Amino Acid Residues β139, β189, and β319 Modulate ADP-Inhibition in Escherichia coli H+-FOF1-ATP Synthase
A. S. Lapashina1,2, T. E. Shugaeva1, K. M. Berezina1, T. D. Kholina1, and B. A. Feniouk1,2,a*
1Lomonosov Moscow State University, Faculty of Bioengineering and Bioinformatics, 119991 Moscow, Russia2Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received October 9, 2018; Revised November 6, 2018; Accepted November 6, 2018
Proton-translocating FOF1-ATP synthase (F-type ATPase, F-ATPase or FOF1) performs ATP synthesis/hydrolysis coupled to proton transport across the membrane in mitochondria, chloroplasts, and most eubacteria. The ATPase activity of the enzyme is suppressed in the absence of protonmotive force by several regulatory mechanisms. The most conserved of these mechanisms is noncompetitive inhibition of ATP hydrolysis by the MgADP complex (ADP-inhibition) which has been found in all the enzymes studied. When MgADP binds without phosphate in the catalytic site, the enzyme enters an inactive state, and MgADP gets locked in the catalytic site and does not exchange with the medium. The degree of ADP-inhibition varies in FOF1 enzymes from different organisms. In the Escherichia coli enzyme, ADP-inhibition is relatively weak and, in contrast to other organisms, is enhanced rather than suppressed by phosphate. In this study, we used site-directed mutagenesis to investigate the role of amino acid residues β139, β158, β189, and β319 of E. coli FOF1-ATP synthase in the mechanism of ADP-inhibition and its modulation by the protonmotive force. The amino acid residues in these positions differ in the enzymes from beta- and gammaproteobacteria (including E. coli) and FOF1-ATP synthases from other eubacteria, mitochondria, and chloroplasts. The βN158L substitution produced no effect on the enzyme activity, while substitutions βF139Y, βF189L, and βV319T only slightly affected ATP (1 mM) hydrolysis. However, in a mixture of ATP and ADP, the activity of the mutants was less suppressed than that of the wild-type enzyme. In addition, mutations βF189L and βV319T weakened the ATPase activity inhibition by phosphate in the presence of ADP. We suggest that residues β139, β189, and β319 are involved in the mechanism of ADP-inhibition and its modulation by phosphate.
KEY WORDS: ATP synthase, F-ATPase, ADP-inhibition, regulation, Escherichia coli, bioenergetics, FOF1DOI: 10.1134/S0006297919040084
Abbreviations: FOF1, FOF1-ATP synthase (F-type ATPase, F-ATPase); Pi, inorganic phosphate; pmf, protonmotive force; SBP, subbacterial inverted membrane particles.
FOF1-ATP synthase (FOF1) is
a membrane-bound multisubunit complex that catalyzes ATP synthesis from
ADP and inorganic phosphate (Pi). The enzyme is found in the
bacterial plasma membrane, inner mitochondrial membrane, and
chloroplast thylakoid membrane. The hydrophilic F1
subcomplex contains nucleotide-binding sites and catalyzes both ATP
synthesis and hydrolysis. The hydrophobic FO subcomplex
translocates protons across the membrane. The F1 subcomplex
of Escherichia coli is composed of five types of subunits in
stoichiometry
α3β3γ1δ1ε1,
and the FO subcomplex consists of three types of subunits
(a1b2c10) [1]. ATP synthesis or hydrolysis catalyzed by
F1 is coupled to the proton transport via the rotary binding
change mechanism [2-4]. Driven
by the protonmotive force (pmf) generated by the respiratory or
photosynthetic electron transport chains, protons are translocated
through FO at the interface of the a subunit and the
c10 ring oligomer. Proton transport induces rotation
of the c10 ring relatively to the
ab2 complex. The ab2 complex, in
turn, is bound to the α3β3δ
complex, and the c10 ring is connected to the γ
and ε subunits. As a result, proton transport is coupled to the
rotation of c10γε “rotor”
relatively to
ab2α3β3δ
“stator”. Rotation of the γ subunit inside the
α3β3 hexamer induces a series of
conformational changes that lead to ADP and Pi binding and
ATP synthesis and release. If the pmf value decreases below the
thermodynamic threshold for ATP synthesis, the reaction is reversed.
The conformational changes in the α3β3
hexamer caused by ATP hydrolysis lead to the rotation of the
γεac10 complex resulting in the
transmembrane proton transport and pmf generation.
ATP hydrolysis by FOF1 may play an important role when the activity of the primary pmf generators decreases. This happens, for example, in plants and photosynthetic bacteria in the dark or in aerobic bacteria and mitochondria when there is insufficient oxygen (ischemia). Under such conditions, FOF1 remains the only enzyme capable of maintaining the pmf necessary for several physiologically important processes. However, if the intracellular ATP concentration is significantly reduced or proton permeability of the membrane increases (e.g., in the presence of protonophores or toxins), ATP hydrolysis catalyzed by FOF1 may deplete the intracellular ATP pool and pose a threat to the cell. In this regard, it is not surprising that the ATPase activity of FOF1 is downregulated by several mechanisms [5].
Noncompetitive inhibition of FOF1-catalyzed ATP hydrolysis by MgADP is the most conserved of these mechanisms; it has been reported for all FOF1-ATP synthases studied so far [6-12]. If MgADP is bound in the catalytic site without Pi, the enzyme may undergo a conformational transition to the inactive state with MgADP trapped in the catalytic site (ADP-inhibition). In mitochondrial, chloroplast, and many bacterial ATP synthases, Pi counteracts this transition. Reactivation of the inhibited FOF1 requires pmf comparable to or exceeding the level of ATP synthesis. Presumably, the pmf-driven rotation of the γ subunit causes transition of the catalytic site with the trapped inhibitory ADP into the open state, promotes ADP release, and re-activates the enzyme. On the other hand, moderate pmf that is not sufficient for enzyme re-activation also counteracts ADP-inhibition by increasing the catalytic site affinity to Pi and thereby reducing the probability of ADP binding in the catalytic site without Pi [13, 14].
However, the pattern of ADP-inhibition in E. coli FOF1 seems to differ from that described above. First, the E. coli enzyme is less susceptible to ADP-inhibition than other studied ATP synthases. Second, in E. coli enzyme, ADP-inhibition is not prevented but rather promoted by Pi [11, 15].
We have previously shown that amino acid residue at the position corresponding to β249 of the E. coli enzyme affects ADP-inhibition. ATP synthases of betaproteobacteria and gammaproteobacteria (including E. coli) contain leucine at this position, while in mitochondrial, chloroplast, and most other eubacterial enzymes, the corresponding residue is glutamine. The replacement of glutamine with leucine dramatically weakens ADP-inhibition in FOF1 from the thermophilic bacterium Bacillus PS3 sp. [16], and the reciprocal βL249Q mutation in the E. coli enzyme promotes ADP-inhibition and, moreover, reverses the effect of Pi: in the mutant FOF1, Pi does not enhance but rather suppresses ADP-inhibition [17].
In this paper, we describe several amino acid positions in the β subunit that differ between FOF1 enzymes of betaproteobacteria/gammaproteobacteria, and ATP synthases of mitochondria, chloroplasts, and most other eubacteria. We have also investigated the influence of amino acid residues in these positions on ADP-inhibition using site-directed mutagenesis.
MATERIALS AND METHODS
Multiple alignment of FOF1 β subunit sequences. We searched 711 fully sequenced genomes of archaea and bacteria in the latest version of the Clusters of Orthologous Groups of proteins (COG) database [18] for ATP synthase subunit sequences. Sequences of mitochondrial (bovine and baker’s yeast) and chloroplast (spinach) enzymes were also added to the database. Hidden Markov model (HMM) profiles were constructed according to the latest version of the COG database as described in [19] and used in the database search with the HMMscan 3.1 software from the HMMer3 package [20]. Since the results of the HMM profile-based search often contain evolutionarily related proteins (e.g., α subunits while using β subunit profile), we sorted the results by the E-value. For β subunit, the hits with the E-values higher than 7.7·10–103 were cut off, since a jump in the E-values was observed after this threshold, separating β subunits from the related ones.
Similar procedure was performed for all remaining FOF1 subunits. The obtained sets of subunits were divided into groups potentially belonging to ATP synthases of the N- and F-type. Operons containing all F1 and FO subunits including δ subunit (that is lacking in the N-type enzyme [26]) were considered the F-type, and β subunits belonging to these operons were aligned with the MUSCLE 3.8 software [21]. Jalview 2.10.5 software [22] was used for visualization and analysis of the resulting multiple alignment.
Site-directed mutagenesis. Mutagenesis was performed using the pFV2 plasmid containing the unc operon (encodes a, b, c, α, β, γ, δ, and ε subunits of E. coli ATP synthase) and ampicillin resistance cassette. In this plasmid, a hexahistidine tag was added to the β subunit N-terminus, and all the cysteine codons except bCys21 were replaced by alanine codons. As shown earlier, these modifications have no significant effect on the enzyme activity [23]. In this work, enzyme expressed from the pFV2 plasmid will be referred to as the wild-type enzyme.
Mutations were introduced into the β subunit by polymerase chain reaction with mutagenic primers using the wild-type pFV2 as a template. Briefly, two primers, one of which contained the desired mutation, were used to synthesize a 200-400 bp oligonucleotide (megaprimer) carrying the mutation and the nearest unique restriction endonuclease site. Then, the third primer was used to expand the fragment flanking the mutation with another unique restriction site. The resulting fragment was cloned into the pBluescript II SK(–) vector, sequenced, and transferred into pFV2. Cloning procedures were performed in E. coli XL-1 Blue cells. For the expression of wild-type or mutant FOF1 variants, the corresponding plasmids were introduced into E. coli BW25113 (ΔatpB-atpC) strain, in which most of the ATP synthase operon was replaced with a kanamycin resistance cassette [16].
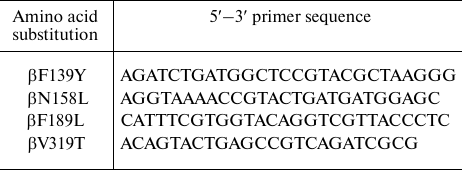
Mutagenic primers (DNA-Sintez, Russia) used in this work are listed in the table.
Primers used in mutagenesis
Preparation of subbacterial inverted membrane particles (SBP). For bacterial growth, 1 ml of an overnight E. coli culture was inoculated into 600 ml of LB medium (Amresco, USA) supplemented with ampicillin (100 mg/liter) and grown for 18-20 h on a rotary shaker at 37°C in 2-liter Erlenmeyer flasks. The total volume of bacterial culture was 3 liters. The cells were harvested by centrifugation (JA-10 rotor; Beckman Coulter, USA) (7000 rpm, 10 min) at room temperature. The wet weight of the harvested bacterial cells was 10-20 g. The cells were washed once with buffer containing 10 mM HEPES-NaOH, pH 7.5, 5 mM MgCl2, and 10% glycerol, resuspended in 25-30 ml of the same buffer, and disrupted by two consecutive passages through a French cell press (SLM Aminco, USA) at 1000 PSI. Cell disruption and all subsequent manipulations were performed at 4°C or on ice. Unbroken cell debris was collected by 30 min centrifugation at 13,700g and discarded. The supernatant was centrifuged for 1 h at 390,000 g, and the SBP pellet was washed with 25 ml of the same buffer and resuspended in the same buffer. The centrifugation was repeated once more, and the SBP pellet was suspended in 1.0-1.5 ml of the same buffer, aliquoted by 50 μl, frozen in liquid nitrogen, and stored at –80°C. Protein concentration in the SBP suspension was determined with a commercial Pierce™ BCA Protein Assay kit (Thermo Scientific, USA) using bovine serum albumin solution as a reference. A typical protein concentration in the SBP suspension was 20-80 mg/ml.
ATPase activity. The ATPase activity of SBP was measured by monitoring the medium acidification with pH indicator phenol red. We measured the absorption at 558 nm (maximum of the deprotonated form) and subtracted the absorption at 477 nm (isosbestic point) [24].
For the measurements with a CLARIOstar plate reader (BMG Labtech, Germany), SBP in 2 mM HEPES containing 1 mM MgCl2, 100 mM KCl, and 30 μM phenol red, pH 8.0 (pheRed buffer) were placed into a 96-well flat-bottom microplate (Greiner Bio-One, USA), 150 μl per well. When the ATPase activity was measured in the presence of Pi, K2HPO4 was added to the pheRed buffer to 6 mM. The measurements were carried out at 37°C. In each well, absorbance was measured at 558 and 477 nm for 3-5 s, then 150 μl of pheRed buffer containing ATP or ATP/ADP mix (total nucleotide concentration, 2 mM) was injected, and absorbance was recorded again for 30-50 s. Then, two subsequent injections of 30 nmol NaOH were made to calibrate the measurement. The ATPase activities were calculated using the CLARIOstar Data Analysis software (BMG Labtech) and the python 2.7 script (Python Software Foundation, USA).
The ATPase activity was also measured with an Aminco DW-2000 dual wavelength spectrophotometer (SLM Aminco). In these experiments, 3 ml of SBP in pheRed buffer was placed into a plastic cuvette, and the difference in absorbance (558 nm – 477 nm) was measured. The baseline was registered for 1 min, then the hydrolysis reaction was started by adding ATP or ATP/ADP mixture, pH 7.9, to a final nucleotide concentration of 1 mM. After that, several calibrating additions of 3 μl of 100 mM NaOH were made. The measurements were also carried out at 37°C. The Origin software package (OriginLab, USA) was used for curve analysis and ATPase activity calculation.
ATP-dependent proton transport. ATP-dependent proton transport was measured using the fluorescent indicator 9-amino-6-chloro-2-methoxyacridine (ACMA; Sigma-Aldrich, USA) that responds to the increase in the transmembrane ΔpH by fluorescence quenching [25]. ACMA fluorescence was excited at 410 nm and registered at 480 nm with a Fluoromax-3 spectrofluorometer (Horiba Jobin Yvon, Japan) in 1-cm acrylic cuvettes. The buffer contained 10 mM HEPES, pH 7.5, 100 mM KCl, 5 mM MgCl2. The ACMA concentration in the cuvette was 0.3 μg/ml. The reaction was initiated by adding ATP to 500 μM. The measurements were carried out at room temperature.
RESULTS
To identify amino acid residues potentially involved in ADP-inhibition, we analyzed β subunit sequences of prokaryotic ATP synthases. A set of 711 archaeal and bacterial genomes in the latest version of the COG database [18] was searched for FOF1 subunits. The sequences of mitochondrial (bovine, baker’s yeast) and chloroplast (spinach) enzymes were also added to the set. The N-type ATPases were removed from the search results, since their role seems to be associated with sodium ions transport rather than ATP synthesis or ATP-dependent pmf generation [26]. As the genomes of archaea and certain eubacteria do not contain any FOF1 genes, the resulting set used for the multiple alignment contained 492 eubacterial, two mitochondrial, and one chloroplast protein sequences. A fragment of the multiple alignment is shown on Fig. 1.
Fig. 1. A fragment of multiple alignment of ATP synthase β subunit amino acid sequences: 1-12) gammaproteobacteria and betaproteobacteria; 13, 14) mitochondria; 15) chloroplasts; 16-31) other eubacteria. Residue numbering (top line) corresponds to E. coli ATP synthase β subunit. Grayscale reflects the extent of residue conservation; black columns are four amino acid positions studied in this work and β249 position investigated earlier.

Analysis of the aligned sequences revealed that β subunits of all betaproteobacteria (34 species) and gammaproteobacteria (62 species plus two E. coli strains) from the set possess highly conserved phenylalanine, asparagine, phenylalanine, and valine residues at positions corresponding to β139, β158, β189, and β319 of the E. coli enzyme, respectively. At the same time, in all other eubacterial, mitochondrial, and chloroplast β subunits analyzed, the conserved residues found at these positions were tyrosine, leucine, leucine, and threonine, respectively.
We hypothesized that these residues may be involved in ADP-inhibition, as it was demonstrated for β249 residue of E. coli FOF1 [17]. We mutated each of these residues to the corresponding amino acid of the mitochondrial/chloroplast enzyme (mutations βF139Y, βN158L, βF189L, and βV319T). In addition, since β139 and β319 residues are located close to each other in the E. coli ATP synthase structure, the double mutant βF139Y+βV319T was also constructed. All mutant enzymes as well as the wild-type enzyme were expressed from the pFV2 plasmid in E. coli strain with deleted ATP synthase operon. The ATPase activity was measured in the SBP obtained from cells expressing the mutant and wild-type enzymes. To investigate the effect of pmf on ATP hydrolysis, the ATPase activity of SBP was measured in the presence of valinomycin and nigericin that dissipate pmf in potassium-containing buffers. The influence of 3 mM phosphate on the ATPase activity was also studied. We did not find any significant differences between the wild-type and βN158L enzymes under all the conditions tested. The ATPase activities of SBP isolated from other mutant strains are shown on Fig. 2.
Fig. 2. ATPase activity of SBP. The ATPase activity of SBP containing wild-type or mutant (βF139Y, βF189L, βV319T, or βF139Y+βV319T) ATP synthase was measured with 1 mM ATP in the presence or absence of the uncoupler (+Val/Nig, valinomycin+nigericin at 500 nM each), potassium phosphate (+Pi, 3 mM), or both. Numbers on top of the bars correspond to average activity calculated from 5-8 traces; error bars represent standard deviations. ATPase activity was expressed in units per mg total membrane protein (U/mg); one activity unit (U) corresponds to 1 μmol ATP hydrolyzed per 1 minute.
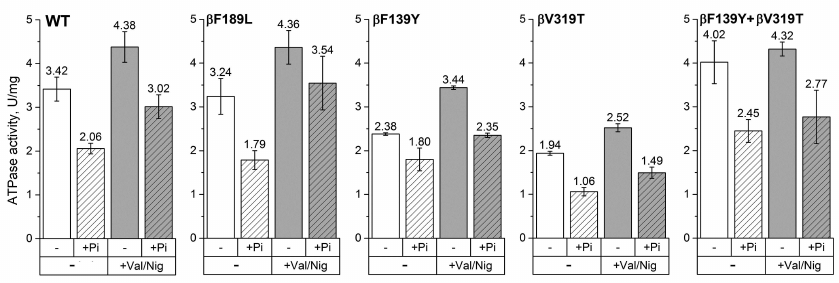
For all the four mutants, the ATPase activities of SBP hydrolyzing 1 mM ATP were comparable to that of the wild-type enzyme. The observed differences in the absolute values (1.94-4.02 U/mg total membrane protein) can be explained by either slight effect of the mutations on the enzyme activity or variations in the FOF1 expression levels in different samples. Phosphate (3 mM) reduced the ATPase activity in all the samples. However, in the βF139Y mutant, this effect was less pronounced (~30%, in contrast to 60-80% in the wild-type enzyme and other mutants). Uncoupling, i.e., increase in the proton permeability of the membrane that leads to the dissipation of pmf, stimulated the ATPase activity of all samples except SBP from the βF139Y+βV319T mutant, for which the effect was within the measurement error. The addition of phosphate together with the uncoupler also reduced the ATP hydrolysis rate in all samples, but the effect was less pronounced for the βF189L mutant (data not provided).
To further clarify the role of the proposed residues in ADP-inhibition of FOF1, the ATPase activity of each mutant was measured in the presence of ADP. The hydrolysis reaction was started by adding either ATP to 1 mM or ATP/ADP mixture to 750/250 µM or 400/600 μM. The effect of pmf and Pi on the ATPase activity in the presence of ADP was also investigated. The results of these experiments are displayed in Figs. 3-6.
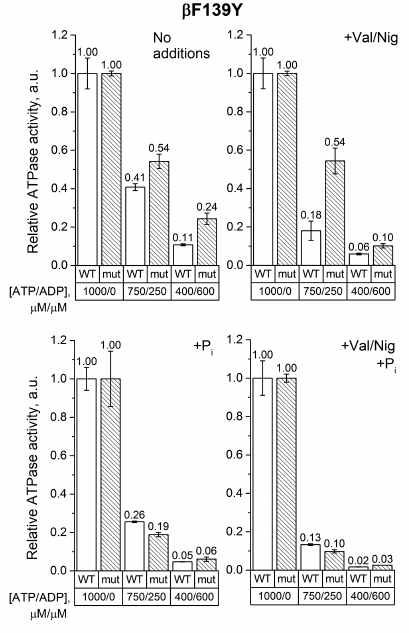
As can be seen from Fig. 3, ADP inhibited the ATPase activity of SBP from βF139Y to a lesser extent than that of the wild-type SBP. This effect was even more pronounced in the absence of pmf. However, in 3 mM phosphate, the mutation did not influence ADP-inhibition in either coupled or uncoupled SBP. It should also be noted that, similarly to the wild-type SBP, phosphate significantly inhibited the ATPase activity of the βF139Y mutant in the presence of ADP.
Fig. 3. Relative ATPase activity of SBP from the wild-type and βF139Y cells. The reaction was initiated by adding ATP to 1 mM or ATP/ADP mixture to 750/250 μM or 400/600 μM, respectively, as described in “Materials and Methods”. The activity was measured in the absence or presence of the uncoupler (+Val/Nig, valinomycin+nigericin at 500 nM each), potassium phosphate (+Pi, 3 mM), or both. Numbers on top of the bars correspond to average activity calculated from 5-8 traces; error bars represent standard deviations. All activities were normalized to the ATPase activity of the corresponding strain with 1 mM ATP without any additions.
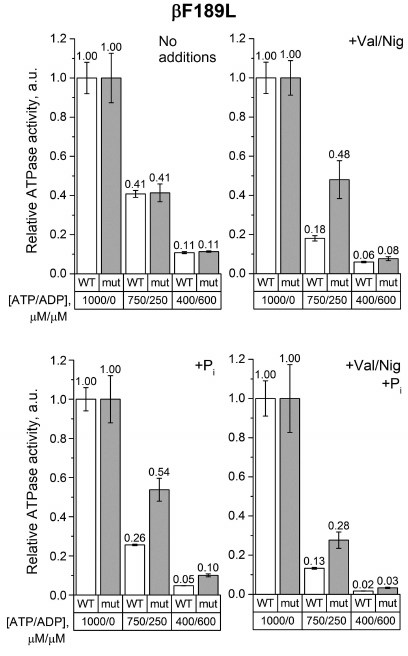
The same measurements were carried out with SBP from the βF189L mutant. In the absence of uncouplers and phosphate, the decrease in the ATP/ADP ratio equally influenced the ATPase activity of the βF189L and wild-type SBP (Fig. 4). However, when the uncoupler and/or 3 mM phosphate were added, the activity of βF189L SBP in a mixture of 750 μM ATP and 250 μM ADP was significantly higher than that of the wild-type SBP. At the same time, at a lower ATP/ADP ratio, this effect was less pronounced.
Fig. 4. Relative ATPase activity of SBP from the wild-type and βF189L cells. Measurements were carried out as indicated in the legend to Fig. 3 for the βF139Y mutant.
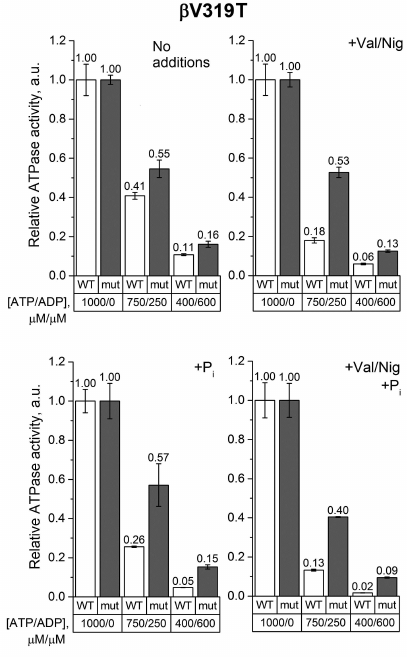
The effect of the ATP/ADP ratio decrease on the ATPase activity of βV319T SBP measured in the absence of phosphate or uncoupler was also similar to that observed in the wild-type samples (Fig. 5). But when the uncoupler and/or 3 mM phosphate were added, an increase in the ADP concentration reduced the ATPase activity of βV319T SBP significantly less than the activity of the wild-type SBP. This effect was even more pronounced at a low ATP/ADP ratio.
Fig. 5. Relative ATPase activity of SBP from the wild-type and βV319T cells. Measurements were carried out as indicated in the legend to Fig. 3 for the βF139Y mutant.
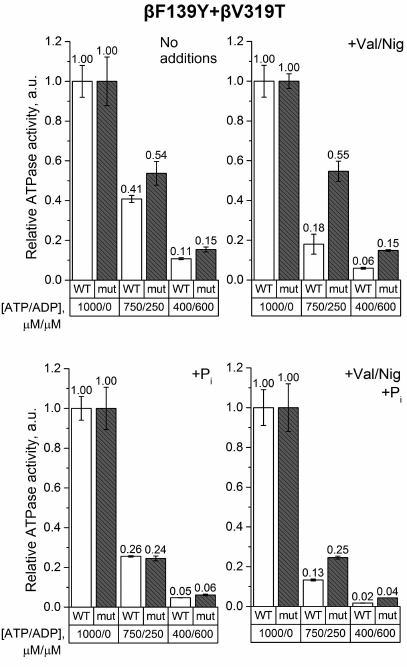
Finally, the experiments on SBP from the βF139Y+βV319T mutant revealed that the ATP/ADP ratio influenced the ATPase activity of the wild-type and the mutant SBP in a similar way regardless of phosphate addition (Fig. 6). However, if pmf was dissipated by uncouplers, the ATPase activity of the wild-type SBP was more sensitive to a decrease in the ATP/ADP ratio than the activity of the mutant SBP.
Fig. 6. Relative ATPase activity of SBP from the wild-type and βF139Y+βV319T cells. Measurements were carried out as indicated in the legend to Fig. 3 for the βF139Y mutant.
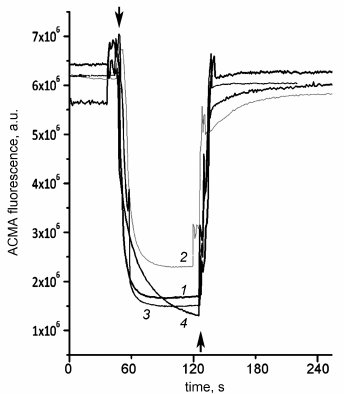
Aside from the effects on the ATPase activity, the studied mutations might influence the coupling between ATP hydrolysis and proton transport. Although addition of the uncoupler stimulated ATPase activity of SBP from the wild-type cells and all single mutants (thus indicating that the enzymes were coupled), we also measured the ATP-dependent proton transport directly. As shown in Fig. 7, ATP addition led to the ACMA fluorescence quenching caused by the formation of pH gradient on the membrane both in the wild-type SBP and all mutant SBP. Addition of the uncoupler led to the gradient dissipation and fluorescence increase to the initial level.
Fig. 7. ATP-dependent proton transport by SBP. pH gradient generation on SBP membranes in response to ATP addition to 500 μM was registered from ACMA fluorescence quenching as described in “Materials and Methods”. Top arrows, ATP addition; bottom arrow, uncoupler additions (valinomycin+nigericin to 500 nM each). 1) Wild-type SBP; 2) βF139Y; 3) βF189L; 4) βV319T.
DISCUSSION
ADP-inhibition in E. coli FOF1 seems to be different from that in other ATP synthases studied. It is relatively weak [12] and rather enhanced than suppressed by Pi [11, 15]. ATP synthases from other organisms have stronger ADP-inhibition which is attenuated by Pi [6, 9, 13, 14]. Previously, we have noticed thet β249 residue in E. coli FOF1 is leucine, while enzymes with stronger ADP-inhibition have a glutamine residue in the corresponding position. The βL249Q substitution in E. coli FOF1 increased ADP-inhibition and reversed the effect of Pi [17].
Our bioinformatic analysis of β subunit sequences from prokaryotic ATP synthases revealed that amino acids in the positions corresponding to β139, β158, β189, β249, and β319 in E. coli FOF1 differ between enzymes from beta- and gammaproteobacteria (including E. coli) and enzymes from other eubacteria, chloroplasts, and mitochondria (Fig. 1).
We suggested that, as in the case of β249, the type of amino acid residue in these positions might influence ADP-inhibition and its modulation by phosphate. To test this hypothesis, amino acid residues in the corresponding positions were replaced by residues typical for other eubacterial, mitochondrial, and chloroplast enzymes.
Our hypothesis was not confirmed in the case of β158 residue: no significant difference between the βN158L mutant and the wild-type E. coli FOF1 was observed under all the conditions tested. Other mutations also had no significant effect on the ATPase activity of E. coli FOF1 at 1 mM ATP (Fig. 2). A slight decrease in the activity of βF139Y and βV319T SBP may be explained by either reduced FOF1 expression in mutant strains or minor negative effect of these mutations on the enzyme catalytic properties. We also found that the ATPase activity of the SBP from the double mutant βF139Y+βV319T was not stimulated by the uncouplers in contrast to the wild-type SBP. It seems likely that passive proton conductivity of SBP membranes of this strain was higher than that of the other SBP samples, and the ATPase activity was not limited by the pmf back-pressure even in the absence of uncouplers.
The effect of the studied mutations on the ATPase activity of FOF1 was revealed when ADP was added to the medium. We measured the ATPase activity in the ATP/ADP mixtures (750/250 μM and 400/600 μM) to model ATP depletion in a living cell, when the total concentration of adenine nucleotides remains constant but the ATP/ADP ratio decreases. Under such conditions, the βF139Y mutation reduced the inhibitory effect of ADP only when Pi was not added (Fig. 3). It is possible that the type of amino acid residue in this position affects the stability of the ADP-inhibited state but does not influence the ability of Pi to modulate the probability of enzyme transition from the active to the ADP-inhibited state. The βF189L substitution, on the contrary, significantly influenced the effect of the ATP/ADP ratio on the ATPase activity in the presence of 3 mM Pi (Fig. 4). In the wild-type SBP in the presence of Pi, a decrease in the ATP/ADP ratio affected the ATPase activity twice as strong as in the mutant sample. Noteworthy, a similar effect was observed in the uncoupled SBP even in the absence of Pi. All effects of the βF189L mutation described above were even more pronounced in SBP from the βV319T mutant (Fig. 5). Possibly, the reduced ATPase activity of this mutant (Fig. 2) could be a result of enhanced ADP-inhibition. Taken together, these results suggest that βF189L and βV319T mutations might, to some extent, prevent the enhancement of ADP-inhibition by Pi specific for E. coli FOF1. Apparently, amino acid residues in these positions affect the pmf-dependent Pi modulation of the enzyme transition from the active to the ADP-inhibited state.
Introduction of the βV319T mutation together with βF139Y had little effect on ATP hydrolysis by SBP in the absence of Pi. The ATPase activity of the double mutant was similar to that of the single mutants. However, in the presence of 3 mM Pi, the effect of βF139Y substitution “dominated”, while at the reduced ATP/ADP ratio, the activity of the double mutant was not higher than that of the wild-type samples, in contrast to the situation with the single βV319T mutant.
To sum up, we found four amino acid positions in the β subunit containing conserved residues that differ between FOF1 enzymes of beta- and gammaproteobacteria (including E. coli) and enzymes from other eubacteria, chloroplasts, and mitochondria. Three of the four positions seem to be involved in ADP-inhibition of ATP synthase. It is possible that the residues in these positions (β139, β189, and β319) together with β249 determine the degree of FOF1 ADP-inhibition and the pattern of its modulation by phosphate. It is tempting to speculate that weak ADP-inhibition enhanced by Pi might be a common feature of ATP synthases of beta- and gammaproteobacteria. Further experiments are necessary to clarify this issue. It should be noted that such studies may be useful both for basic and applied research, since several dangerous pathogens belong to this class of bacteria (Fig. 1). It might be possible to develop low-molecular-weight compounds that would selectively affect ADP-inhibition and suppress the activity of ATP synthase of pathogenic gammaproteobacteria without affecting the eukaryotic mitochondrial enzyme. For the proton-translocating complex FO, this approach already proved successful and resulted in the development of a new antibiotic drug that efficiently and selectively inhibits mycobacterial ATP synthase [27, 28].
Acknowledgements
Authors thank Alexey Eliseev for his assistance in experiments with the βN158L mutant.
Funding
This work was supported by the Russian Science Foundation (project no. 14‐14‐00128; Molecular mechanisms of energy transformations in bacterial oxidative phosphorylation).
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
1.Sobti, M., Smits, C., Wong, A. S., Ishmukhametov,
R., Stock, D., Sandin, S., and Stewart, A. G. (2016) Cryo-EM structures
of the autoinhibited E. coli ATP synthase in three rotational
states, Elife, 5, e21598.
2.Stewart, A. G., Laming, E. M., Sobti, M., and
Stock, D. (2014) Rotary ATPases – dynamic molecular machines,
Curr. Opin. Struct. Biol., 25, 40-48.
3.Watanabe, R. (2013) Rotary catalysis of
FOF1-ATP synthase, Biophysics, 9,
51-56.
4.Junge, W., and Nelson, N. (2015) ATP synthase,
Annu. Rev. Biochem., 84, 631-657.
5.Feniouk, B. A., and Yoshida, M. (2008) Regulatory
mechanisms of proton-translocating FOF1-ATP
synthase, Results Probl. Cell Differ., 45, 279-308.
6.Carmeli, C., and Lifshitz, Y. (1972) Effects of
Pi and ADP on ATPase activity in chloroplasts, Biochim.
Biophys. Acta, 267, 86-95.
7.Minkov, I. B., Fitin, A. F., Vasilyeva, E. A., and
Vinogradov, A. D. (1979) Mg2+-induced ADP-dependent
inhibition of the ATPase activity of beef heart mitochondrial coupling
factor F1, Biochem. Biophys. Res. Commun., 89,
1300-1306.
8.Yoshida, M., and Allison, W. S. (1983) Modulation
by ADP and Mg2+ of the inactivation of the
F1-ATPase from the thermophilic bacterium, PS3, with
dicyclohexylcarbodiimide, J. Biol. Chem., 258,
14407-14412.
9.Turina, P., Rumberg, B., Melandri, B. A., and
Graber, P. (1992) Activation of the H+-ATP synthase in the
photosynthetic bacterium Rhodobacter capsulatus, J. Biol.
Chem., 267, 11057-11063.
10.Zharova, T. V., and Vinogradov, A. D. (2004)
Energy-dependent transformation of FOF1-ATPase in
Paracoccus denitrificans plasma membranes, J. Biol.
Chem., 279, 12319-12324.
11.Fischer, S., Graber, P., and Turina, P. (2000)
The activity of the ATP synthase from Escherichia coli is
regulated by the transmembrane proton motive force, J. Biol.
Chem., 275, 30157-30162.
12.Lapashina, A. S., and Feniouk, B. A. (2018)
ADP-inhibition of H+- FOF1-ATP
synthase, Biochemistry (Moscow), 10, 1141-1160.
13.Zharova, T. V., and Vinogradov, A. D. (2006)
Energy-linked binding of Pi is required for continuous
steady-state proton-translocating ATP hydrolysis catalyzed by
FOF1 ATP synthase, Biochemistry,
45, 14552-14558.
14.Feniouk, B. A., Suzuki, T., and Yoshida, M.
(2007) Regulatory interplay between proton motive force, ADP,
phosphate, and subunit ε in bacterial ATP synthase, J. Biol.
Chem., 282, 764-772.
15.D’Alessandro, M., Turina, P., and Melandri,
B. A. (2008) Intrinsic uncoupling in the ATP synthase of Escherichia
coli, Biochim. Biophys. Acta, 1777, 1518-1527.
16.Feniouk, B. A., Wakabayashi, C., Suzuki, T., and
Yoshida, M. (2012) A point mutation, betaGln259Leu, relieves MgADP
inhibition in Bacillus PS3 ATP synthase, Biochim. Biophys.
Acta – Bioenergetics, 1817, S13.
17.Lapashina, A. S., Prikhodko, A. S., Shugaeva, T.
E., and Feniouk, B. A. (2018) Residue 249 in subunit beta regulates ADP
inhibition and its phosphate modulation in Escherichia coli ATP
synthase, Biochim. Biophys. Acta – Bioenergetics; doi:
10.1016/j.bbabio.2018.12.003 [Epub ahead of print].
18.Galperin, M. Y., Makarova, K. S., Wolf, Y. I.,
and Koonin, E. V. (2015) Expanded microbial genome coverage and
improved protein family annotation in the COG database, Nucleic
Acids Res., 43, D261-D269.
19.Dibrova, D. V., Konovalov, K. A., Perekhvatov, V.
V., Skulachev, K. V., and Mulkidjanian, A. Y. (2017) COGcollator: a web
server for analysis of distant relationships between homologous protein
families, Biol. Direct., 12, 29.
20.Finn, R. D., Clements, J., and Eddy, S. R. (2011)
HMMER web server: interactive sequence similarity searching, Nucleic
Acids Res., 39, W29-W37.
21.Edgar, R. C. (2004) MUSCLE: multiple sequence
alignment with high accuracy and high throughput, Nucleic Acids
Res., 32, 1792-1797.
22.Waterhouse, A. M., Procter, J. B., Martin, D. M.
A., Clamp, M., and Barton, G. J. (2009) Jalview version 2 – a
multiple sequence alignment editor and analysis workbench,
Bioinformatics, 25, 1189-1191.
23.Ishmukhametov, R. R., Galkin, M. A., and Vik, S.
B. (2005) Ultrafast purification and reconstitution of His-tagged
cysteine-less Escherichia coli F1FO ATP
synthase, Biochim. Biophys. Acta, 1706, 110-116.
24.Nishimura, M., Ito, T., and Chance, B. (1962)
Studies on bacterial photophosphorylation. III. A sensitive and rapid
method of determination of photophosphorylation, Biochim. Biophys.
Acta, 59, 177-182.
25.Casadio, R., and Melandri, B. A. (1985)
Calibration of the response of 9-amino acridine fluorescence to
transmembrane pH differences in bacterial chromatophores, Arch.
Biochem. Biophys., 238, 219-228.
26.Dibrova, D. V., Galperin, M. Y., and
Mulkidjanian, A. Y. (2010) Characterization of the N-ATPase, a
distinct, laterally transferred Na+-translocating form of
the bacterial F-type membrane ATPase, Bioinformatics, 26,
1473-1476.
27.Hards, K., Robson, J. R., Berney, M., Shaw, L.,
Bald, D., Koul, A., Andries, K., and Cook, G. M. (2015) Bactericidal
mode of action of bedaquiline, J. Antimicrob. Chemother.,
70, 2028-2037.
28.Koul, A., Dendouga, N., Vergauwen, K.,
Molenberghs, B., Vranckx, L., Willebrords, R., Ristic, Z., Lill, H.,
Dorange, I., Guillemont, J., Bald, D., and Andries, K. (2007)
Diarylquinolines target subunit c of mycobacterial ATP synthase,
Nat. Chem. Biol., 3, 323-324.