Construction of Artificial TNF-Binding Proteins Based on the 10th Human Fibronectin Type III Domain Using Bacterial Display
L. N. Shingarova1,a*, L. E. Petrovskaya1, A. V. Zlobinov1,2, S. Sh. Gapizov1,2, E. A. Kryukova1, K. R. Birikh1, E. F. Boldyreva1, S. A. Yakimov1, D. A. Dolgikh1,2, and M. P. Kirpichnikov1,2
1Shemyakin–Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, 117997 Moscow, Russia2Lomonosov Moscow State University, Faculty of Biology, 119234 Moscow, Russia
* To whom correspondence should be addressed.
Received December 22, 2017; Revision received February 21, 2018
Construction of antibody mimetics on the base of alternative scaffold proteins is a promising strategy for obtaining new products for medicine and biotechnology. The aim of our work was to optimize the cell display system for the 10th human fibronectin type III domain (10Fn3) scaffold protein based on the AT877 autotransporter from Psychrobacter cryohalolentis K5T and to construct new artificial TNF-binding proteins. We obtained a 10Fn3 gene combinatorial library and screened it using the bacterial display method. After expression of the selected 10Fn3 variants in Escherichia coli cells and analysis of their TNF-binding activity, we identified proteins that display high affinity for TNF and characterized their properties.
KEY WORDS: tumor necrosis factor, TNF-binding proteins, 10th human fibronectin type III domain, bacterial display, autotransporter from Psychrobacter cryohalolentis K5TDOI: 10.1134/S0006297918060081
Abbreviations: ASP, alternative scaffold protein; AT, autotransporter; bioTNF, biotinylated TNF; CDR, complementarity-determining region; 10Fn3, 10th human fibronectin type III domain; IPTG, isopropyl β-D-1-thiogalactopyranoside; MTT, 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide; PBS, phosphate buffered saline; PMSF, phenylmethylsulfonyl fluoride; TMB, 3,3′,5,5′-tetramethylbenzidine; TNF, tumor necrosis factor.
Tumor necrosis factor (TNF) is a multifunctional cytokine and one of the
major factors in inflammation, innate immunity, and other essential
functions of the human body [1, 2]. Many autoimmune and inflammatory processes are
accompanied by TNF overproduction. Thus, systemic and local increases
in TNF concentrations are typical for rheumatoid arthritis, psoriasis,
Crohn disease, septic shock, and multiple sclerosis [3, 4]. In some disorders,
inhibition of TNF activity produces clearly pronounced clinical effect;
that is why identification of TNF blockers is a promising direction in
protein engineering and biotechnology [2, 5, 6]. The major method for TNF
neutralization is the use of specific antibodies (e.g., infliximab) and
the immunoadhesin Etanercept composed of the ligand-binding region of
human TNF receptor and the Fc region of human immunoglobulin G1 [2]. Another type of TNF blockers are dominant-negative
TNF mutants (DN-TNFs) that bind to specific cell receptors and suppress
further signaling in the cells [7], single-domain
variable fragments of camelid antibodies [8], and
TNF-binding proteins of poxviruses [9, 10].
Despite obvious advantages of antibodies, such as high affinity, specificity, high elimination half-time, and biological safety, their clinical application might be with significant drawbacks, in particular, high production cost and, consequently, high cost of the final product, because of the necessity for expensive mammalian cell expression systems. Moreover, the high molecular mass of the antibodies prevents their penetration into the treated tissues and limits the efficiency of antibody use in the treatment of various disorders [11].
One approach to solving these problems is the use of alternative scaffold proteins (ASPs) capable of high-affinity binding of the corresponding antigens. ASPs are stable, have low molecular mass, and can be produced in a bacterial expression system [12-14], which makes them good candidates for creating artificial binding molecules by the methods of protein engineering.
The 10th human fibronectin type III domain (10Fn3) is one of the most commonly used ASPs. 10Fn3 is a small protein with extremely high solubility and thermostability. Its molecule lacks cysteine residues, which enables efficient expression of this protein in the cytoplasm of Escherichia coli cells. The presence of 10Fn3 in normal serum at a concentration of 0.1 mg/ml ensures its low immunogenicity and opens wide possibilities for its use in the development of new therapeutic agents. Three of the exposed 10Fn3 loops – BC, DE, and FG – are structural analogs of the antigen-binding regions in antibody molecules. Several 10Fn3-based artificial binding proteins have been obtained that displayed high specificities and dissociation constants in the pico- and nanomolar ranges [16, 17]. Genes for 10Fn3 antigen-binding variants are often selected from combinatorial libraries by the methods of cell-free (mRNA and ribosomal) display or cell (phage and yeast) display [17].
Earlier, we cloned a gene for the putative AT877 autotransporter (AT) from Psychrobacter cryohalolentis K5T and studied the properties of its passenger domain possessing esterase activity. We also demonstrated the possibility of the use of the type Va secretion system for the transport of cold-active esterase EstPc to the surface of E. coli cells [18]. The efficiency of the 10Fn3 exposure was significantly increased by its incorporation into a chimeric passenger domain containing EstPc at the N-terminus [19].
In this work, to estimate the efficiency of the AT877-based bacterial display system, we screened the combinatorial library for the TNF-binding variants of 10Fn3 that were exposed at the cell surface in the content of the AT877 autotransporter hybrid passenger domain. The resulting proteins were characterized by high solubility and capacity for TNF binding.
MATERIALS AND METHODS
Reagents used in the study were from Sigma (USA), BioRad (USA), Merck (Germany), and Panreac (Spain); components of bacterial culture media were from Difco (USA); full 199 medium, calf embryonic serum, and DMSO were from Biolot (Russia). All solutions were prepared in deionized water (MilliQ, USA). Recombinant human TNF was obtained as described in [20]. Biotinylated TNF (bioTNF) was obtained using an EZ-Link NHS-PEG4-Biotin reagent (Thermo Fisher, USA) as recommended by the manufacturer.
Recombinant DNAs were cloned in E. coli XL-1 Blue cells (Stratagene, USA) using enzymes from Fermentas (Latvia) according to standard procedures. Oligonucleotides (Table 1) were synthesized by Evrogen (Russia), except Bcd and Fgd that were synthesized from trimer phosphoramidites at the Department of Pharmaceutical Chemistry, University of Kuopio (Finland).
Table 1. Primers used for creating
10Fn3 gene combinatorial library
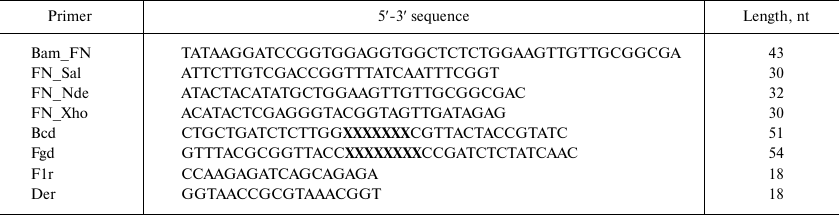
Note: X, nucleotide triplets corresponding to 19 amino acids at the
ratio described in [24].
Gene amplification by PCR was performed using Pfu polymerase as recommended by the enzyme manufacturer. The reaction regime included template denaturation for 3 min at 95°C; 25 cycles of denaturation at 45°C for 45 s, primer annealing at 52-55°C for 45 s, and elongation at 72°C for 45°C; and elongation at 72°C for 5 min.
Library construction for 10Fn3 gene variants in the pEFN877 plasmid. First, we obtained three PCR fragments using the pETNF3 plasmid (pET32a with inserted 10Fn3 gene) as a template and primer pairs Bam_FN + F1r (fragment F1), Bcd + Der (fragment F2), and Fgd + FN_Sal (fragment F3). The amplified fragments were purified by electrophoresis in 1.5% agarose gel and ligated by PCR using the Bam_FN and FN_Sal primers. The resulting product (294 bp) containing the 10Fn3 gene library was treated with BamHI and SalGI restriction endonucleases and cloned into the pEFN877 plasmid [19] by the BamHI/SalGI sites. Bacterial clones obtained by transformation of E. coli XL-1 Blue cells were washed off the LB agar plates and used for isolation of plasmid DNA using a QIAprep Spin miniprep kit (Qiagen, USA).
Screening for TNF-binding 10Fn3 variants by the bacterial display method. To obtain bacterial strains with surface expression of 10Fn3 variants, E. coli BL21(DE3)pLysS cells (Stratagene) were transformed with the total plasmid DNA containing the 10Fn3 gene library and grown at 37°C in 20 ml of LB medium containing 100 μg/ml ampicillin to optical density A560 = 0.6-0.8. Protein expression was induced by adding 0.1 mM IPTG, and the cells were incubated for 1 h at 22°C on a shaker at 220 rpm. The cells were then pelleted by centrifugation at 2000g for 15 min at 4°C, and the pellet was resuspended in 1.5 ml of cold PBS.
To eliminate clones with 10Fn3 variants that bound streptavidin nonspecifically, 150 μg of Dynabeads M-280 Streptavidin magnetic beads (Invitrogen, USA) was added to the cell suspension. After incubation for 30 min at 4°C under constant stirring, the beads were removed with a magnet. To select 10Fn3 variants specifically binding TNF (positive selection), 50 nM bioTNF was added to the cell supernatant, and the mixture was incubated for 1 h at 4°C with constant stirring. The cells were then collected by centrifugation at 2000g at 4°C for 15 min, resuspended in 1.5 ml cold PBS, and mixed with 150 μg of magnetic beads pretreated with the BB buffer (3% BSA in PBS). The mixture was incubated for 30 min at 4°C under constant stirring. The magnetic beads with the bound cells were collected with a magnet, washed twice with 750 μl cold PBS, transferred to 5 ml LB medium containing ampicillin, chloramphenicol, and glucose, and grown overnight. The next two rounds of selection were performed using the same procedure. After three rounds of selection, total plasmid DNA containing the library of TNF-binding 10Fn3 variants was isolated.
Confirmation of selection efficiency by ELISA. The efficiency of selection of the TNF-binding 10Fn3 variants exposed at the E. coli cell surface was determined by ELISA after each round of selection. Escherichia coli BL21(DE3)pLysS cells expressing the 10Fn3 gene library at their surface were pelleted by centrifugation as described above and washed with cold PBS. The resulting suspension was incubated for 1 h at 4°C with 100 nM bioTNF; the cells were then centrifuged, washed again with cold PBS, and treated with neutravidin–HRP (horseradish peroxidase) conjugate (Thermo Scientific) for 30 min. To remove unbound conjugates, the cells were pelleted, washed with PBS, resuspended in 500 μl PBS, and transferred into wells of an ELISA plate (50 μl/well). Immune complexes were visualized by reaction with the single-component substrate tetramethyl benzidine (TMB; 50 μl/well). After 5 min, the reaction was stopped by adding 50 μl 10% H2SO4. Absorbance in the wells was measured with a Model 680 plate reader (BioRad) at 450 nm. Each experiment was repeated five times; cells treated according to the same procedure in the absence of bioTNF were used as a negative control.
Cloning of the 10Fn3 gene library into expression vector. Total DNA coding for the library of TNF-binding 10Fn3 variants after three rounds of selection was used as a template in PCR with the FN_Xho and FN_Nde primers. The resulting PCR products were treated with XhoI and NdeI and cloned into the pET32a vector.
Analysis of the clone library. Competent E. coli XL-1 cells were transformed with ligase mixture containing plasmids bearing a library of the TNF-binding 10Fn3 variants and plated onto L-agar medium containing 100 μg/ml ampicillin. Grown clones were washed off with 5 ml of the medium and used for isolation of total plasmid DNA. An aliquot of the plasmid DNA was used for the transformation of E. coli BL21(DE3) cells (Novagen, USA) that were then plated on the same medium. Individual colonies were transferred into wells of a 96-deep-well plate (Greiner, Germany) containing 1 ml of the medium for autoinduction [22] and grown overnight at 30°C.
The plate was centrifuged for 20 min at 4000 rpm. The pellets were resuspended in 100 μl CelLytic lysis solution (Sigma), incubated for 10 min at room temperature with constant stirring, and then centrifuged under the same conditions. The supernatant (10 μl) was used in the analysis of TNF binding by ELISA.
Protein purification. Escherichia coli BL21(DE3) cells transformed with one of the plasmids were grown in 200 ml LB medium containing 100 μg/ml ampicillin at 37°C to optical density A560 = 0.5-0.7, and protein expression was induced by adding 0.2 mM IPTG. The cells were incubated for another 4 h at 27°C and pelleted by centrifugation. To purify the recombinant protein from the soluble cell fraction, the cells were resuspended in buffer A (50 mM Tris-HCl, pH 8.0, 1 mM PMSF, 200 mM NaCl) and disrupted by sonication (6 × 30-s pulses). The lysate was clarified by centrifugation; 10 mM imidazole was added to the resulting supernatant, which was loaded on a Ni-Sepharose FastFlow column (0.5 × 2 cm; GE Healthcare, Sweden) equilibrated with buffer A. The column was then washed with buffer B (20 mM Tris-HCl, pH 8.0, 0.5 M NaCl, 20 mM imidazole), and bound proteins were eluted with buffer C (20 mM Tris-HCl, pH 8.0, 200 mM NaCl, 300 mM imidazole). Fractions containing purified protein were combined and dialyzed against buffer D (20 mM Tris-HCl, pH 8.0, 50 mM NaCl).
Protein concentration was determined with a Qubit device (Life Technologies, USA) as recommended by the manufacturer.
ELISA and TNF cytotoxicity assay were performed as described in [21].
Immunoblotting. Proteins separated by gel electrophoresis were transferred onto nitrocellulose membranes (BioRad). The membranes were preincubated in 1% BSA in TBS for 1 h at 37°C to block nonspecific interactions and then were treated with the conjugate of anti-His6-tag antibodies with HRP (1 μg/ml; Invitrogen, USA) in 1% BSA in TBS for 1 h at room temperature. The membrane was then stained using precipitating TMB as a substrate (Clinical Science Products, USA).
Analytical gel filtration was peformed on a Superdex 75 10/300 GL column (10 × 30 mm; GE Healthcare) at a flow rate of 0.4 ml/min in 100 mM Tris-HCl (pH 8.0), 150 mM NaCl. Protein peaks were detected from absorbance at 230 nm.
Dissociation constant Kd of the 10Fn3 complex with TNF was determined by competitive ELISA as suggested by Martineau [23]. Two-fold TNF dilutions in 150 μl PBS containing 1% BSA were prepared; the initial protein concentration was 600 nM (as calculated per protein monomer). Each dilution was supplemented with 150 μl PBS/1% BSA containing 80 nM 10Fn3 variant and incubated at 37°C for 1 h to establish equilibrium in the 10Fn3 binding to TNF. The reaction mixture samples (100 μl; n = 3) were transferred to the wells of a 96-well plate with immobilized TNF (0.1 μg/well) and incubated for 1 h at 37°C under constant stirring. The wells were then washed three times with PBS containing 0.1% Tween-20, and primary monoclonal murine anti-His-tag antibodies in 100 μl PBS containing 1% BSA (1 : 10,000 dilution) were added to the wells. The reaction mixtures were incubated as described above, and the wells were then washed. To visualize immune complexes, secondary anti-mouse HRP-conjugated IgG antibody (Sigma) was used at 1 : 5000 dilution. Staining with TMB was performed as described above. The absorbance values for each of the dilutions were used to determine the dependence of absorbance on TNF concentration. The resulting experimental dependence was approximated with OriginPro v. 9.1 software in accordance with the equation:
where A is absorbance in a single experiment for each TNF concentration; Amax is absorbance in the control experiment in the absence of TNF; A0 is the minimal absorbance in the experiment; x is total TNF concentration (both bound and free TNF); a is initial 10Fn3 concentration; Kd is dissociation constant of the TNF complex with 10Fn3.
RESULTS
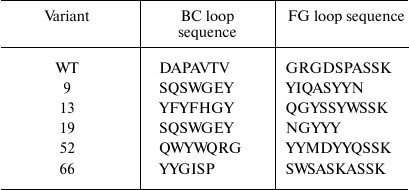
Construction and expression of the combinatorial library of 10Fn3 gene variants in the pEFN877 plasmid. To obtain ASPs based on 10Fn3 by randomization of fragments coding for the BC and FG loops, we constructed a combinatorial library by amplifying DNA fragments coding for BC and FG loops with randomized nucleotide sequence using primers synthesized from trimer phosphoramidites [24]. The ratio between the codons corresponded to the so-called magic formula that reflects amino acid composition typical for complementarity-determining regions (CDRs) of natural antibodies (Tyr – 20%; Ser and Gly – 15% each; all other amino acids except Cys – 3.1% each) [25]. The absence of stop codons and the reading frame shifts resulting from nucleotide deletions or insertions significantly increased the effective library size, whereas the use of codons most typical for E. coli and the lack of codons coding for Cys promoted high expression levels of the recombinant proteins. The Bcd primer provided randomization of seven amino acids that form the BC loop; the Fgd primer did the same for eight residues of the FG loop (Fig. 1a and Table 2). According to published data, the length of the FG loop in 10Fn3 varies from 7 to 11 amino acid residues [26]. In this study, we left the DE loop sequence intact, because, according to the results of structural studies, it forms a rigid β-hairpin structure, while the BC and FG loops display conformational mobility [27, 28]. The resulting PCR fragments with randomized regions were then combined with a fragment coding for the 10Fn3 N-terminal region, resulting in the creation of the combinatorial library.
Fig. 1. a) Positions of primers used for constructing the 10Fn3 gene combinatorial library. BC, DE, and FG are sequences coding for the 10Fn3 loops; X, randomized fragments. b) Structure of the hybrid autotransporter in the content of the pEFB877 plasmid. SS, PelB signal sequence; 10Fn3, 10th human fibronectin type III domain; EstPc, P. cryohalolentis K5T esterase; α, α-helical linker; β-barrel, AT877 translocator domain; His6, hexahistidine tag. Sites for restriction endonucleases used for cloning are shown.
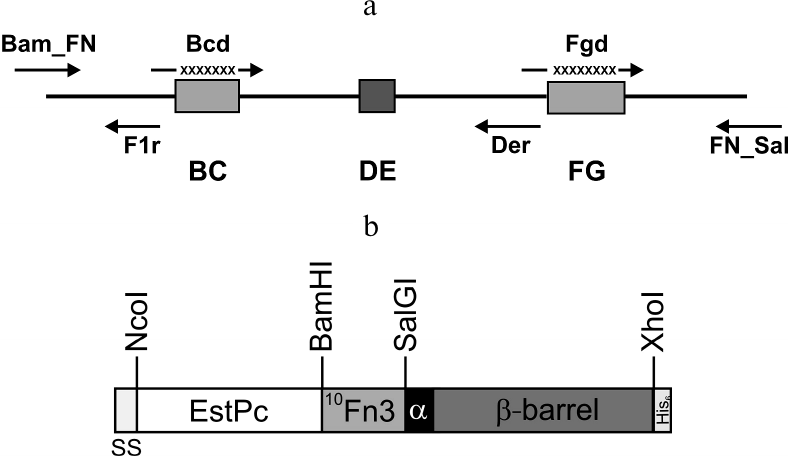
Table 2. Amino acid sequences of BC and FG
loops in TNF-binding 10Fn3 variants
Earlier, we constructed the pEFN877 plasmid containing the hybrid EstFn877 gene including the sequences for P. cryohalolentis K5T EstPc esterase, 10Fn3, α-linker, and P. cryohalolentis K5T AT translocator domain in the same reading frame (Fig. 1b) [19]. The PelB sequence at the N-terminus of the hybrid protein and the C-terminal His6-tag were provided by the pET20b plasmid.
For further selection of the TNF-binding 10Fn3 variants, we cloned the combinatorial library into the pEFN877 vector by replacing the wild-type 10Fn3. The size of the library was determined at the ligation stage: we used 3 μg of the vector, so that the size of the resulting library was ~4.8·109 variants.
The molecular diversity of the library was confirmed by sequencing of 20 random clones. All the sequenced genes contained different nucleotide sequences, three of which (15%) contained single-nucleotide deletions (probably due to errors in primers synthesis). Based on the efficiency of E. coli cell transformation and the proportion of correct nucleotide sequences, the actual library repertoire was 2.5·108 variants, which was considerably less than the theoretically calculated size. The resulting total DNA coding for the hybrid AT was used for transformation of E. coli BL21(DE3)pLysS cells. Gene expression was induced by adding IPTG to the cells at the logarithmic growth phase followed by cell incubation for 1 h at 22°C to prevent cell death from excessive biosynthesis of the membrane protein. Analysis of the cell lysates by electrophoresis and immunoblotting with antibodies against the His6-tag showed that IPTG induced biosynthesis of a protein with electrophoretic mobility that corresponded to the calculated molecular mass of the hybrid autotransporter (78 kDa) (Fig. 2, a and b, lane 2).
Fig. 2. Analysis of expression of the 10Fn3 gene combinatorial library by electrophoresis in 12% polyacrylamide gel (a) and immunoblotting with anti-His6 antibodies (b) in lysates of BL21(DE3)pLysS cells transformed with total plasmid DNA (library cloned in the pEFN877 vector) before induction (lane 1), after induction before selection (lane 2), and after one (lane 3) and two (lane 4) rounds of selection. M, molecular mass markers (Fermentas); arrow, position of the hybrid AT.

Screening of the combinatorial library by the bacterial display method. To select E. coli cells expressing TNF-binding 10Fn3 variants at the cell surface, bioTNF was immobilized on streptavidin-coated magnetic beads. To eliminate sequences that code for proteins that bind streptavidin nonspecifically, induced cells were preincubated with uncoated magnetic beads. The cells that did not bind to the uncoated beads were incubated with 50 nM bioTNF and then mixed with the streptavidin-coated beads. The formed conjugates were incubated in the cultivation medium for amplification, then the next induction/selection rounds were performed. It should be noted that this selection method does not require elution of the bound clones, which is one of the advantages of bacterial display.
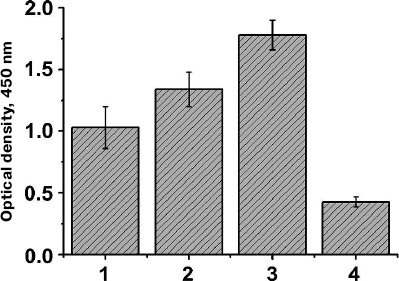
Analysis of cell lysates by immunoblotting showed that clones with a considerably higher expression level of the hybrid AT could be isolated even after the first selection round (Fig. 2, a and b, lanes 3 and 4). To confirm the efficiency of the clone selection, E. coli cells with the library of surface-expressed 10Fn3 variants were analyzed by ELISA after each selection round. We found that the efficiency of TNF binding by the selected cells increased after each selection round (Fig. 3). These data also proved that the AT877 AT translocator domain ensures exposure of the hybrid passenger domain including the 10Fn3 combinatorial library at the surface of E. coli cells and allows selection of the TNF-binding clones.
Fig. 3. Analysis of TNF-binding activity of BL21(DE3)pLysS cells expressing at their surface the 10Fn3 gene library using ELISA. Mean values of the cell culture optical density (n = 5) are shown for the cells after successive selection rounds (1-3) and for control cells of the same strain transformed with the pEFN877 plasmid (4).
After the third selection round, the genes for the TNF-binding 10Fn3 variants in the combination with the AT were recloned into the pET32a plasmid resulting in the library for cytoplasmic expression of the corresponding proteins in E. coli BL21(DE3) cells. After growing individual clones in plate wells with the autoinduction medium and analysis of the supernatants by ELISA, we selected five clones with the highest TNF-binding activity. Analysis by SDS-PAGE confirmed that >90% of grown clones expressed proteins of the required size (10-12 kDa) in the soluble fraction of the cells. Sequencing of these clones revealed several variants of the 10Fn3 gene containing amino acid variations in the BC and FG loops (Table 2). It should be noted the most common residues in the loops were serine and threonine, which correlates with the library design and is in good agreement with published data on the use of these residues for creation of binding surfaces in proteins [25, 29].
Purification and characterization of TNF-binding proteins. To increase the solubility of the expressed proteins, bacterial cell cultures were grown at 27°C after induction. Analysis of the recombinant protein localization after cell disruption by sonication showed that all these proteins were completely or mostly soluble and occurred in the cell cytoplasmic fraction (Fig. 4a). The TNF-binding variants FN9, FN13, FN19, FN52, and FN66 were isolated by metal-affinity chromatography (1-2 mg) with a high degree of purity (>90%). The protein yield was 20-30 mg/liter bacterial culture (Fig. 4b).
Fig. 4. Electrophoresis in 14% SDS-polyacrylamide gel of pellets (p) and supernatants (s) of cells expressing various 10Fn3 variants (a) and purified 10Fn3 proteins (b). c) Gel filtration of purified TNF-binding 10Fn3 variants: 1) FN52; 2) FN9; 3) FN19.
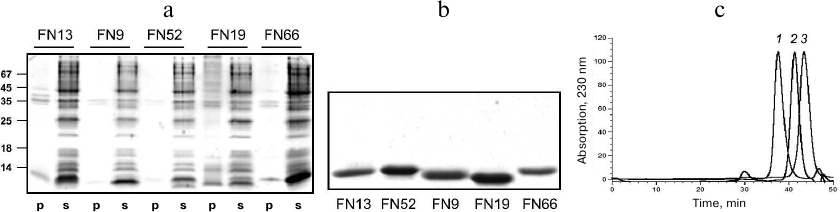
The homogeneity of the obtained fractions was determined by analytical gel filtration on a calibrated Superdex 75 column (Fig. 4c). Most preparations predominately contained monomeric and dimeric forms of the proteins; although some fractions contained high molecular mass aggregates. In our further experiments, we used the aggregate-free preparations.
The TNF-binding activity of the proteins was assayed by ELISA. Plates with adsorbed 10Fn3 variants were incubated with serial dilutions of bioTNF. The bioTNF binding was detected using neutravidin–HRP conjugates. The wild-type fibronectin domain did not bind bioTNF; none of the analyzed 10Fn3 variants bound BSA (negative control). The variant with the highest TNF-binding activity was FN52 (Fig. 5a).
Fig. 5. a) Analysis of TNF binding to 10Fn3 variants by ELISA: 1) FN52; 2) FN66; 3) FN19; 4) 10Fn3. b) Cytotoxic activity of TNF (1) and TNF mixture with FN52 (2) in L929 cells.
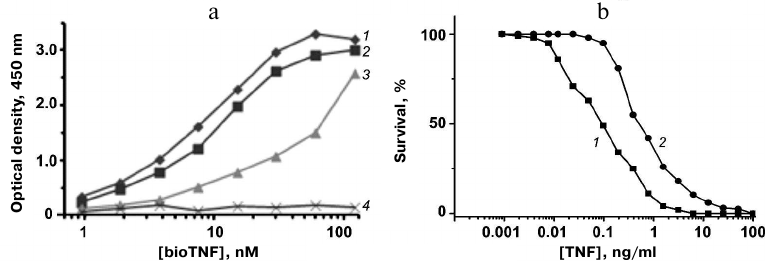
The affinity of the TNF binding by FN52 was estimated by competitive ELISA based on determination of the free protein concentration in the presence of TNF. The obtained value Kd was 35 ± 7 nM, indicating relatively high affinity of FN52 toward TNF.
The neutralizing activity of FN52 was studied in the standard TNF toxicity test using mouse fibroblast L929 cells expressing type I TNF receptor. L929 cells were cultured in the presence of the 10× molar excess of FN52 and different TNF dilutions. We found that addition of FN52 increased 4-fold the TNF concentration causing 50% cell death (Fig. 5b). Therefore, FN52 acted as a TNF antagonist.
DISCUSSION
Exposure of combinatorial libraries of binding proteins, in particular 10Fn3, at the surface of microbial cells (cell display) is an efficient approach that allows selection of protein variants with high affinity toward a target ligand [30]. The first attempts on the selection of the 10Fn3-based ASPs used phage display [17], which was later replaced by the yeast display in which proteins are expressed as hybrids with the Aga2p protein subunit [31]. The disadvantage of the latter method is low efficiency of yeast transformation compared to E. coli cells, resulting in a considerably smaller library size (number of variants). At the same time, E. coli-based bacterial display systems allow screening much larger libraries; moreover, different methods for exposing proteins at the cells surface have been developed for E. coli cells [32]. The number of molecules that could be exposed at the bacterial cell surface is much higher (up to 1.5·105) than in the phage-based system [19].
One of the most popular approaches to constructing cell display systems is using the Va type secretion pathway that involves AT proteins of the outer membrane in Gram-negative bacteria. The autotransporter molecule consists of the N-terminal passenger domain and the C-terminal translocator domain connected by an α-helical linker [33, 34]. By replacing the passenger domain with a recombinant protein by genetic engineering methods, it is possible to obtain hybrid ATs that would expose the target protein at the bacterial cell surface [35]. Here, we for the first time demonstrated the possibility of designing TNF-binding 10Fn3 proteins using E. coli bacterial display. For this purpose, a combinatorial 10Fn3 gene library was cloned into the pEFN877 plasmid also containing nucleotide sequences for P. cryohalolentis esterase and AT translocator domain in the same reading frame. As shown earlier, the transport of the 10Fn3-containing passenger domain to the cell surface in the absence of the esterase is extremely inefficient [19].
Numerous studies on the translocation of AT molecules to the bacterial cell surface showed that the efficiency of this process is determined by many factors, including the passenger domain structure. Still, the mechanisms of AT transport are not completely understood. According to the hairpin model, translocation of the unfolded passenger domain through a pore formed by the outer membrane-inserted β–barrel occurs in the C- to N-terminus direction [34, 36]. The C-terminal fragments of most passenger domains contain the so-called autochaperone that has stable structure and high folding rate [37]. Supposedly, folding of this autochaperone at the cell surface determines the directionality of the passenger domain transport and makes it irreversible. At the same time, the N-terminal fragment of the passenger domain is less stable, which creates an increasing gradient in the protein stability from the N-terminus to the C-terminus [38].
10Fn3 demonstrates high folding rate and stable spatial structure [39] that might hinder its secretion by the Va type mechanism. To overcome this obstacle, we placed the EstPc esterase from P. cryohalolentis K5T at the N-terminus of the passenger domain. EstPc is a cold-active enzyme that is less stable than other proteins with higher temperature optima [40]. Therefore, the generated recombinant AT protein possessed a stability gradient, which considerably increased the efficiency of 10Fn3 translocation to the bacterial cell surface [19].
Here, we demonstrated that the cell display system developed by us ensures efficient exposure of the 10Fn3 combinatorial library at the surface of E. coli cells, thereby allowing selection of the TNF-binding variants of this scaffold protein. It should be noted that most TNF-binding 10Fn3 variants obtained earlier by screening of the same library by the cell-free CIS-display method formed insoluble inclusion bodies when expressed in bacterial cells (unpublished data). In this study, the selected recombinant proteins were highly soluble, proving the advantage of the bacterial display method. We believe that selection of rapidly folding 10Fn3 variants was facilitated by the structure of the hybrid AT passenger domain.
After performing three rounds of the 10Fn3 gene library screening by the cell display method and analysis of the TNF-binding activity of the selected clones with the cytoplasmically expressed recombinant proteins, we obtained a high-affinity 10Fn3 variant that formed a complex with TNF with dissociation constant of ~35 nM and neutralized TNF cytotoxicity in L929 cells. Earlier, TNF-binding 10Fn3 variants with Kd of 1-24 nM have been obtained by combinatorial library screening by the mRNA display method [41]. The library used in [41] was significantly larger (~1012 clones). In this work, we developed a cell display system based on the P. cryohalolentis K5T AT and demonstrated the possibility of export of the recombinant 10Fn3 variants to the surface of E. coli cells. The developed system can be successfully used for obtaining high-affinity TNF-binding proteins that would be soluble and expressed at high levels, which also could be used later for selection of proteins capable of binding ligands other than TNF.
Acknowledgments
We thank A. V. Azhayev (Kuopio University) for kindly providing oligonucleotides for production of the combinatorial library.
This work was supported by the President of the Russian Federation grant (NSH-8384.2016.4), Molecular and Cell Biology Program of the Russian Academy of Sciences, and Russian Foundation for Basic Research (project 16-04-00717a).
REFERENCES
1.Kalliolias, G. D., and Ivashkiv, L. B. (2016) TNF
biology, pathogenic mechanisms and emerging therapeutic strategies,
Nat. Rev. Rheumatol., 12, 49-62.
2.Efimov, G. A., Kruglov, A. A., Shavarev, D. S.,
Drutskaya, M. S., and Nedospasov, S. A. (2014) New trends in
anticytokine therapy, Russ. Zh. Immunol., 8, 706-710.
3.Korneev, K. V., Atretkhany, K.-S. N., Drutskaya, M.
S., Grivennikov, S. I., Kuprash, D. V., and Nedospasov, S. A. (2017)
TLR-signaling and proinflammatory cytokines as drivers of
tumorigenesis, Cytokine, 89, 127-135.
4.Kruglov, A. A., Kuchmiy, A., Grivennikov, S. I.,
Tumanov, A. V., Kuprash, D. V., and Nedospasov, S. A. (2008)
Physiological functions of tumor necrosis factor and the consequences
of its pathologic overexpression or blockade: mouse models, Cytokine
Growth Factor Rev., 19, 231-244.
5.Zelova, H., and Hosek, J. (2013) TNF-alpha
signalling and inflammation: interactions between old acquaintances, J.
Inflamm. Res.'', 62, 641-651.
6.Astrakhantseva, I. V., Efimov, G. A., Drutskaya, M.
S., Kruglov, A. A., and Nedospasov, S. A. (2014) Modern anti-cytokine
therapy of autoimmune diseases, Biochemistry (Moscow),
79, 1308-1321.
7.Steed, P. M., Tansey, M. G., Zalevsky, J.,
Zhukovsky, E. A., Desjarlais, J. R., Szymkowski, D. E., Abbott, C.,
Carmichael, D., Chan, C., Cherry, L., Cheung, P., Chirino, A. J.,
Chung, H. H., Doberstein, S. K., Eivazi, A., Filikov, A. V., Gao, S.
X., Hubert, R. S., Hwang, M., Hyun, L., Kashi, S., Kim, A., Kim, E.,
Kung, J., Martinez, S. P., Muchhal, U. S., Nguyen, D.-H. T.,
O’Brien, C., O’Keefe, D., Singer, K., Vafa, O., Vielmetter,
J., Yoder, S. C., and Dahiyat, B. I. (2003) Inactivation of TNF
signaling by rationally designed dominant-negative TNF variants,
Science, 301, 1895-1898.
8.Coppieters, K., Dreier, T., Silence, K., Haard, H.
D., Lauwereys, M., Casteels, P., Beirnaert, E., Jonckheere, H., Wiele,
C. V. D., and Staelens, L. (2006) Formatted anti-tumor necrosis factor
α VHH proteins derived from camelids show superior potency and
targeting to inflamed joints in a murine model of
collagen‐induced arthritis, Arthritis Rheum., 54,
1856-1866.
9.Tregubchak, T. V., Shekhovtsov, S. V.,
Nepomnyashchikh, T. S., Peltek, S. E., Kolchanov, N. A., and
Shchelkunov, S. N. (2015) TNF-binding domain of the variola virus CrmB
protein synthesized in Escherichia coli cells effectively
interacts with human TNF, Dokl. Biochem. Biophys., 462,
176-180.
10.Shchelkunova, G. A., and Shchelkunov, S. N.
(2016) Immunomodulating drugs based on poxviral proteins,
BioDrugs, 30, 9-16.
11.Vazquez-Lombardi, R., Phan, T. G., Zimmermann,
C., Lowe, D., Jermutus, L., and Christ, D. (2015) Challenges and
opportunities for non-antibody scaffold drugs, Drug Discov.
Today, 20, 1271-1283.
12.Kariolis, M. S., Kapur, S., and Cochran, J. R.
(2013) Beyond antibodies: using biological principles to guide the
development of next-generation protein therapeutics, Curr. Opin.
Biotechnol., 24, 1072-1077.
13.Stahl, S., Kronqvist, N., Jonsson, A., and
Lofblom, J. (2013) Affinity proteins and their generation, J. Chem.
Technol. Biotechnol., 88, 25-38.
14.Deyev, S., Lebedenko, E., Petrovskaya, L.,
Dolgikh, D., Gabibov, A., and Kirpichnikov, M. (2015) Man-made
antibodies and immunoconjugates with desired properties: function
optimization using structural engineering, Russ. Chem. Rev.,
84, 1-26.
15.Skrlec, K., Strukelj, B., and Berlec, A. (2015)
Non-immunoglobulin scaffolds: a focus on their targets, Trends
Biotechnol., 33, 408-418.
16.Koide, S., Koide, A., and Lipovsek, D. (2012)
Target-binding proteins based on the 10th human fibronectin type III
domain (10Fn3), Methods Enzymol., 503,
135-156.
17.Lipovsek, D. (2011) Adnectins: engineered
target-binding protein therapeutics, Protein Eng. Des. Sel.'',
24, 3-9.
18.Petrovskaya, L., Novototskaya-Vlasova, K.,
Kryukova, E., Rivkina, E., Dolgikh, D., and Kirpichnikov, M. (2015)
Cell surface display of cold-active esterase EstPc with the use of a
new autotransporter from Psychrobacter cryohalolentis K5T,
Extremophiles, 19, 161-170.
19.Petrovskaya, L. E., Zlobinov, A. V., Shingarova,
L. N., Boldyreva, E. F., Gapizov, S. Sh., Novototskaya-Vlasova, K. A.,
Rivkina, E. M., Dolgikh, D. A., and Kirpichnikov, M. P. (2018) Fusion
with the cold-active esterase facilitates autotransporter-based surface
display of the 10th human fibronectin domain in Escherichia
coli, Extremophiles, 22, 141-150.
20.Shingarova, L., Sagaidak, L., Turetskaia, R.,
Nedospasov, S., Esipov, D., and Korobko, V. (1996) Human tumor necrosis
factor mutants: preparation and some properties, Bioorg. Khim.,
22, 243-251.
21.Petrovskaya, L. E., Shingarova, L. N., Kryukova,
E. A., Boldyreva, E. F., Yakimov, S. A., Guryanova, S. V.,
Novoseletsky, V. N., Dolgikh, D. A., and Kirpichnikov, M. P. (2012)
Construction of TNF-binding proteins by grafting hypervariable regions
of F10 antibody on human fibronectin domain scaffold, Biochemistry
(Moscow), 77, 62-70.
22.Studier, F. W. (2005) Protein production by
auto-induction in high-density shaking cultures, Protein Expr.
Purif., 41, 207-234.
23.Martineau, P. (2010) Affinity measurements by
competition ELISA, in Antibody Engineering (Kontermann, R., and
Dubel, S., eds.) Springer-Verlag, Berlin-Heidelberg, pp. 657-665.
24.Brockmann, E. C., Akter, S., Savukoski, T.,
Huovinen, T., Lehmusvuori, A., Leivo, J., Saavalainen, O., Azhayev, A.,
Lovgren, T., Hellman, J., and Lamminmaki, U. (2011) Synthetic
single-framework antibody library integrated with rapid affinity
maturation by VL shuffling, Protein Eng. Des. Sel.'', 24,
691-700.
25.Fellouse, F. A., Esaki, K., Birtalan, S., Raptis,
D., Cancasci, V. J., Koide, A., Jhurani, P., Vasser, M., Wiesmann, C.,
Kossiakoff, A. A., Koide, S., and Sidhu, S. S. (2007) High-throughput
generation of synthetic antibodies from highly functional minimalist
phage-displayed libraries, J. Mol. Biol., 373,
924-940.
26.Hackel, B. J., Kapila, A., and Wittrup, K. D.
(2008) Picomolar affinity fibronectin domains engineered utilizing loop
length diversity, recursive mutagenesis, and loop shuffling, J. Mol.
Biol., 381, 1238-1252.
27.Main, A. L., Harvey, T. S., Baron, M., Boyd, J.,
and Campbell, I. D. (1992) The three-dimensional structure of the tenth
type III module of fibronectin: an insight into RGD-mediated
interactions, Cell, 71, 671-678.
28.Dickinson, C. D., Veerapandian, B., Dai, X.-P.,
Hamlin, R. C., Xuong, N.-h., Ruoslahti, E., and Ely, K. R. (1994)
Crystal structure of the tenth type III cell adhesion module of human
fibronectin, J. Mol. Biol., 236, 1079-1092.
29.Gilbreth, R. N., Esaki, K., Koide, A., Sidhu, S.
S., and Koide, S. (2008) A dominant conformational role for amino acid
diversity in minimalist protein–protein interfaces, J. Mol.
Biol., 381, 407-418.
30.Lofblom, J. (2011) Bacterial display in
combinatorial protein engineering, Biotechnol. J., 6,
1115-1129.
31.Chen, T. F., de Picciotto, S., Hackel, B. J., and
Wittrup, K. D. (2013) Engineering fibronectin-based binding proteins by
yeast surface display, Methods Enzymol., 523,
303-326.
32.Van Bloois, E., Winter, R. T., Kolmar, H., and
Fraaije, M. W. (2011) Decorating microbes: surface display of proteins
on Escherichia coli, Trends Biotechnol., 29,
79-86.
33.Leo, J. C., Grin, I., and Linke, D. (2012)
Type V secretion: mechanism(s) of autotransport through the
bacterial outer membrane, Philos. Trans. R. Soc. Lond. B. Biol.
Sci., 367, 1088-1101.
34.Leyton, D. L., Rossiter, A. E., and Henderson, I.
R. (2012) From self sufficiency to dependence: mechanisms and factors
important for autotransporter biogenesis, Nat. Rev. Microbiol.,
10, 213-225.
35.Nicolay, T., Vanderleyden, J., and Spaepen, S.
(2015) Autotransporter-based cell surface display in Gram-negative
bacteria, Crit. Rev. Microbiol., 41, 109-123.
36.Noinaj, N., Kuszak, A. J., Gumbart, J. C.,
Lukacik, P., Chang, H., Easley, N. C., Lithgow, T., and Buchanan, S. K.
(2013) Structural insight into the biogenesis of β-barrel membrane
proteins, Nature, 501, 385-390.
37.Junker, M., Besingi, R. N., and Clark, P. L.
(2009) Vectorial transport and folding of an autotransporter virulence
protein during outer membrane secretion, Mol. Microbiol.,
71, 1323-1332.
38.Braselmann, E., and Clark, P. L. (2012)
Autotransporters: the cellular environment reshapes a folding mechanism
to promote protein transport, J. Phys. Chem. Lett., 3,
1063-1071.
39.Plaxco, K. W., Spitzfaden, C., Campbell, I. D.,
and Dobson, C. M. (1997) A comparison of the folding kinetics and
thermodynamics of two homologous fibronectin type III modules, J.
Mol. Biol., 270, 763-770.
40.Novototskaya-Vlasova, K., Petrovskaya, L.,
Yakimov, S., and Gilichinsky, D. (2012) Cloning, purification, and
characterization of a cold adapted esterase produced by
Psychrobacter cryohalolentis K5T from Siberian cryopeg, FEMS
Microbiol. Ecol., 82, 367-375.
41.Xu, L., Aha, P., Gu, K., Kuimelis, R. G., Kurz,
M., Lam, T., Lim, A. C., Liu, H., Lohse, P. A., and Sun, L. (2002)
Directed evolution of high-affinity antibody mimics using mRNA display,
Chem. Biol., 9, 933-942.