We introduced a new technology for the structural analysis of viral genomes and established the primary structures of VP1 genes in several strains of the foot and mouth disease virus circulating in the USSR during the late 1970s. Some of these genomes occurred to be products of reactivation of the strains used as inactivated vaccines [34]. These results received a great resonance, and not only in the scientific circles. As a result, the companies producing vaccines against the foot and mouth disease have modified the methodology of viral inactivation and established more strict control of technological processes.
We were among the first who introduced structural analysis of viral genomes into epidemiological studies. Methodological developments of our Vector Center have been actively implemented at other virology centers. We have collaborated with the Influenza Institute (Academy of Medical Sciences of the USSR), Ivanovsky Institute of Virology (Academy of Medical Sciences of the USSR) and Institute of Foot and Mouth Disease (Ministry of Agriculture of the USSR) in a series of studies in a new area of epidemiology later called molecular epidemiology. From the early 1980s and until introduction of automated sequencers in scientific practice, Russia had been the leader in this field of science.
Conflict of interest. The author declares no conflict of interest.
Ethical norm compliance. This article does not contain studies with participation of humans or animals.
REFERENCES
1.Boutorin, A. S., Remy, P., Ebel, J. P., and
Vasilenko, S. K. (1982) Comparison of the hydrolysis patterns of
several tRNAs by cobra venom ribonuclease in different steps of the
aminoacylation reaction, Eur. J. Biochem., 121,
587-595.
2.Favorova, O. O., Fasiolo, F., Keith, G., Ebel, J.
P., and Vasilenko, S. K. (1981) Complexes of aminoacyl-tRNA synthetases
and tRNA. Studies by partial cobra venom RNase hydrolysis,
Biochemistry, 20, 1006-1010.
3.Boutorin, A. S., Clark, B. F. C., Ebel, J. P.,
Kruse, T. A., Petersen, H. U., Remi, P., and Vasilenko, S. K. (1981) A
study of the interaction of E. coli elongation factor Tu with
aminoacyl-tRNAs by partial digestion with cobra venom RNase, J. Mol.
Biol., 152, 593-608.
4.Vasilenko, S. K., Carbon, F., Ebel, J. P., and
Ehresman, Sh. (1981) Topography of 16S RNA in 30S subunits and in 70S
subunits. Accessibility to cobra venom ribonuclease, J. Mol.
Biol., 152, 699-721.
5.Vasilenko, S. K. (1985) Ribonucleases of Snake
Venoms: Enzymatic Properties and Use for Studying Macrostructure of RNA
and RNP: Dis. Doct. Biol. Sci. [in Russian], Novosibirsk, pp.
235-286.
6.Vasilenko, S. K. (1963) Isolation of
phosphodiesterase and phosphomonoesterase from venom of the gyurza
Vipera lebetina using chromatography on sulfoethyl cellulose,
Biokhimiya, 28, 602-605.
7.Babkina, T. G., and Vasilenko, S. K. (1964)
Nuclease activity of the Central Asian snake venoms, Biokhimiya,
29, 268-272.
8.Vasilenko, S. K., and Babkina, G. T. (1965)
Isolation and properties of ribonuclease from the cobra Naja
oxiana venom, Biokhimiya, 30, 705-712.
9.Vasilenko, S. K., and Rait, V. K. (1975) Method for
isolation of high purity ribonuclease from the Central Asian cobra
Naja oxiana venom, Biokhimiya, 40, 578-583.
10.Vasilenko, S. K., Serbo, N. A.,
Ven’yaminova, A. G., Boldyreva, L. G., Budker, V. G., and Kobets,
N. D. (1976) Preparative preparation of 5′-oligonucleotides with
ribonuclease from cobra venom, Biokhimiya, 41,
260-263.
11.Vasilenko, S. K. (1985) Ribonucleases of Snake
Venoms: Enzymatic Properties and Use for Studying Macrostructure of RNA
and RNP: Dis. Doct. Biol. Sci. [in Russian], Novosibirsk, pp.
56-71.
12.Vasilenko, S. K. (1985) Ribonucleases of Snake
Venoms: Enzymatic Properties and Use for Studying Macrostructure of RNA
and RNP: Dis. Doct. Biol. Sci. [in Russian], Novosibirsk, pp.
73-91.
13.Magaril, S. A., Vasilenko, S. K., and Rait, V. K.
(1981) Specific features of synthetic polyribonucleotide hydrolysis
with endoribonuclease from the cobra Naja oxiana venom,
Biokhimiya, 45, 408-413.
14.Nicholson, A. (2014) Ribonuclease III mechanisms
of double-stranded RNA cleavage, Wiley Interdiscip. Rev. RNA,
5, 31-48.
15.Ebel, J. P., Remy, P., Baltzinger, M., Ehrlich,
R., Jefevre, J. F., Renaud, M., Fasiolo, F., Keith, G., Favorova, O.
O., Vasilenko, S. K., Giege, R., Dietrich, A., Kern, D., Moras, D.,
Thierry, J. C., Jocrot, B., and Zaccai, G. (1980) Aminoacyl-tRNA
synthetase, interaction with their ligands, in Enzyme Regulation and
Mechanism of Action, Proc. FEBS Special Meeting on Enzymes, Cavtat,
Dubrovnik, 1979 (Eildner, P., and Rien, B., eds.), Pergamon Press, N.
Y., pp. 211-221.
16.Ebel, J. P., Renaud, M., Dietrich, A., Fasiolo,
F., Keith, F., Favorova, O. O., Vassilenko, S. K., Baltzinger, M.,
Ehrlich, R., Remy, P., Bonnet, J., and Giege, R. (1979) Interaction
between tRNA and aminoacyl-tRNA synthetase in the valine and
phenylalanine system from yeast, in Transfer RNA,
Structure, Properties, and Recognition (Shimmel,
S., and Soll, D., eds.) Cold Spring Harbor Laboratory Press, N. Y., pp.
325-343.
17.Vasilenko, S. K., Butorin, A. S., Maev, S. P.,
Vityugov, F. I., Boldyreva, L. G., and Rait, V. K. (1983) Specificity
of nuclease from the cobra Naja naja oxiana venom to
macrostructure of polyribonucleotides, Mol. Biol. (Moscow),
17, 818-823.
18.Vasilenko, S. K. (1963) Information Bulletin
of the Council on Molecular Biology of Academy of Sciences of the
USSR (Moscow), No. 2, p. 19.
19.Vasilenko, S. K., Demushkin, V. P., Bydovsky, E.
I., and Knorre, D. G. (1965) Determination of nucleotide sequence in
oligonucleotides, DAN USSR, 162, 604-607.
20.Stanley, J., and Vasilenko, S. K. (1978) A
different approach to RNA sequencing, Nature, 274,
87-89.
21.Petrov, N. A., Netesov, S. V., Blinov, V. M., and
Vasilenko, S. K. (1986) Evolution of the hemagglutinin H3 subtype gene
of human influenza A virus, Mol. Genet. (Moscow), 11,
7-14.
22.Beklemishev, A. B., Blinov, V. M., Petrov, N. A.,
and Vasilenko, S. K. (1986) The primary structure of full-size copy of
hemagglutinin gene of the influenza A/Kiev 8/59/79(H1N1) virus,
Bioorg. Khim., 12, 375-383.
23.Petrov, N. A., Grinev, A. A., Vasilenko, S. K.,
Zhilinskaya, I. N., Paramonova, M. S., and Golubev, D. B. (1988) The
primary structure of influenza virus hemagglutinin at serial passages
on chicken embryos, Bull. Exp. Biol. Med., 11,
596-598.
24.Petrov, N. A., Kiselev, O. I., Grinbaum, E. B.,
Luzyanina, T. I., Polezhaev, F. I., and Vasilenko, S. K. (1990) A
possible circulation of the influenza A virus vaccine strains in the
biosphere, Dokl. Akad. Nauk SSSR, 315, 725-728.
25.Petrov, N. A., Vasilenko, S. K., Grinev, A. A.,
Kiselev, O. I., Grinbaum, E. B., and Luzyanina, T. I. (1988) The
primary structure of the influenza A/SSSR/2/85(H3N2) virus
hemagglutinin gene variants, in Problems of Constructing Influenza
Vaccines (Korneeva, E. P., ed.) [in Russian], Institute of
Influenza, St. Petersburg, pp. 54-60.
26.Petrov, N. A., and Vasilenko, S. K. (1990)
Current variety of human influenza A viruses on the molecular level,
Mol. Genet. (Moscow), 12, 3-10.
27.Sandakhchiev, L. S., Petrov, N. A., Yakhno, M.
A., Luzyanina, T. I., Grinev, A. A., Vasilenko, S. K., Grinbaum, E. B.,
Golubev, D. B., Govotkova, E. A., and Zhdanov, V. M. (1988) The primary
structure of genomes of the current Hong Kong-like strains of the
influenza A(H3N2) viruses, Dokl. Akad. Nauk SSSR, 302,
1494-1497.
28.Zhdanov, V. M., Petrov, N. A., Grinev, A. A.,
Yakhno, M. A., Isachenko, V. A., Gorbunov, Y. A., Vtorushina, N. A.,
Netesov, S. V., Vasilenko, S. K., and Sandakhchiev, L. S. (1989) The
primary structure of hemagglutinin of the influenza A(H3N2) viruses
isolated in the USSR in 1985, Vopr. Virusol. (Moscow), 2,
155-160.
29.Sandakhchiev, L. S., Petrov, N. A., Vasilenko, S.
K., Luzyanina, T. Y., Grinbaum, E. B., Gorbunov, Y. A., Vtorushina, I.
A., and Golubev, D. B. (1989) The primary structure of influenza
A(H3N2) atypical viruses in 1982-1985, Dokl. Akad. Nauk SSSR,
308, 477-480.
30.Petrov, N. A., Grinbaum, E. B., Litvinova, O. M.,
Luzyanina, T. Y., Vasilenko, S. K., Kiselev, O. I., and Sandakhchiev,
L. S. (1990) Structure of the influenza A/Leningrad/22/81/(H1N1) virus
genome, Mol. Genet. (Moscow), 7, 17-21.
31.Petrov, N. A., Vasilenko, S. K., Gorbunov, Y. A.,
Kiselev, O. I., and Sandakhchiev, L. S. (1990) The primary structure of
the hemagglutinin gene of the influenza A(H3N2) drift variant strain
A/Riga/9977/86, Vopr. Virusol. (Moscow), 5, 372-373.
32.Medvedeva, M. N., Petrov, N. A., and Vasilenko,
S. K. (1990) Characteristics of hemagglutinin of the influenza
A/Victoria/35/72(H3N2) virus persistent variants, Vopr. Virusol.
(Moscow), 5, 374-376.
33.Petrov, N. A. (1991) Molecular and Etiological
Analysis of the Current Variety of the Human Influenza A Viruses:
Dis. Doct. Biol. Sci. [in Russian], Moscow, pp. 24-34.
34.Onishchenko, A. M., Petrov, N. A., Blinov, V. M.,
Vasilenko, S. K., Sandakhchiev, L. S., Burdov, A. P., Ivanyushchenkov,
V. P., and Perevozchikova, N. A. (1986) The primary structure of
DNA-copy of the VP1 protein gene of the mouth and foot disease
A22 virus, Bioorg. Khim., 12, 416-419.