Identification and Characterization of MicroRNAs in Skin of Chinese Giant Salamander (Andrias davidianus) by the Deep Sequencing Approach
Yong Huang1,a,b* and Wang Bao Gong2
1College of Animal Science and Technology, Henan University of Science and Technology, 471023 Luoyang, China2Key Laboratory of Tropical and Subtropical Fishery Resource Application and Cultivation, Ministry of Agriculture, Pearl River Fisheries Research Institute, Chinese Academy of Fishery Sciences, Guangzhou, China
* To whom correspondence should be addressed.
Received November 9, 2017; Revision received January 14, 2018
MicroRNAs (miRNA) play a pivotal role in regulating a broad range of biological processes, acting by cleaving mRNAs or by translational repression. However, the miRNAs from skin of Andrias davidianus have not been reported. In this study, a small-RNA cDNA library was constructed and sequenced from skin of A. davidianus. A total of 513 conserved miRNAs belonging to 174 families were identified. The remaining 108 miRNAs we identified were novel and likely to be skin tissue-specific but were expressed at low levels. The presence of randomly selected 15 miRNAs identified and their expression in eight different tissues from A. davidianus were validated by stem-loop qRT-PCR. For better understanding the functions of miRNAs, 129,791 predicated target genes were analyzed by GO and their pathways illustrated by KEGG pathway analyses. The results show that these identified miRNAs from A. davidianus skin are involved in a broad range of physiological functions including metabolism, growth, development, and immune responses. This study exhaustively identifies miRNAs and their target genes, which will ultimately pave the way for understanding their role in skin of A. davidianus and other amphibians. Further studies are necessary to better understand miRNA-mediated gene regulation.
KEY WORDS: Andrias davidianus, microRNA, skin, deep sequencing, target geneDOI: 10.1134/S0006297918060147
Abbreviations: GO analysis, gene ontology enrichment analysis; KEGG analysis, Kyoto Encyclopedia of Genes and Genomes pathway analysis; miRNA, microRNA.
MicroRNAs (miRNAs) are small noncoding transcripts of ~22 nucleotides
(nt) in length that can regulate expression at the posttranscriptional
level, which leads to the inhibition of translation or the degradation
of mRNAs [1, 2]. Mature miRNA
genes are transcribed from pri-miRNAs that can form a fold-back,
hairpin structure. In a sequential process, the pre-miRNAs are exported
into the cytoplasm and cleaved by RNase III Dicer and form a
double-stranded RNA [3, 4]. One
of the duplex strands of the mature miRNAs can be incorporated into the
RISCs; thereafter, they bind to the 3′ UTR of the target mRNAs to
regulate expression of target mRNAs [5]. An
increasing number of studies have demonstrated that miRNAs play
important regulatory roles in numerous biological processes, including
organ development, cell differentiation, proliferation, apoptosis,
homeostasis, metabolism, tumorigenesis, bacterial and viral infections,
immunological regulation, etc. in plants, animals, and even viruses [6-10].
The Chinese giant salamander (Andrias davidianus) belonging to the family Cryptobranchidae Fitzinger 1826 is the largest amphibian in the world [11]. It has a fair-sounding Chinese name, “babyfish (wawa-yu)”, and it mainly inhabits mountain brooks or interior water areas in South-Western and Eastern China. Andrias davidianus is renowned as a living fossil, since it has been in existence for more than 350 million years, representing the transitional type of animal from aquatic to terrestrial life [12, 13]. So, A. davidianus is also considered to be a valuable model of scientific value, medical value, edible value, view value, and so on [14]. However, due to habitat loss, climate change, environment pollution, overfishing, and disease (such as bacterial, fungal, and viral infections), their populations have dramatically declined, making it an endangered species [15, 16]. It is now estimated to be critically endangered by the International Union for Conservation of Nature and Nature Resources and is a State Special Protected Animal (category II) under Chinese Conservation Law, and in Appendix I of the Convention on International Trade in Endangered Species of Wild Fauna and Flora.
In recent years, with the rapid development of artificial cultivation, more than two million A. davidianus are bred annually, which become an important commercial aquaculture species in China. However, with the increase of intensive culture of this species, emerging infectious diseases have been increasing and have caused major impacts to A. davidianus industry. The skin of A. davidianus is a very important immune organ and is considered to be the first line of defense against external pathogenic microbes or predators, belonging to the nonspecific immune defense system [17, 18]. Therefore, understanding the mechanisms of the immune systems of A. davidianus in response to bacterial and viral infection is essential for managing disease outbreaks and may contribute to the sustained development of A. davidianus culture. So, identification, expression analysis, and target gene predication of the miRNAs in the skin should facilitate our understanding of the involvement of miRNA in immune system functions. There are reports concerning the miRNAs of A. davidianus [14, 19, 20]. In these previous reports, researchers identified miRNAs from the muscle, spleen, liver, testis, ovary, and mixed tissues of A. davidianus, but no study on miRNAs from skin of A. davidianus has been reported. Therefore, in the present study, we constructed a small-RNA cDNA library from A. davidianus skin. Through deep sequencing of the small RNA library and subsequent bioinformatics analysis, miRNAs in skin of A. davidianus have belen identified.
MATERIALS AND METHODS
Animal samples collection. Healthy A. davidianus individuals were obtained from a farm located in Luan Chuan (Henan Province, China). Skin tissue was collected from a five-year-old A. davidianus salamander immediately after dissection and washed in sterile PBS. A sample for RNA extraction was snap-frozen in liquid nitrogen and stored at −80°C before use. The present study involving animals was conducted according to the Regulations for the Administration of Affairs Concerning Experimental Animals (Ministry of Science and Technology, China, revised in June 2004) and approved by the Institutional Animal Care and Use Committee in the College of Animal Science and Technology, Henan University of Science and Technology, Henan, China.
Small RNA library construction and deep sequencing. Total RNA was isolated from the above-described sample using an RNAiso Plus kit (Takara, China) according to the manufacturer’s instructions. The integrity of the total RNA was analyzed using an Agilent 2100 Bioanalyzer system and RNA6000 Nano LabChip Kit (Agilent, USA) with RNA integrity value (RIN) >8.0. Then a small RNA library was constructed respectively using TruSeq Small RNA Sample Prep Kits (Illumina, USA). Briefly, about total 5 μg RNA was resolved on denaturing PAGE gels, and then fractions 18 to 26 nt in size were collected. Two adaptors were sequentially ligated to the 5′ and 3′ ends of purified small RNAs. The ligation products were reverse-transcribed using SuperScript Reverse Transcriptase II (Life Technologies, USA) and amplified with 15 PCR cycles. Then, 8% (w/v) PAGE was used to purify the amplification products. Finally, the library was used for clustering and sequencing using an Illumina Genome Analyzer II (Hangzhou, China). After image analysis, sequence quality evaluation and summarization of data production were performed with the Illumina Pipeline. The raw small RNA transcriptome sequencing data from the skin of A. davidianus was submitted to the NCBI Sequence Read Archive under accession number SRP110836.
Sequence analysis and miRNA identification. The raw sequences were filtered to remove the adapter sequences and contaminated reads. The retained mappable reads were further analyzed following the work flow using ATCG101-miRsoftware (version 4.2; LC Sciences). The unique small RNA sequences were firstly mapped to known pre-miRNAs deposited in the miRBase database (release 21.0) (http://www.mirbase.org/) using the mapping program Bowtie (http://bowtie-bio.sourceforge.net/index.shtml). After that, the reads matched to rRNA, tRNA, snRNA, snoRNA and other noncoding RNA sequences deposited in Rfam (http://rfam.janelia.org) and the repeat-RepBase (http://www.girinst.org/repbase) were deducted. The remaining reads were mapped onto the reference transcriptomic dataset of A. davidianus [21]. Sequences without mismatch were retained for novel miRNA prediction. Finally, candidate novel miRNAs were identified by prediction of the secondary structures with the Mfold program (http://mfold.rna.albany.edu/?q=mfold/RNA-Folding-Form). The criteria for secondary structure prediction were: (i) number of nucleotides in one bulge in stem, ≤12; (ii) number of base pairs in the stem region of the predicted hairpin, ≥16; (iii) cutoff of free energy (kcal/mol), ≤15; (iv) length of hairpin (up and down stems + terminal loop), ≥50; (v) length of hairpin loop, ≤20; (vi) number of nucleotides in one bulge in mature region, ≤8; (vii) number of biased errors in one bulge in mature region, ≤4; (viii) number of biased bulges in mature region, ≤2; (ix) number of errors in mature region, ≤7; (x) number of base pairs in the mature region of the predicted hairpin, ≥12. The naming system of miRNAs identified in this study is as follows: the miRNA name is composed of the first known miR name in a cluster, an underscore, and a matching annotation: L – n means the miRNA_seq (detected) is n base less than known rep_miRSeq in the left side; R – n means the miRNA_seq (detected) is n base less than known rep_miRSeq in the right side; L + n means the miRNA_seq (detected) is n base more than known rep_miRSeq in the left side; R + n means the miRNA_seq (detected) is n base more than known rep_miRSeq in the right side; 2ss5TC13TA means 2 substitutions (ss), which are T to C at position 5 and T to A at position 13. If there is no matching annotation, the miRNA_seq (detected) is exactly the same as known rep_miRSEq. Novel identified 5′/3′ sequences are annotated as p5/p3 to distinguish from the reported 5′/3′ sequences. The expression of the miRNA is normalized to get the expression of transcript per million. The normalization formula is:
Normalized expression = actual miRNA count/total count of clean reads × 1,000,000.
Verification and expression analyses by stem-loop qRT-PCR. For qPCR studies of validated miRNAs, the total RNAs from A. davidianus fresh tissues were reverse transcribed to cDNAs using RT primers. Primers for the stem-loop RT were designed using methods described previously [22-24]. The qPCR reactions were carried out in a Bio-RadiCycler real-time PCR machine (Bio-Rad, USA) with the following cycling conditions: 95°C for 15 min followed by 40 cycles of 94°C for 15 s, 55°C for 30 s, and 70°C for 30 s. The threshold for detection was set as a cycle threshold (Ct) <35. All reactions were run in triplicate. The quantification of each miRNA relative to 18S rRNA gene was calculated using the 2−ΔΔCt method. Differences in miRNA expression were determined by one-way analysis of variance (ANOVA) using R software. The primers for the validated miRNAs are listed in Table S1 (see Supplement to this paper on the site of the journal http://protein.bio.msu.ru/biokhimiya).
Prediction of miRNA targets, GO enrichment, and KEGG pathway analyses. Two miRNA target prediction algorithms, miRanda (http://www.microrna.org/microrna/home.do) and TargetScan (http://www.targetscan.org/), were used to identify the target genes of miRNAs from the transcriptome of A. davidianus in our previous work by Huang et al. TargetScan was used to search for miRNA seed matches (nt 2-8 from the 5′ end of miRNA) in the sequences. miRanda was used to match the entire miRNA sequences. The miRanda and TargetScan parameters were set as free energy <–20 kcal/mol and score >50. Finally, the results predicted by the two algorithms were combined, and the overlaps were calculated. Then, enrichment analysis of the predicted target genes was conducted with the GO (http://www.geneontology.org/) and KEGG pathways (http://www.genome.jp/kegg/).
RESULTS AND DISCUSSION
Overview of deep sequencing data. Deep sequencing enables sequencing of the entire small RNA transcriptome in a sample, providing the complete set of miRNAs transcribed at a given time. In the present study, a total of 9,835,446 raw reads were obtained from the skin of A. davidianus using an Illumina HiSeq2500 sequencing platform. After removing the reads with a 3ADT&length filter, junk reads, RFam (rRNA, tRNA, snRNA, snoRNA, and other Rfam RNAs), and RepBase sequences, there were 1,689,352 mappable (clean) reads obtained for this library (table and Fig. 1). Furthermore, the length distribution based on both total abundances and unique reads were very similar in this library, ranging from 18 to 26 nt after the filtering process. The length of these reads was not evenly distributed. Most clean reads had lengths of 21-23 nt, and the most abundant size class in the small RNA sequence distribution was 22 nt followed by 21 and 23 nt (Fig. 2). Small RNAs were commonly ~20 nt in size, which is consistent with the typical size of miRNA from Dicer digestion products in animals including cattle [25], fish [26], silkworm [27], goat [28], African clawed frog [29], and chicken [30]. Our results have also a similar trend. This phenomenon suggests that the length distribution may be similar in closely related animal species.
Analysis of small RNA sequences from skin of A. davidianus
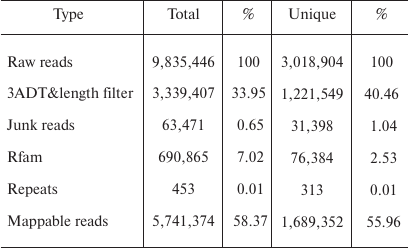
Fig. 1. Small RNA reads were blasted against the Rfam database noncoding RNA to annotate rRNA, tRNA, snoRNA, snRNA, and other RNAs. Left, total reads; right, unique reads.
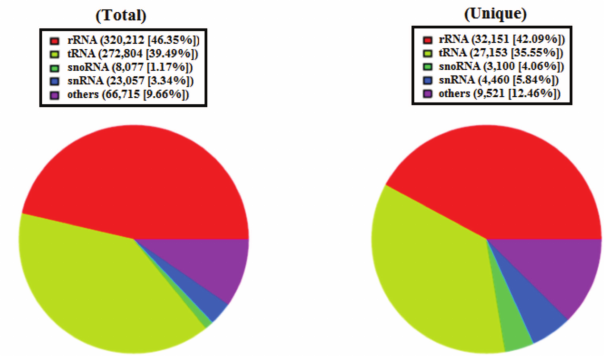
Fig. 2. Length distribution analysis of sequenced small RNAs in skin of A. davidianus. The X axis represents the size distribution and frequency of miRNAs in both types of sequences. The Y axis represents the small RNA abundance in both types.
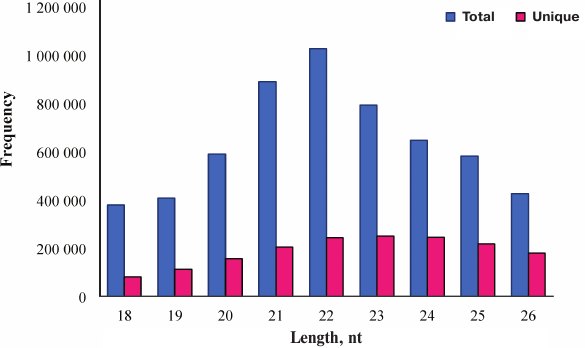
Identification of conserved miRNAs in skin of A. davidianus. Conserved miRNAs are found in many animal species and have important functions in animal development and physiological processes. To identify conserved miRNAs in skin of A. davidianus, clean reads of >17-nt- and <27-nt-length from the small RNA library was subjected to homology search against metazoan mature miRNAs in the miRBase database (Release 21), in which miRNAs of A. davidianus had not been identified. Following a set of strict filtering criteria and further sequence analysis, sequence analyses used the Blastn program (E value ≤e–4) with mature miRNAs and allowed no more than two mismatches between sequences. A total of 513 conserved miRNAs were identified from skin of A. davidianus (Table S2, see Supplement). Most identified miRNAs are conserved across 53 metazoan species (Fig. 3 and Table S3, see Supplement). These identified miRNAs were found belonging to 174 families with an average of approximately 2.95 miRNA members per family (Table S4, see Supplement). Among all the miRNA families, let-7 was the largest family, with 24 members; mir-10, mir-15, mir-17, mir-154, mir-30, mir-130, mir-221, mir-8 and mir-467 had 17, 15, 14, 13, 11, 10, 10, 9, and 9 members, respectively. The remaining miRNA families have 1-8 members. The deep-sequencing approach allows us to determine the relative abundance of various miRNAs by calculating read frequencies. In this study, the most highly expressed miRNA in skin of A. davidianus from let-7 family was xtr-let-7e_1ss17TA (122,055 reads), followed by hsa-miR-21-5p from mir-21 family (90,334 reads) and aca-miR-99b-5p_R-1 from mir-10 family (87,974 reads). On the contrary, some miRNAs such as aca-miR-129b-5p_R+2, fru-miR-122_R+2 and gga-miR-122-5p_R+1_1ss23TA showed less than two reads. The different reads of miRNAs often reflect the different roles in a particular tissue or development stage as well as corresponding to biological mechanisms.
Let-7 is a highly significant miRNA family that was first discovered in Caenorhabditis elegans, and plays a key role in regulating late developmental events [31]. Highly expressed miRNAs are likely to have essential and broad regulatory functions. Let-7 regulates cell proliferation and differentiation and is also highly conserved across animal species [32]. For example, there were reports that demonstrated the importance of let-7 in different biological functions of brain, liver, and gonads [33-35]. Current results are similar to those reported in a previous study of miRNAs in different A. davidianus tissues [20], in which let-7 showed high expression in liver, spleen, and muscle, respectively. These findings suggest that let-7 gene families were important regulators of fundamental biological processes [36, 37]. Previous studies in mammals have also indicated that the let-7 gene family can regulate the expression of major cytokine inducible proteins in response to microbial challenge [38, 39]. Swaminathan et al. confirmed that the let-7 family can regulate the expression of IL-10 at the posttranscriptional level in CD4+ T cells infected with HIV-1 [40]. A recent study showed that miR-21 could inhibit cell proliferation [41], suggesting that it might be a negative regulatory factor for animal growth. In another report, miR-21 is also involved in immune response in teleost fish, and it has also been reported as miRNAs associated with immune response in higher vertebrates [42-44].
Fig. 3. Conservation profile of identified miRNAs in animals. The X axis represents conserved miRNAs numbers. The Y axis represents queried species.
Identification of novel miRNAs in skin of A. davidianus. One of the greatest advantages of deep sequencing can also be employed to detect novel miRNAs with low expression in the small RNA transcriptome [45]. To predict the novel miRNAs from the skin of A. davidianus, sequencing reads not aligning to the miRBase were further analyzed to discover novel miRNAs using the Mfold software. Novel miRNAs are identified based on alignment, secondary structure, and free energy and location on the precursor arm. Since the genome sequence of A. davidianus is not available in public databases, the A. davidianus mRNA transcriptome was used to identify novel miRNAs and their precursors. A total of 108 novel miRNA gene candidates were identified (Table S5, see Supplement). The lengths of novel miRNAs varied from 18 to 26 nt, which is also typical of Dicer digestion products. Among these, the predicted novel miRNAs displayed an MFE for miRNA precursor hairpin structures ranging from –110.9 to –18.2 kcal/mol, with an average of –48.3 kcal/mol, which was higher than that in tRNA (–227.5 kcal/mol) and rRNA (–233 kcal/mol) [46]. Notably, these novel miRNAs were either –5p or –3p arms of proposed precursors, and the lengths of pre-miRNA sequences ranged between 50 and 145 nt with an average length of 88 nt. In addition, the number of reads for most predicted novel miRNAs was low, and much less than that for the conserved miRNAs. Of these, 18 were found to be weakly expressed, as indicated by a normalized sequencing read of less than 1 in this library, apart from PC-5p-4717_453 that was had relative high reads of more than 1000 (1081 reads). It has been reported that miRNAs may be expressed at very low levels and may be present only in particular cell types and/or under particular circumstances [45, 47].
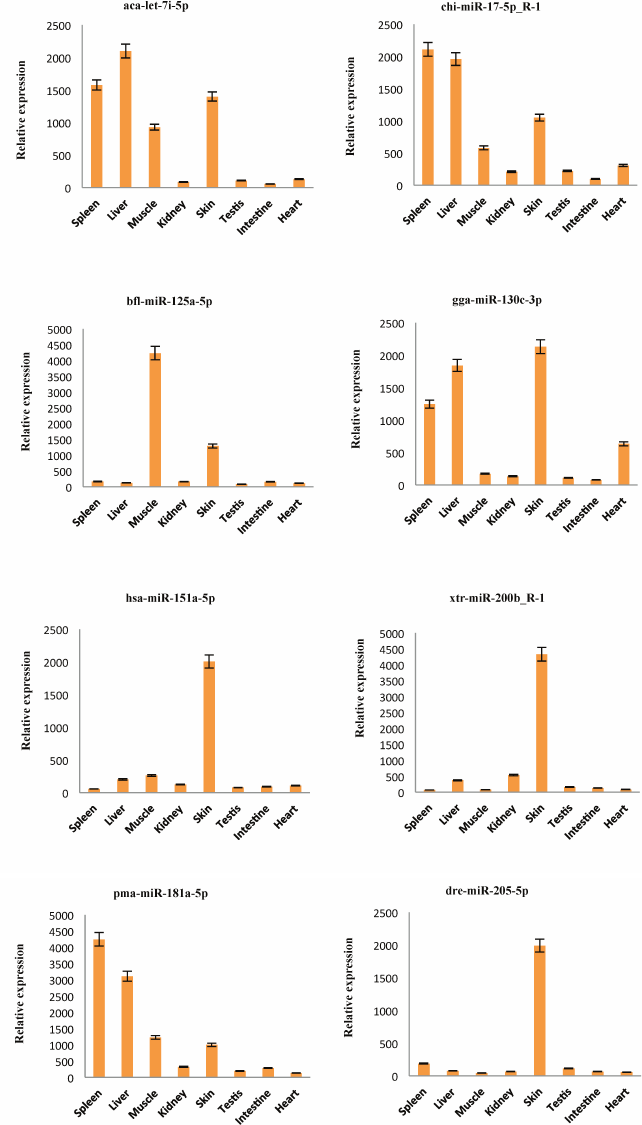
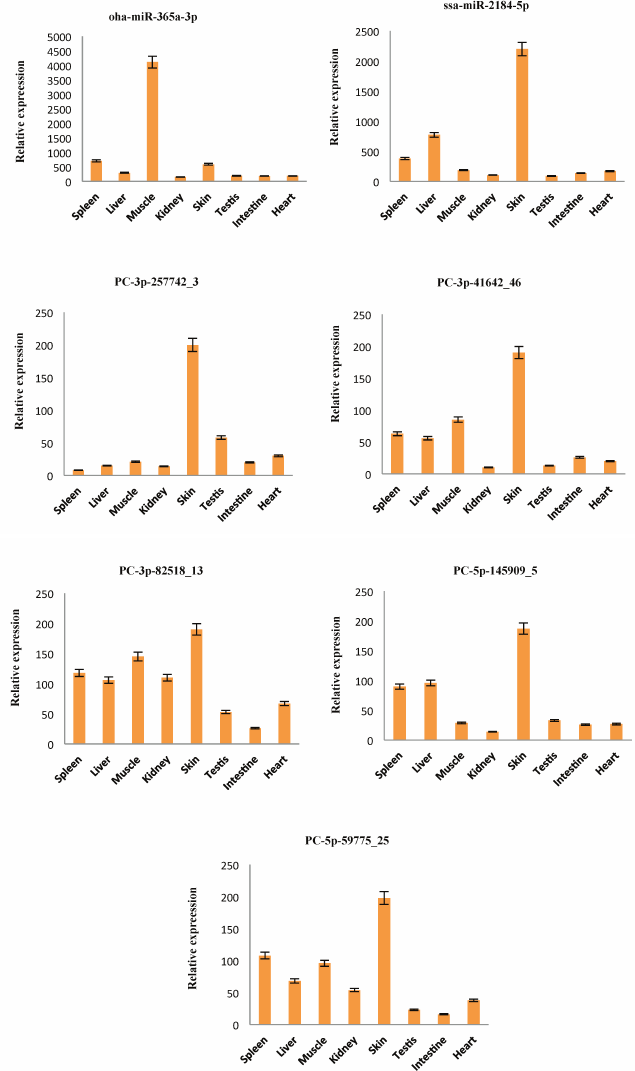
Detection of the expression of miRNAs using stem-loop qRT-PCR. To confirm the expression pattern of the identified miRNAs from skin of A. davidianus, 10 conserved miRNAs (aca-let-7i-5p, chi-miR-17-5p_R-1, bfl-miR-125a-5p, gga-miR-130c-3p, hsa-miR-151a-5p, xtr-miR-200b_R-1, pma-miR-181a-5p, dre-miR-205-5p, oha-miR-365a-3p, and ssa-miR-2184-5p) with more than 1000 reads and five novel miRNAs (PC-3p-257742_3, PC-3p-41642_46, PC-3p-82518_13, PC-5p-145909_5, and PC-5p-59775_25) with more than 100 reads were randomly selected for stem-loop qRT-PCR analysis in eight different tissues of A. davidianus including spleen, liver, muscle, kidney, skin, testis, intestine, and heart. As illustrated in Fig. 4, 15 miRNAs were expressed in all tissues examined. Although five novel miRNAs were detected in relatively low read numbers with deep sequencing, and these could also be detected through stem-loop qRT-PCR. Aca-let-7i-5p was more abundantly expressed in liver, spleen, skin, and moderately in muscle and least expressed in intestine. Currently, let-7 was found to control glucose homeostasis and insulin sensitivity in liver of mice [34]. The result indicated that let-7 gene may play important regulation roles in liver of animals. Chi-miR-17-5p_R-1 was expressed abundantly in spleen, liver, and skin and weakly in kidney and intestine, whereas bfl-miR-125a-5p showed high expression in muscle, followed by skin, and was weakly expressed in other six tissues. The five miRNAs (gga-miR-130c-3p, hsa-miR-151a-5p, xtr-miR-200b_R-1, dre-miR-205-5p, and ssa-miR-2184-5p) were expressed predominantly in the skin, suggesting that these conserved miRNAs may play crucial roles for gene regulation in physiological activity of skin. Expression of pma-miR-181a-5p was abundant in spleen and liver, followed by muscle and skin, and it was weak in testis and heart. Oha-miR-365a-3p exhibited high expression in muscle and was weakly expressed in seven other tissues. The remaining five novel miRNAs (PC-3p-257742_3, PC-3p-41642_46, PC-3p-82518_13, PC-5p-145909_5, and PC-5p-59775_25) all exhibited preferential high expression in skin, indicating these miRNAs may play particular roles in skin tissue. These results showed that the expression pattern of these miRNAs varies dramatically between different tissues, some miRNAs exhibiting preferential expression in certain tissues and showing lower expressions in other tissues, whereas a few miRNAs were highly expressed in multiple tissues. Taken together, the miRNA expression pattern of stem-loop qRT-PCR was also consistent with deep-sequencing reads.
Fig. 4. Relative expression of 15 miRNAs in eight A. davidianus tissues using stem-loop qRT-PCR. The analysis was done for three biological samples with technical replicates. Error bars represent standard deviation from the mean. Significant difference at p < 0.05.
Prediction of target genes of miRNAs, GO annotation and KEGG pathway enrichment analysis. In total, 129,791 target sequences corresponded to 3589 individual mRNAs were identified for 513 putative conserved and 108 novel A. davidianus miRNAs, with an approximate average of 209 targets per miRNA (Table S6, see Supplement). More than 29.6% (129,791 out of 437,784) of the target genes could be annotated. Most of the miRNAs have >2 targets. The found potential targets were involved in diverse biological functions and relate to animal growth and development, metabolism, signal transduction, and so on. Meanwhile, many target genes were involved in immune processes. Immunity is a complex biological system in multicellular organisms to defend themselves from invading pathogens. The fine tuning of the innate immune response by miRNAs is a concept now supported by a rapidly growing body of evidence [48]. In the present study, A. davidianus miRNAs from skin are involved in targeting immune-related proteins; interferon-induced guanylate-binding protein 1-like, interferon-inducible GTPase 5-like, immunoglobulin (CD79A) binding protein 1, interferon-related developmental regulator 2-like, interferon-related developmental regulator 1, immunoglobulin-like domain containing receptor 1, interferon-induced transmembrane protein 5-like, interferon regulatory factor 2 binding protein-like, and many others (Table S7, see Supplement). Of them, interferon-induced guanylate-binding protein 1-like is targeted by 14 different miRNAs from let-7 family, but also by a novel miRNA (PC-5p-4717_453). These results suggest that these miRNAs may be involved in regulation of the immunity system of the skin of A. davidianus.
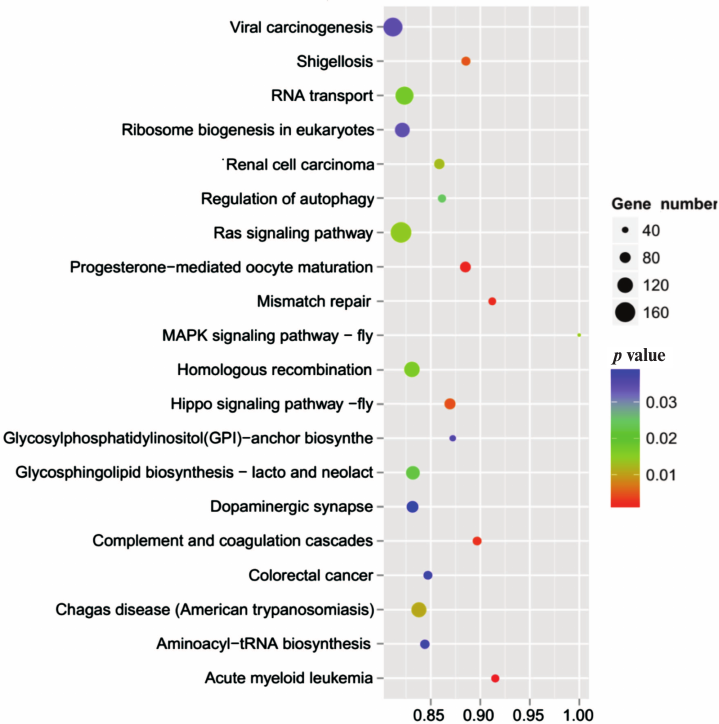
To further understand the physiological role of annotated targets of the conserved and novel miRNAs identified in this study, these mRNAs were subjected to GO analysis to evaluate their potential functions. This has shown that these target genes may be involved in 2781 molecular functions, 6897 biological processes, and 1070 cellular components (Fig. 5 and Table S8, see Supplement), indicating that these potential targets are associated with a broad range of biological functions. The most common target-related GO-terms were nucleus, integral component of membrane, and cytoplasm (in biological processes), nucleus, integral component of membrane, and cytoplasm (in cell components), and metal ion binding, zinc ion binding, and ATP binding (in molecular function). Many of the target genes were also involved in innate immune response, regulation of immune response, immune system development, adaptive immune response and humoral immune response, and so on. Biological interpretations of the miRNA target genes were further analyzed using the KEGG pathway database. These target genes were found to be enriching in a broad range of pathways (Table S9, see Supplement). Further, KEGG analysis revealed the potentially enriched functional groups and classified them into 20 top pathways (Fig. 6). The three most enriched pathways were involved the Viral carcinogenesis, Ras signaling pathway, and RNA transport, which may regulate the cellular immunity, growth, and development. In addition to Viral carcinogenesis pathways, some target genes were also involved in the other two immune-related pathways including Chagas disease and Acute myeloid leukemia. However, the validation of the existence of a relationship between miRNAs and predicted target mRNAs in the regulation of specific physiological processes during A. davidianus development needs further experimental evidence.
Fig. 5. Functional classification of miRNA target genes according to GO category.
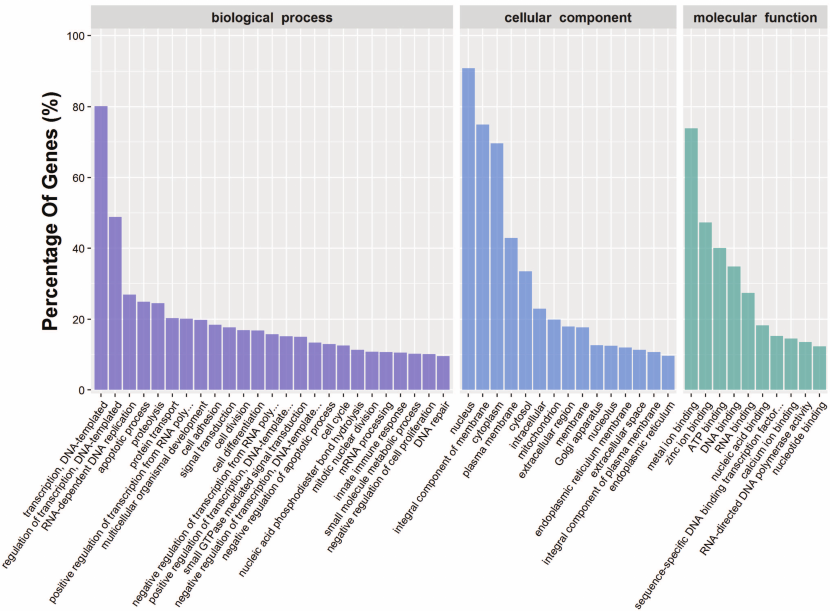
Fig. 6. KEGG pathway analysis of the target genes of miRNAs.
In the present study, we report miRNAs from skin of A. davidianus for the first time by employing deep sequencing. The expression of these small RNAs in A. davidianus skin provides evidence for their regulatory activities. Furthermore, we also analyzed potential functions of these identified miRNAs by GO enrichment analysis and KEGG pathway analysis of the predicted target genes. This study provides a starting point for future studies aimed at understanding the roles of miRNAs in major physiological process such as growth, development, and immunity. In summary, miRNAs from the A. davidianus skin repertoire provides a novel resource to advance genomic research in this species, and the findings also advance the understanding of amphibian biology.
Acknowledgments
This work was supported financially by the Natural Science Foundation of Henan Province of China (162300410069) and the Educational Commission of Henan Province of China (16A240002).
REFERENCES
1.Bartel, D. P. (2009) MicroRNAs: target recognition
and regulatory functions, Cell, 136, 215-233.
2.Chekulaeva, M., and Filipowicz, W. (2009)
Mechanisms of miRNA-mediated post-transcriptional regulation in animal
cells, Curr. Opin. Cell Biol., 21, 452-460.
3.Johanson, T. M., Lew, A. M., and Chong, M. M.
(2013) MicroRNA-independent roles of the RNase III enzymes Drosha and
Dicer, Open Biol., 3, 130144.
4.Gregory, R. I., Chendrimada, T. P., Cooch, N., and
Shiekhattar, R. (2005) Human RISC couples microRNA biogenesis and
posttranscriptional gene silencing, Cell, 123,
631-640.
5.Tang, G. (2005) siRNA and miRNA: an insight into
RISCs, Trends Biochem. Sci., 30, 106-114.
6.Shukla, G. C., Singh, J., and Barik, S. (2011)
MicroRNAs: processing, maturation, target recognition and regulatory
functions, Mol. Cell Pharmacol., 3, 83-92.
7.Huang, Y., Shen, X. J., Zou, Q., Wang, S. P., Tang,
S. M., and Zhang, G. Z. (2011) Biological functions of microRNAs: a
review, J. Physiol. Biochem., 67, 129-139.
8.Shi, C., Zhang, X., Li, X., Zhang, L., Li, L., Sun,
Z., Fu, X., Wu, J., Chang, Y., Li, W., Chen, Q., and Zhang, M. (2016)
Effects of microRNA-21 on the biological functions of T-cell acute
lymphoblastic lymphoma/leukemia, Oncol. Lett., 12,
4173-4180.
9.Wang, L., Li, G., Yao, Z. Q., Moorman, J. P., and
Ning, S. (2015) MicroRNA regulation of viral immunity, latency, and
carcinogenesis of selected tumor viruses and HIV, Rev. Med.
Virol., 25, 320-341.
10.Martin, R. C., Liu, P. P., Goloviznina, N. A.,
and Nonogaki, H. (2010) MicroRNA, seeds, and Darwin?: diverse function
of miRNA in seed biology and plant responses to stress, J. Exp.
Bot., 61, 2229-2234.
11.Murphy, R. W., Fu, J., Upton, D. E., de Lema, T.,
and Zhao, E. M. (2000) Genetic variability among endangered Chinese
giant salamanders, Andrias davidianus, Mol. Ecol.,
9, 1539-1547.
12.Gao, K. Q., and Shubin, N. H. (2003) Earliest
known crown-group salamanders, Nature, 422, 424-428.
13.Gao, K. Q., and Shubin, N. H. (2001) Late
Jurassic salamanders from Northern China, Nature, 410,
574-577.
14.Huang, Y., Yang, Y. B., Gao, X. C., Ren, H. T.,
and Sun, X. H. (2017) Identification and characterization of the
Chinese giant salamander (Andrias davidianus) miRNAs by deep
sequencing and predication of their targets, 3 Biotech.,
7, 235.
15.Pounds, J. A., Bustamante, M. R., Coloma, L. A.,
Consuegra, J. A., Fogden, M. P., Foster, P. N., La Marca, E., Masters,
K. L., Merino-Viteri, A., Puschendorf, R., Ron, S. R.,
Sa’nchez-Azofeifa, G. A., Still, C. J., and Young, B. E. (2006)
Widespread amphibian extinctions from epidemic disease driven by global
warming, Nature, 439, 161-167.
16.Fisher, M. C., Henk, D. A., Briggs, C. J.,
Brownstein, J. S., Madoff, L. C., McCraw, S. L., and Gurr, S. J. (2012)
Emerging fungal threats to animal, plant and ecosystem health,
Nature, 484, 186-194.
17.Simmaco, M., Mignogna, G., and Barra, D. (1998)
Antimicrobial peptides from amphibian skin: what do they tell us?
Biopolymers, 47, 435-450.
18.Li, F., Wang, L., Lan, Q., Yang, H., Li, Y., Liu,
X., and Yang, Z. (2015) RNA-Seq analysis and gene discovery of
Andrias davidianus using Illumina short read sequencing, PLoS
One, 10, e0123730.
19.Chen, R., Du, J., Ma, L., Wang, L. Q., Xie, S.
S., Yang, C. M., Lan, X. Y., Pan, C. Y., and Dong, W. Z. (2017)
Comparative microRNAome analysis of the testis and ovary of the Chinese
giant salamander, Reproduction, 154, 169-179.
20.Huang, Y., Ren, H. T., Xiong, J. L., Gao, X. C.,
and Sun, X. H. (2017) Identification and characterization of known and
novel microRNAs in three tissues of Chinese giant salamander base on
deep sequencing approach, Genomics, 109, 258-264.
21.Huang, Y., Gao, X. C., Xiong, J. L., Ren, H. T.,
and Sun, X. H. (2017) Sequencing and de novo transcriptome
assembly of the Chinese giant salamander (Andrias davidianus),
Genom. Data, 12, 109-110.
22.Hurley, J., Roberts, D., Bond, A., Keys, D., and
Chen, C. (2012) Stem-loop RT-qPCR for microRNA expression profiling,
Methods Mol. Biol., 822, 33-52.
23.Yang, L. H., Wang, S. L., Tang, L. L., Liu, B.,
Ye, W. L., Wang, L. L., Wang, Z. Y., Zhou, M. T., and Chen, B. C.
(2014) Universal stem-loop primer method for screening and
quantification of microRNA, PLoS One, 9,
e115293.
24.Huang, Y., Cheng, J. H., Luo, F. N., Pan, H.,
Sun, X. J., Diao, L. Y., and Qin, X. J. (2016) Genome-wide
identification and characterization of microRNA genes and their targets
in large yellow croaker (Larimichthys crocea), Gene,
576, 261-267.
25.Sun, J., Zhang, B., Lan, X., Zhang, C., Lei, C.,
and Chen, H. (2014) Comparative transcriptome analysis reveals
significant differences in microRNA expression and their target genes
between adipose and muscular tissues in cattle, PLoS One,
9, e102142.
26.Fu, Y., Shi, Z., Wu, M., Zhang, J., Jia, L., and
Chen, X. (2011) Identification and differential expression of microRNAs
during metamorphosis of the Japanese flounder (Paralichthys
olivaceus), PLoS One, 6, e22957.
27.Yu, X., Zhou, Q., Cai, Y., Luo, Q., Lin, H., Hu,
S., and Yu, J. (2009) A discovery of novel microRNAs in the silkworm
(Bombyx mori) genome, Genomics, 94, 438-444.
28.Ji, Z., Wang, G., Xie, Z., Zhang, C., and Wang,
J. (2012) Identification and characterization of microRNA in the dairy
goat (Capra hircus) mammary gland by Solexa deep-sequencing
technology, Mol. Biol. Rep., 39, 9361-9371.
29.Ambady, S., Wu, Z., and Dominko, T. (2012)
Identification of novel microRNAs in Xenopus laevis metaphase II
arrested eggs, Genesis, 50, 286-299.
30.Sun, G. R., Li, M., Li, G. X., Tian, Y. D., Han,
R. L., and Kang, X. T. (2012) Identification and abundance of miRNA in
chicken hypothalamus tissue determined by Solexa sequencing, Genet.
Mol. Res., 11, 4682-4694.
31.Reinhart, B. J., Slack, F. J., Basson, M.,
Pasquinelli, A. E., Bettinger, J. C., Rougvie, A. E., Horvitz, H. R.,
and Ruvkun, G. (2000) The 21-nucleotide let-7 RNA regulates
developmental timing in Caenorhabditis elegans, Nature,
403, 901-906.
32.Roush, S., and Slack, F. J. (2008) The let-7
family of microRNAs, Trends Cell Biol., 18, 505-516.
33.Lehmann, S. M., Kruger, C., Park, B., Derkow, K.,
Rosenberger, K., Baumgart, J., Trimbuch, T., Eom, G., Hinz, M., Kaul,
D., Habbel, P., Kaelin, R., Franzoni, E., Rybak, A., Nguyen, D., Veh,
R., Ninnemann, O., Peters, O., Nitsch, R., Heppner, F. L., Golenbock,
D., Schott, E., Ploegh, H. L., Wulczyn, F. G., and Lehnardt, S. (2012)
An unconventional role for miRNA: let-7 activates Toll-like receptor 7
and causes neurodegeneration, Nat. Neurosci., 15,
827-835.
34.Frost, R. J., and Olson, E. N. (2011) Control of
glucose homeostasis and insulin sensitivity by the let-7 family of
microRNAs, Proc. Natl. Acad. Sci. USA, 108,
21075-21080.
35.Toledano, H., D’Alterio, C., Czech, B.,
Levine, E., and Jones, D. L. (2012) The let-7-Imp axis regulates ageing
of the Drosophila testis stem-cell niche, Nature,
485, 605-610.
36.Zhu, H., Shyh-Chang, N., Segre, A. V., Shinoda,
G., Shah, S. P., Einhorn, W. S., Takeuchi, A., Engreitz, J. M., Hagan,
J. P., Kharas, M. G., Urbach, A., Thornton, J. E., Triboulet, R.,
Gregory, R. I., DIAGRAM Consortium, MAGIC Investigators, Altshuler, D.,
and Daley, G. Q. (2011) The Lin28/let-7 axis regulates glucose
metabolism, Cell, 147, 81-94.
37.Su, J. L., Chen, P. S., Johansson, G., and Kuo,
M. L. (2012) Function and regulation of let-7 family microRNAs,
MicroRNA, 1, 34-39.
38.Mondol, V., and Pasquinelli, A. E. (2012)
Let’s make it happen: the role of let-7 microRNA in development,
Curr. Top. Dev. Biol., 99, 1-30.
39.Wang, X., Cao, L., Wang, Y., Liu, N., and You, Y.
(2012) Regulation of let-7 and its target oncogenes (review), Oncol.
Lett., 3, 955-960.
40.Swaminathan, S., Suzuki, K., Seddiki, N., Kaplan,
W., Cowley, M. J., Hood, C. L., Clancy, J. L., Murray, D. D., Mendez,
C., Gelgor, L., Anderson, B., Roth, N., Cooper, D. A., and Kelleher, A.
D. (2012) Differential regulation of the let-7 family of microRNAs in
CD4+ T cells alters IL-10 expression, J. Immunol.,
188, 6238-6246.
41.Lin, L., Gan, H., Zhang, H., Tang, W., Sun, Y.,
Tang, X., Kong, D., Zhou, J., Wang, Y., and Zhu, Y. (2014) MicroRNA21
inhibits SMAD7 expression through a target sequence in the 3′
untranslated region and inhibits proliferation of renal tubular
epithelial cells, Mol. Med. Rep., 10, 707-712.
42.Forster, S. C., Tate, M. D., and Hertzog, P. J.
(2015) MicroRNA as type I interferon-regulated transcripts and
modulators of the innate immune response, Front. Immunol.,
6, 334.
43.Zhou, R., O’Hara, S. P., and Chen, X. M.
(2011) MicroRNA regulation of innate immune responses in epithelial
cells, Cell. Mol. Immunol., 8, 371-379.
44.Andreassen, R., and Hoyheim, B. (2017) miRNAs
associated with immune response in teleost fish, Dev. Comp.
Immunol., 75, 77-85.
45.Chen, X., Li, Q., Wang, J., Guo, X., Jiang, X.,
Ren, Z., Weng, C., Sun, G., Wang, X., Liu, Y., Ma, L., Chen, J. Y.,
Wang, J., Zen, K., Zhang, J., and Zhang, C. Y. (2009) Identification
and characterization of novel amphioxus microRNAs by Solexa sequencing,
Genome Biol., 10, R78.
46.Zhang, B. H., Pan, X. P., Cox, S. B., Cobb, G.
P., and Anderson, T. A. (2006) Evidence that miRNAs are different from
other RNAs, Cell. Mol. Life Sci., 63, 246-254.
47.Huang, L., Yin, Z. J., Feng, Y. F., Zhang, X. D.,
Wu, T., Ding, Y. Y., Ye, P. F., Fu, K., and Zhang, M. Q. (2016).
Identification and differential expression of microRNAs in the ovaries
of pigs (Sus scrofa) with high and low litter sizes, Anim.
Genet., 47, 543-551.
48.Gantier, M. P. (2010) New perspectives in
microRNA regulation of innate immunity, J. Interferon Cytokine
Res., 30, 283-289.
Supplementary Tables S1-S9 (Excel)