REVIEW: Unsolvable Problems of Biology: It Is Impossible to Create Two Identical Organisms, to Defeat Cancer, or to Map Organisms onto Their Genomes
E. D. Sverdlov
Shemyakin–Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, 117997 Moscow, Russia; E-mail: edsverd@gmail.com
Received November 12, 2017
The review is devoted to unsolvable problems of biology. 1) Problems unsolvable due to stochastic mutations occurring during DNA replication that make it impossible to create two identical organisms or even two identical complex cells (Sverdlov, E. D. (2009) Biochemistry (Moscow), 74, 939-944) and to “defeat” cancer. 2) Problems unsolvable due to multiple interactions in complex systems leading to the appearance of unpredictable emergent properties that prevent establishment of unambiguous relationships between the genetic architecture and phenotypic manifestation of the genome and make impossible to predict with certainty responses of the organism, its parts, or pathological processes to external factors. 3) Problems unsolvable because of the uncertainty principle and observer effect in biology, due to which it is impossible to obtain adequate information about cells in their tissue microenvironment by isolating and analyzing individual cells. In particular, we cannot draw conclusions on the properties of stem cells in their niches based on the properties of stem cell cultures. A strategy is proposed for constructing the pattern most closely approximated to the relationship of genotypes with their phenotypes by designing networks of intermediate phenotypes (endophenotypes).
KEY WORDS: stochastic mutations, emergent properties, heterogeneity, biological uncertainty principle, phenotype, genotype, selected functionDOI: 10.1134/S0006297918040089
The techniques have galloped ahead of the concepts.
We have moved away from studying the complexity
of the organism; from processes and organization
to composition.
James Black, Nobel Prize for Medicine in 1988
“One of the most striking aspects of physics is the simplicity of its laws. Maxwell’s equations, Schrödinger’s equation, and Hamiltonian mechanics can each be expressed in a few lines. The ideas that form the foundation of our worldview are also very simple indeed: The world is lawful, and the same basic laws hold everywhere. Everything is simple, neat, and expressible in terms of everyday mathematics, either partial differential or ordinary differential equations. Everything is simple and neat — except, of course, the world. Every place we look – outside the physics classroom – we see a world of amazing complexity” [1].
Goldenfeld and Kadanoff [1] gave some recommendations for studying the complex world. These recommendations are as simple as the laws of physics: “To extract physical knowledge from a complex system, one must focus on the right level of description… For example, some computational biologists try to simulate protein dynamics by following each and every small part of the molecule. The result? Most of the computer cycles are spent watching little OH groups wiggling back and forth. Nothing biological significant occurs in the time they can afford. Use the right level of description to catch the phenomena of interest. Don’t model bulldozers with quarks… As science turns to complexity, one must realize that complexity demands attitudes quite different from those heretofore common in physics. Up to now, physicists looked for fundamental laws true for all times and all places. But each complex system is different; apparently, there are no general laws for complexity. Instead, one must reach for “lessons” that might, with insight and understanding, be learned in one system and applied to another.”
On the other hand, such an outstanding physicist as Niels Bohr formulated the incognizability of life, because “we should doubtless kill an animal if we tried to carry out an investigation of its organs so far that we could tell the part played by single atoms in vital functions… The minimal freedom which we must allow the organism will be just large enough to permit it, so to say, to hide its ultimate secrets from us. On this view, the very existence of life must be considered in biology as an elementary fact, just as the existence of the quantum of action has to be taken as the basic fact that cannot be derived from ordinary mechanical physics” [2]. This is the biology’s uncertainty principle that is similar to the uncertainty principle in physics.
Even if we do not kill a living system, our intrusion with an investigation device so distorts its properties that we investigate not this system but the product of its interaction with the device at the interaction site. Because it is impossible in principal to exclude the interaction between an electron and a device used by us to study the properties of the electron, we cannot determine the electron velocity and position simultaneously. The interaction between the device and the object that distorts properties of the object is called the observer effect.
The uncertainty principle is one of the fundamental laws of physics. Formulated by Bohr, it is as simple in biology as in physics. Being probably unable to formulate positive fundamental laws, such as the Schrödinger equation or Newton laws, we can formulate prohibition laws for biology.
A remarkable Soviet astrophysicist Shklovsky has expressed his point of view as: “Science is a sum of taboos. It’s impossible to create a perpetuum mobile. One cannot transmit a signal with a speed higher than the speed of light in vacuum, and there is no way to simultaneously measure the speed and coordinates of the electron” [3]. This is a very elegant definition, although it is certainly insufficient. Nevertheless, it gives a possibility to define some basic fundamental laws not only for physics. The prohibition laws. The “it’s impossible” laws.
And then one can ask if prohibitions exist in biology. The knowing of such taboos would allow us to refuse to perform investigations that fall under prohibition laws, which would result in enormous economy of resources. Fundamentally unsolvable problems are not worth time and money. Just as creation of perpetuum mobile.
I tried to answer this question in my review “Fundamental taboos of biology” published in Biochemistry (Moscow) in 2009 [4], wherein as the main taboo I put forward the impossibility of existence of two identical complex living systems because of stochastic mutations occurring during each cell division.
In this review, I have proposed broader generalizations and tried to illustrate them, in particular, by the impossibility of the concept of unambiguous relations between the genetic architecture and phenotypic manifestations of the genome.
Some of the concepts expressed here have been published earlier [5, 6].
CATEGORIES OF UNSOLVABLE PROBLEMS
I. Problems unsolvable because of stochastic mutations occurring during DNA replication: 1) it is impossible to create two identical entities, including two identical complex cells [4]; 2) it is impossible to defeat cancer.
I would also like very much to formulate one more prohibition: it is impossible to defeat old age and natural death, but I won’t discuss it because of the limited volume of this review and complexity of the problem. Instead, I refer the readers to the recent reviews [7-9] and leave the problem for their own consideration.
II. Problems unsolvable because of the relationships in complex systems leading to unpredictable emergent properties: 1) based on properties of a trait, it is impossible to establish its cause(s) (inverse problem); 2) based on known causes, it is impossible to establish unambiguously the properties of a trait, if these causes interact with each other resulting in emergent properties (direct problem); 3) it is impossible to predict with certainty a response of a complex system to an external factor.
III. Problems unsolvable because of the uncertainty principle and the observer effect in biology. 1) It is impossible to obtain an adequate information about cells in their tissue microenvironment by isolating and analyzing single cells (transcriptome, proteome, etc.). In particular, it is impossible to deduce conclusions about properties of stem cells in their niches based on the cultures of these cells. 2) It should be remembered that a probe introduced into a system for observation changes the properties of this system, at least, at the site of probe introduction (the observer effect).
I have mentioned this problem in the introduction. Apparently, Niels Bohr was the first who formulated it. I also will not discuss this problem here and refer the readers to the reviews [10, 11].
This system of prohibitions, in particular, the impossibility of the existence of identical organisms due to inevitable stochastic mutations leading to an extreme heterogeneity within an organism and between organisms, appeals for caution, or more exactly, limits prospects of personalized medicine [12].
Here, I will not discuss the problems that have been already discussed in my recent reviews, except the latest data; instead, I refer the readers to these reviews and literature cited therein. The main focus of the present review is problems I and II.
I. 1) Problems unsolvable because of stochastic mutations during DNA replication: it is impossible to create two identical entities, including two identical complex cells
These prohibitions are associated with various mutations continuously occurring during DNA replication. As it often happens in biology, there is no precise definition of mutation. Mutations are changes in the nucleotide sequence of the organism’s genome. They emerge because of unrepaired DNA damages (usually caused by radiation or chemical mutagens), errors during DNA replication, or insertion/deletion of DNA segments by mobile genetic elements. Mutations can lead or not lead to marked changes in the observed traits (phenotype) of an organism. The word mutation is derived from the Latin word mutare – to change.
The first mutations are transmitted to us with the parents’ sex cells and then are accumulated in DNA, starting with the first divisions of the zygote and throughout the whole life.
Genes and chromosomes can mutate in any somatic or germ tissue, causing somatic and germinal mutations, respectively. Somatic mutations are not transmitted to the progeny, whereas germinal mutations can be transmitted.
Germinal mutations occur in the germ lines. If a mutant sex cell participates in fertilization, the mutations will be passed to the next generation. Germinal mutations will be present in all somatic cells, whereas a post-zygotic somatic mutation will be detected only when the mutant cell gives rise to a cell line constituting a significant portion of a selected population of the cells [13].
If a somatic mutation emerges in a single cell during somatic tissue development, this cell becomes a progenitor of a population of identical mutant cells. A population of cells originated from a single precursor cell is called clone. The earlier the mutation occurs in the development, the greater will be the proportion of mutant cells in the organism.
Recently developed next generation high-throughput sequencing methods significantly contributed to the progress in the estimation of mutation rates.
Current estimations of mutation rates. In normal tissues, estimates of mutation rates significantly vary: usually from 10—9 to 10—10 mutations per base pair per cell division [14-16]. For example, the average mutation rate in the cell line involved in the retina development was determined as 0.99·10—9 per base pair per cell division [15]. Similar mutation rates (0.27·10—9, 1.47·10—9, and 0.34·10–9) were found for the intestinal epithelial cells, B and T lymphocytes, and the HPRT gene in the fibroblast cultures, respectively. The ratio of base substitutions to small insertions/deletions, and mutations occurring during recombination was found to be ~0.9 : 0.08 : 0.04. These values are in a good agreement with the later estimates [17].
As to adult stem cells (ASCs) and embryonic stem cells, a small number of which form all embryonic tissues, they supposedly should have reliable mechanisms for DNA repair and prevention of DNA damage [18-20]. The maintenance of genome integrity is a strongly regulated decisive factor both for precursors and stem cells in their natural environment, but not for isolated stem cells. For example, stem cell niches play an essential role in maintaining ASCs or preventing tumorigenesis by providing signals that inhibit proliferation and differentiation [21-23]. During specification of cell lines in the blastocyst, cells of the inner cell mass are also under a strict control of signals from other cells and of contact/position cell–cell relations [16, 24].
The mutation rate in the human germ cell line is ~6·10—11 per base pair per cell division, which is similar to the lowest values observed in unicellular organisms and is 10-100 times lower than in human somatic tissues [13, 25-28]. The mutation rates in both germ and somatic cells in mice are significantly higher than in humans [13].
Mutations in germ cells often lead to hereditary diseases (see reviews [29-33]).
It is important to note that units of measure for mutation rates in somatic and germ cells are usually different. For somatic cells, it is usually the number of mutations per base pair per cell division, whereas for germ cells, the number of mutations per base pair per genome per generation is more common. In population biology and demography, generation time is the average time between two consecutive generations in the lineages of a population. In human populations, generation time typically ranges from 22 to 33 years. Generation time can be also defined as the time necessary for birth, puberty and reproduction.
Skipping many nuances associated with differences in the generation time of germ cells (e.g., spermatozoa and oocytes) (the reader can find them in the database or in the good blog), below are approximate calculations for the heterogeneity of somatic cells, since they are the primary base for examining differences between individuals.
An adult human body consists of approximately 1014 cells. If we neglect the facts that different tissues terminally differentiate for different time periods and that cells can die, the number of cell divisions to form a terminally differentiated cell equals to N ~ 46 (1014 = 2N, Nlog2 = 14). Assuming that the mutation rate is 10–9 per bp per division and the genome length is 3·109 bp, a differentiated somatic cell will possess ~120 mutations distinguishing it from its precursor. Neighboring cells will receive the same number of mutations, but they will be located at other genomic sites (stochastics!). Therefore, each two cells in an adult organism will differ from each other by more than 200 substitutions. The probability of the existence of two cells with coinciding 100 mutations is extremely low. Thus, each individual is a mosaic of different cells. This theoretical conclusion has received its experimental confirmation with the emergence of the whole-genome sequencing [34, 35]. Let us add to the above the stochastics of epigenetic changes [36] and we’ll come to the conclusion that there cannot be two genetically and epigenetically identical individuals. Each person is unique from the standpoint of genome and epigenome.
Hence, the prohibition (I. 1): it is impossible to create two identical individuals or two identical complex cells. Identical (homozygous) twins are not identical [37-39].
I. 2) It is impossible to defeat cancer. As long as genes continue to mutate spontaneously, cancer will never be eradicated completely. It will arise constantly. The treatment of cancer is problematic
This problem has two sides: inevitability of cancer arising in a population and problems with its prognostic diagnosis and treatment.
Inevitability of cancer arising in a population. In 1996, a well-known interview was published [40] with the prominent oncologist Alfred G. Knudson, in which he stated: “Cancer will never be completely eradicated as long as genes continue to mutate spontaneously. To think otherwise is unrealistic … But we can hope that over the next quarter-century we will minimize cancer mortality for those under the age of 60.
Now it is axiomatic that the major cause of cancer is gene damage that later leads to the evolution of a complex system of cancer tumor [41] (see below). Available experiments justify this conclusion by the following findings: preservation of a malignant phenotype of cancer cells through numerous cell divisions; mutagenicity of various agents capable of inducing cancer; the presence of chromosomal abnormalities in cancer cells; cancer as a family disease; and predisposition to cancer in case of inherited impairments of DNA repair. Very recently, brilliant works of C. Tomasetti and B. Vogelstein have shown that the probability of tumor emergence in a tissue is proportional to the rate of stem cell division in this tissue, and thus to the mutation rate [42]. All this taken together indicates that cancer is underlain by modified genome.
Studies on RNA-containing oncoviruses were the first to demonstrate that normal cellular genes, e.g., protooncogenes, could be a potential cause of tumorigenicity. Protooncogenes can transform into oncogenes due to acquisition of a genetically dominant ability to accelerate cell proliferation. Later, it was found that the emergence of cancer cells could be also promoted by recessive defects in tumor suppressor genes whose homozygotic deficiencies contribute to tumorigenesis. Combinations of activated oncogenes and defective tumor suppressor genes are found in the majority, if not all, human cancer tumors. They gradually accumulate and stimulate clonal development and diversification resulting in malignancy (see reviews [43-48]).
Human organism plays with the fire of evolution. Evolutionary inevitability of cancer tumors [49]. Cancer tumor is a result of the evolutionary produced process of organism’s development that requires a renewal of tissues during the functioning of a multicellular organism.
Such constant renovation of tissues is essential for normal functioning of multicellular organisms. A mechanism has evolved in evolution that includes the death of old cells and their replacement with the new ones. This process requires cell division to continue throughout an organism’s lifetime. However, every cell division is associated with the emergence of mutations in daughter cells [50] and can initiate evolutionary events leading to fatal malignancy. Therefore, cancer is the price for multicellularity [49]. All vertebrates suffer from cancer tumors, and, seemingly, it was always like that, since traces of tumors or metastasizing cancers have been found in the fossil remains of dinosaurs. Invertebrates are also susceptible to tumor-like formations [51-54]. It is reasonable to think that the probability of cancer increases with the increase in the number of divisions in the organism. Hence, the probability of cancer increases with an increase in lifespan. Norwegian scientist Jarle Breivik formulated this as follows [55]: “Cancer is a natural consequence of aging, and the better medical science gets at keeping people alive, the more cancer there will be in the population”.
During evolution, organisms had to evolve mechanisms for efficient suppression of cancer (and have evolved indeed) [56] and, therefore, there is no direct correlation between the cell number in a body and organism’s susceptibility to cancer (the Peto’s paradox), but this is beyond the scope of our review.
Treatment of cancer is problematic. The fact that the mutation rate in cancer cells is higher than in normal ones has been well established (for the latest reviews see [50, 57]). This means that the heterogeneity of a cancer tumor is higher than of the normal tissue.
By the moment when a tumor can be detected (109 cells, 1 g), the tumor cell might contain up to 10,000 mutations in its genome. Dr. Glazier (see the quotation in [50]) evaluated the possible number of different cells possessing this amount of randomly distributed mutations as ~1068000! Therefore, there are no two identical cells within the same tumor, and there are no two identical cells in different tumors. Moreover, tumors of the same type are different in different patients [58]. A tumor is heterogeneous both genetically and epigenetically [59]. All cells in the tumor are genetically distinct, which means that some of these cells may be resistant to virtually any treatment [60]. When treated, sensitive cells die, but resistant ones persist and give rise to a new tumor resistant to the therapeutic agent used. The so-called molecular target therapy based on targeting molecules or groups of molecules altered in tumor cells is thus inadequate to the multilayer complexity of cancer.
It seems that the deeper we understand intimate molecular details of cancer, the more we focus on particular targets and the more our treatment approaches become inadequate to the complexity of the problem. Exhaustive genotyping will probably be of help for only a small part of patients [61]. Many tumors are not associated with any well-studied mutations that could be targeted in therapy. For example, only ~15-20% of all lung tumors have mutations that are suitable for targeted therapy [61].
Intratumor heterogeneity also imposes serious limitations on the identification of mutant molecules or signaling pathways based on molecular analysis of tumor biopsy. A molecular analysis of a tumor biopsy sample will not be necessarily reproduced with other samples of the same tumor. Therefore, the treatment based on such analysis may not be very useful, because cells in other parts of the tumor with different molecular characteristics can remain active and resistant to the treatment [12].
To continue this review, it might be useful to give a short description of complex systems mentioned above and that include any living organism and a majority of its pathologies.
A short excursion into unsolvable problems caused by the complexity of biological systems
A problem of complex systems has been examined in detail in my previous reviews [5, 6]. To be objective, I also recommend a recent review [62] that also describes complex systems. Here, I will present only a very brief summary of the major points.
A complex system is a multicomponent system consisting of interacting subunits whose interactions result in the appearance of the so-called emergent properties. These properties are inherent to the whole system and unpredictable based on properties of initial subunits (see below). Emergent properties are the most important features of complex systems. They cannot be ascribed to individual interacting components but are properties of the whole system. A complex system itself can consist of hierarchical levels, each of which has its own emergent properties [63-67].
Complex systems are nonlinear and extremely sensitive to initial conditions [68]. This means that the trajectory of the system [63], defined as changes in its state (for example, in time) is unpredictable. For example, changes of temperature, blood pressure, blood formula, and other parameters that determe patient’s condition in time can not be prognosed. The dependence on initial conditions means that two systems that function according to the same rules and have very close initial states will stll have different trajectories over time. The immune system, for example, consists of various elements (macrophages, T and B cells, etc.) that interact with each other by an exchange of signals (in particular, cytokines). Even under the action of quite identical stimuli, the immune system (like other complex systems, including cancer) may respond very differently.
Complex systems are nonlinear, i.e., their response to the sum of external signals is not equal to the sum of responses to all these signals taken separately [69]. Small changes in some influencing factors will not necessarily induce small responses of the system, and vice versa. Quite often, a small impact can cause an unexpectedly large response.
In complex systems, it is impossible to exactly predict the effect of environmental factors. In an organism, such effect (like the influence of stochastic factors) begins in utero and continues throughout the entire life of the individual [39]. Complex systems, as a whole, cannot be simulated with a computer [68, 70].
Cancer is a complex system with a large number of interactions with the environment generating unpredictable properties. A tumor represents a complex and varying in time and space diversity of cells. Each of these cells has its own signal cascades, replication, transcription etc. and undergoes numerous alterations during transformation into a cancer cell. The tumor has a complexity of a growing and developing system with all its traits and properties that allows it to withstand anticancer agents and to induce intratumor cell heterogeneity, which makes each tumor unique [58] for each patient. In this regard, cancer is different from all other diseases [71].
However, the tumor complexity is far from being limited by a set of cancer genes and cells that influence tumor progression. In their latest list of cancer hallmarks, Hanahan and Weinberg [72] have pointed out that tumors exhibit another dimension of complexity: tumors use in their evolution a wide repertoire of normal cells which they adapt for their needs. These cells promote the acquisition of distinctive features and create the “microenvironment” of the tumor and its ecologic niche that plays a crucial role in both primary tumor evolution and its metastasis. It could be now assumed with certainty that the major complexity of tumor is caused by the enormous number of interactions between cancer (usually epithelial) cells and various stroma cells of the tumor microenvironment (TME) [73].
Therapeutic approaches can be directed not at cancer cells but at the disruption of interactions within the evolving tumor. In the last years, a fundamentally new approach has received a big resonance in the scientific community. Instead of treating mutations in cancer cells, the new approach focuses on disrupting complex interactions between cancer cells and immune components of the stroma that determine the success of the cancerous organism evolution. These interactions allow cancer cells to inhibit immune cells in their environment and to avoid destruction by the immune system. Successful application of inhibitors of such interactions in clinical practice during the last 5 years [74-76] has demonstrated that the immune system can recognize cancer and suppress or even eliminate tumors.
Although these therapeutic approaches considerably increased the lifespan of many cancer patients, a large number of patients with malignancies do not respond to therapy [77-79]. Moreover, such treatment is often accompanied by numerous adverse autoimmune effects [80, 81]. In general, the effect of therapy for an individual patient is unpredictable. Perhaps, future studies will discover new promising immunological targets or combine old targets with new immunotherapeutic approaches, chemo- and radiotherapy, and therapy using oncolytic viruses and small molecules.
However, the results obtained once more demonstrate that complexity remains complexity with its unpredictability, and its response to various effects is unpredictable.
Unsolved problem of type II. Complex relationships between the genome and the body generate unsolvable problems in studying genotype interconnections with phenotype and in decoding genome functional architecture. I shall start with quoting an article published by the electronic resource “Evolution News@DiscoveryCSC” (https://evolutionnews.org/2017/02/encode_team_con/) on February 13, 2017: “With the fresh funding, the ENCODE team continues to destroy the myth of “junk” DNA”. In this article, it was said that the “National Institutes of Health (NIH) just recently funded five centers to explore what the “dark matter” of genome (the non-protein-coding part) is doing”. The “search for a function” strategy continues to be fruitful… This approach states that “If it’s there, it’s probably doing something important”. The investments for 2017 were $31.5 million.
The ENCODE Project Consortium [82] published in 2012 a huge list of “functional elements” in the human genome under the title “The Encyclopedia of DNA Elements”. The most striking was the statement that 80% of human genome is transcribed, i.e., has a “biochemical function”, which severely contradicts the traditional concept that more than 90% of the human genome is non-functional “junk”. This contradiction has provoked hot debates mainly focused at the proportion of “junk” in the human genome. Here lies the point of the close intertwining of three extremely urgent problems: strategy for the financing of science, choice of an adequate scientific methodology of the development of biological science – “hypothesis-driven science” vs. “Big Data-driven science”, i.e., science based on the analysis of large sets of data, and, finally, “evolution vs. intelligent design of life” (Intelligent Design, ID).
ENCODE is a typical example of a big-science style of research. Bruce Alberts, the former Editor-in-Chief of Science magazine, on occasion of the release in 2012 of 30 articles of the ENCODE project (Consortium, 2012 #121), issued the article “The End of “Small Science?”, where he warned: “Each of these big-science efforts drives the development of valuable new methodologies, as required to bring each type of investigation to scale. But the scale also creates a constituency that makes these projects difficult to stop, even when there are clear signs of diminishing returns. In this time of very tight resources, it becomes increasingly critical to make objective, tough decisions about what kind of projects stand the best chance of producing the results needed for deeply understanding, rather than merely describing biological systems”. Alberts had forcible arguments to say: “…clear signs of diminishing returns…”. For example, 10 years after the Human Genome Project had finished, Science published a paper entitled “Deflating the Genomic Bubble”, where the authors asked: “…what became of all the genomic medicine we were promised?” [84]. Since that time, little has changed.
In this part of the review, I will consider the unsolvable problem of exact mapping of genomic elements that determine organism’s phenotype, in particular, the problem of functionality of non-coding and non-regulating genomic elements (junk) and put forward a hypothesis about the most promising approaches for identification of functional regions of the genome.
Listen! If stars are lit, then someone must need them, of course? These are the words of Vladimir Mayakovsky’s poem “Listen!” It reflects a feature of the human thinking: everything that exists is expedient and strictly determined. A human is searching for order, even if it does not exist [10]. This is the so-called view of Pangloss, who is a character in the novel “Candide” by Voltaire, and presumably a caricature of the philosopher Gottfried Leibniz, who theorized that we lived in the best of all worlds. The term “Panglossian paradigm” was introduced by Stephen Gould and Richard Levontin as a point of view in biology which says that all properties of living beings are adaptations for definite purposes. This feature of human mind has led, in particular, to a flow of mathematical models describing complex phenomena in simplified (mainly linear) terms [10]. These models can work rather well but can entice researchers into the so-called “likelihood trap”, which has nothing in common with the reality. The best example of such trap is the geocentric model by Ptolemy. As to biology, the most dangerous trap is the “statistical significance p ≤ N (where N can vary from 0.05 to less than 10-20, depending on the subject of study)”. In the latter case, cause-effect relation is substituted with correlation. System biology that promises an immense success of “personalized medicine” is another example of such trap [6].
Scientists studying functional genomic elements have formed two almost equal and irreconcilable camps, similar to Lilliputian fractions of Big-Endians and Little-Endians in the Jonathan Swift’s novel that could not agree on the correct practice of egg breaking. Within the scope of the Panglossian paradigm, many researchers believe that, e.g., very low-level transcripts represent a huge world of functional RNAs only because they exist. Their opponents believe that there is a reason to doubt this Panglossian point of view. No doubt that many functional coding and non-coding RNAs can be found among such transcripts, but it is even more probable that the overwhelming majority of these transcripts are simply junk.
So, who is right? To answer this question, we have, first of all, to define the term “function”, that is what we mean under this term, and to determine the level of the living system organization at which the concept of “function” becomes meaningful.
Function or activity? Use the right level of description to catch the phenomena of interest. Don’t model bulldozers out of quarks. This subtitle is a paraphrase of the recommendation by American physicists Kodanoff and Goldenfeld in their remarkable article “Simple lessons from complexity” which I have mentioned earlier [1]. The level of description of “function” that the authors of ENCODE used in 2012 to ascribe biochemical functions to 80% of the human genome [82] was not “right”. “Biochemical function” was not defined correctly by ENCODE. The definition was given to a “functional element” which is “a discrete genome segment that encodes a defined product (for example, protein or non-coding RNA) or displays a reproducible biochemical signature (for example, protein binding site or specific chromatin structure)” [82].
Nevertheless, the figure 80% was enthusiastically accepted by determinists (and by the believers in the intelligent design), because it seemed to support the lack of non-functional elements in the genome, and thus supported intelligent design (ID).
However, this interpretation has been strongly criticized by supporters of the evolutionary origin of organisms and their genomes: If an element possesses some biochemical activity, it does not necessarily prove its significance for functioning of the cell and, especially, of the whole multicellular organism. According to [85] and other authors, the term “function” in biology can have two major meanings: selected effect and causal role [86-89].
The function of selected effect, which is also called genetic function, explains the origin, etiology, and subsequent evolution of a trait. Such functionality is protected by the natural selection; if this protection does not work, functional elements will accumulate deleterious mutations and lose their functional activity with time [85]. “Distinguishing what an element (genetic or otherwise) does (its causal role) from why it exists (its selective effect) is at the heart of biology” [90].
For an element to have a causal role function, e.g., transcription, it is necessary and sufficient for it to be transcribed. All selected effect functions also have causal roles. On the contrary, most of functional elements with causal roles do not have selected effect function. Therefore, the term “function” should be limited to selected effect function, and causal role should be defined as “activity” [87].
Based on the extent of mutation load tolerable for mammalian species, the Nobel Prize winner, Hermann Joseph Muller, evaluated in 1967 the upper limit for the number of genes in the human genome as 30,000, which is very close to the currently accepted number (cited from [91]). Note that this was a purely holistic evaluation.
Graur et al. [89] also used a holistic approach to demonstrate that the maximum proportion of functional elements in the human genome does not exceed 25%, and the rest is junk DNA. The proportion of the genome possessing the selective effect function was evaluated based on the known rates of deleterious mutations, replacement fertility rates, mutation load, and, as a consequence, on the reduction in the reproductive success caused by deleterious mutations. Graur suggested that only functional parts of the genome could be damaged by harmful mutations, whereas mutations in the non-functional parts should be neutral.
Because of deleterious mutations, each couple in each generation must produce more than two children in order to maintain a constant population size. The larger the proportion of the functionally important part in the genome, the more descendants should be produced by each couple to maintain the size of the population. Graur found that if 80% of the genome were functional, unacceptably high birth rates would be required.
Recent findings that 8.2% (7.1-9.2%) of the human genome is a subject of negative selection and presumably functional [92] corroborate these conclusions.
Graur speculates: “There is no need to sequence everything under the sun. We need only to sequence the sections we know are functional” (ScienceDaily, July 14, 2017; www.sciencedaily.com/releases/2017/07/170714140234.htm). This is very similar to the thoughts of Alberts: “…the grand challenges that remain in attaining a deep understanding of the chemistry of life will require going beyond detailed catalogs. Ensuring a successful future for the biological sciences will require restraint in the growth of large centers and -omics-like projects, so as to provide more financial support for the critical work of innovative small laboratories striving to understand the wonderful complexity of living systems” [83].
Deducing genetic causes of functions from the behavior of a complex system is the inverse problem that cannot be solved. Deducing functions from the components comprising the system is the direct problem that also cannot be solved because of emergent properties.
The estimations of Muller and Graur are extremely valuable, especially from the evolutionary point of view; however, they do not indicate specific functional elements of the genome and specific functions performed by them.
This question cannot be answered based on the analysis of phenotypes, because it would require solving the so-called inverse problem that cannot be solved in common case [93]. For complex systems, especially for such a complex system as an organism, it is also impossible to solve the direct problem – to deduce properties of the phenotype from the genomic structures and other molecular components involved in the phenotype formation. This impossibility is caused by the interactions of these components that lead to the generation of unpredictable emergent properties.
The simplest paradigmatic example of the direct problem: it is impossible to predict all properties of water, such as boiling temperature, surface tension, solvent properties, specific weight, freezing with the formation of snowflakes of various shapes, etc., based on the properties of hydrogen and oxygen molecules. The “phenotype” of water arises from the interactions between hydrogen and oxygen. The inverse problem: it is impossible to deduce the constituents of water and to predict their properties from the properties (“phenotype”) of water.
To return to the relations between an organism and its genome, I will quote [with my notes in square brackets] one of the most respected modern scientists and philosophers of science and a Nobel Prize winner Sydney Brenner, who introduced into science such a remarkable model organism as Caenorhabditis elegans. The first part of the section title is a paraphrase of his article [93]. (In one of his lectures Brenner has illustrated the inverse problem approximately as follow: “Can you imagine details of a drum by hearing only its sound?”).
“Sequencing the human genome was once likened to sending a man to the moon. The comparison turns out to be literally correct because sending a man to the moon is easy; it is getting him back that is difficult and expensive. Today the human genome sequence is, so to speak, stranded on a metaphorical moon and it is our task to bring it back to Earth and give it the life it deserves. Everybody understood that getting the sequence would be really easy, only a question of 3M Science—enough Money, Machines and Management. Interpreting the sequence to discover the functions of its coding and regulatory elements and understanding how these are integrated into the complex physiology of a human being was always seen as a difficult task, but since it is easier to go on collecting data the challenge [interpretation, ES] has not really been seriously taken up” [93].
“The genome must therefore form the kernel of any theory we construct but since transforming the information in a genome into the final living organism involves many complicated processes mediated by molecules specified in the genome, all of this will need to be known in considerable detail before we can read and understand genomes. There is no simple way to map organisms onto their genomes once they have reached a certain level of complexity. Thus, while the genome sequence is central, it is a level of abstraction which is too cryptic to be used for the organization of data and the derivation of theoretical models. Proposals to base everything on the genome sequence by annotating it with additional data [direct task, ES] will only increase its opacity” [93].
Thus, we fall into the “scissors of impossibility”. Brenner would not be Brenner if he had not offered a solution based not on the genome, but on the cell, as the center in our approach to deciphering functions. He called his algorithm “CellMap”. Although I also think that it is a cell that should to be in the center of further efforts of the scientific community (see [94] for example), I disagree with some of his statements. But not having a space for presenting his concepts, I do not have an opportunity to argue. The readers may draw their own conclusions from a very interesting article of Brenner [93].
Exact solutions are impossible. Where to search for the best approximations? I would not suggest anything new by proposing the “evolution–development–diseases” triad, whose components are closely connected through common regulatory systems, as a promising field to search for approximations. The principle of Dobzhansky “Nothing makes sense in biology except in the light of evolution” has been an axiom for a long time. We could draw connections between the selected effect functions and genetic elements based on a broad comparative analysis of development regulation and diseases arising because of dysregulations of development or homeostasis.
There is a large arsenal of models and analytical methods that give us a hope for success. To solve this problem, scientific community has to initiate and support programs and consortia that would be different from “-omics” projects by focusing their attention not on the development of high-throughput technologies but on searching for associations between phenotypes and functional genomic elements. A key role in this search could belong to the construction of networks of intermediate phenotypes (endophenotypes) that are closer to the main gene products than the observed final phenotype (figure).
Using endophenotypes for establishing relationships between the genotype and the phenotype
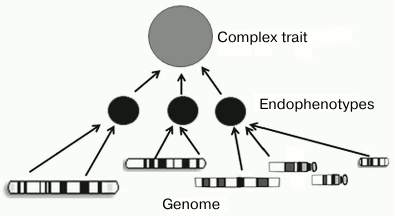
The concept of endophenotype suggests that being more proximal to the main gene products than the observed final phenotype, endophenotypes possess less complex network of causative genetic interactions (genetic architecture), which will make easier identification of specific genetic factors underlying the complex trait. In particular, many wide-spread diseases, as well as physiological and pathophysiological processes, such as sporadic cancer, diabetes mellitus, ischemic heart disease, or autoimmune disorders, are complex phenotypes and have endophenotypes. Thus, the susceptibility to ischemic heart disease is affected by such intermediate phenotypes as arterial pressure, hypercholesterolemia, and susceptibility to proatherogenic agents [95].
An endophenotype has to fit a set of requirements [96]. In particular, it has to be heritable, and its heritability has to agree with the heritability of the external phenotype. It also has to be reliably measurable, etc. [95-97].
Searching for and constructing networks of interactions involving endophenotypes will be a difficult and very routine work. But the era of premature sensations is over, and it is time to gather stones.
Acknowledgments
I am grateful to Dr. B. Glotov and T. V. Vinogradova for critical analysis and help in the preparation of the review.
The work was supported by the Russian Science Foundation (project 14-50-00131).
REFERENCES
1.Goldenfeld, N., and Kadanoff, L. P. (1999) Simple
lessons from complexity, Science, 284, 87-89.
2.Bohr, N. (1961) Atomic Physics and Human
Knowledge [Russian translation], IL Publishers, Moscow, pp.
22-23.
3.Shklovsky, I. (1991) Echelon [in Russian],
Novosti, Moscow, p. 109.
4.Sverdlov, E. D. (2009) Fundamental taboos of
biology, Biochemistry (Moscow), 74,
939-944.
5.Sverdlov, E. D. (2016) Multidimensional complexity
of cancer. Simple solutions are needed, Biochemistry
(Moscow), 81, 731-738.
6.Sverdlov, E. D. (2014) Systems biology and
personalized medicine: to be or not to be? Ross. Fiziol. Zh. im. I.
M. Sechenova, 100, 505-541.
7.Granger, A., Mott, R., and Emambokus, N. (2016) Is
aging as inevitable as death and taxes? Cell Metab.,
23, 947-948.
8.Vijg, J., and Le Bourg, E. (2017) Aging and the
inevitable limit to human life span, Gerontology, 63,
432-434.
9.Le Bourg, E., and Vijg, J. (2017) The future of
human longevity: time for a reality check, Gerontology,
63, 527-528.
10.Zbilut, J. P., and Giuliani, A. (2008) Biological
uncertainty, Theory Biosci., 127, 223-227.
11.Strippoli, P., Canaider, S., Noferini, F.,
D’Addabbo, P., Vitale, L., Facchin, F., Lenzi, L., Casadei, R.,
Carinci, P., Zannotti, M., and Frabetti, F. (2005) Uncertainty
principle of genetic information in a living cell, Theor. Biol. Med.
Model., 2, 40.
12.Tannock, I. F., and Hickman, J. A. (2016) Limits
to personalized cancer medicine, N. Engl. J. Med., 375,
1289-1294.
13.Milholland, B., Dong, X., Zhang, L., Hao, X.,
Suh, Y., and Vijg, J. (2017) Differences between germline and somatic
mutation rates in humans and mice, Nat. Commun.,
8, 15183.
14.Prindle, M. J., Fox, E. J., and Loeb, L. A.
(2010) The mutator phenotype in cancer: molecular mechanisms and
targeting strategies, Curr. Drug Targets, 11,
1296-1303.
15.Lynch, M. (2010) Rate, molecular spectrum, and
consequences of human mutation, Proc. Natl. Acad. Sci. USA,
107, 961-968.
16.Sverdlov, E. D., and Mineev, K. (2013) Mutation
rate in stem cells: an underestimated barrier on the way to therapy,
Trends Mol. Med., 19, 273-80.
17.Narasimhan, V. M., Rahbari, R., Scally, A.,
Wuster, A., Mason, D., Xue, Y., Wright, J., Trembath, R. C., Maher, E.
R., Heel, D. A. V., Auton, A., Hurles, M. E., Tyler-Smith, C., and
Durbin, R. (2017) Estimating the human mutation rate from autozygous
segments reveals population differences in human mutational processes,
Nat. Commun., 8, 303.
18.Hong, Y., Cervantes, R. B., Tichy, E.,
Tischfield, J. A., and Stambrook, P. J. (2007) Protecting genomic
integrity in somatic cells and embryonic stem cells, Mutat.
Res., 614, 48-55.
19.Luo, L. Z., Gopalakrishna-Pillai, S., Nay, S. L.,
Park, S. W., Bates, S. E., Zeng, X., Iverson, L. E., and
O’Connor, T. R. (2012) DNA repair in human pluripotent stem cells
is distinct from that in non-pluripotent human cells, PLoS One,
7, e30541.
20.Rebuzzini, P., Pignalosa, D., Mazzini, G., Di
Liberto, R., Coppola, A., Terranova, N., Magni, P., Redi, C. A.,
Zuccotti, M., and Garagna, S. (2012) Mouse embryonic stem cells that
survive gamma-rays exposure maintain pluripotent differentiation
potential and genome stability, J. Cell. Physiol.,
227, 1242-1249.
21.Li, L., and Neaves, W. B. (2006) Normal stem
cells and cancer stem cells: the niche matters, Cancer Res.,
66, 4553-4557.
22.Plaks, V., Kong, N., and Werb, Z. (2015) The
cancer stem cell niche: how essential is the niche in regulating
stemness of tumor cells? Cell. Stem. Cell, 16,
225-238.
23.Ye, J., Wu, D., Wu, P., Chen, Z., and Huang, J.
(2014) The cancer stem cell niche: cross talk between cancer stem cells
and their microenvironment, Tumour Biol., 35,
3945-3951.
24.Gasperowicz, M., and Natale, D. R. (2011)
Establishing three blastocyst lineages — then what? Biol.
Reprod., 84, 621-630.
25.Lynch, M., Ackerman, M. S., Gout, J. F., Long,
H., Sung, W., Thomas, W. K., and Foster, P. L. (2016) Genetic drift,
selection and the evolution of the mutation rate, Nat. Rev.
Genet., 17, 704-714.
26.Otte, J., Wruck, W., and Adjaye, J. (2017) New
insights into human primordial germ cells and early embryonic
development from single-cell analysis, FEBS Lett., 591,
2226-2240.
27.Chen, C., Qi, H., Shen, Y., Pickrell, J., and
Przeworski, M. (2017) Contrasting determinants of mutation rates in
germline and soma, Genetics, 207, 255-267.
28.Scally, A. (2016) The mutation rate in human
evolution and demographic inference, Curr. Opin. Genet. Dev.,
41, 36-43.
29.Rahbari, R., Wuster, A., Lindsay, S. J.,
Hardwick, R. J., Alexandrov, L. B., Turki, S. A., Dominiczak, A.,
Morris, A., Porteous, D., Smith, B., Stratton, M. R., and Hurles, M. E.
(2016) Timing, rates and spectra of human germline mutation, Nat.
Genet., 48, 126-133.
30.De Ligt, J., Veltman, J. A., and Vissers, L. E.
(2013) Point mutations as a source of de novo genetic disease,
Curr. Opin. Genet. Dev., 23, 257-263.
31.Arnheim, N., and Calabrese, P. (2016) Germline
stem cell competition, mutation hot spots, genetic disorders, and older
fathers, Annu. Rev. Genom. Hum. Genet., 17, 219-243.
32.Campbell, C. D., and Eichler, E. E. (2013)
Properties and rates of germline mutations in humans, Trends
Genet., 29, 575-584.
33.Segurel, L., Wyman, M. J., and Przeworski, M.
(2014) Determinants of mutation rate variation in the human germline,
Annu. Rev. Genom. Hum. Genet., 15, 47-70.
34.Freed, D., Stevens, E. L., and Pevsner, J. (2014)
Somatic mosaicism in the human genome, Genes (Basel), 5,
1064-1094.
35.Campbell, I. M., Shaw, C. A., Stankiewicz, P.,
and Lupski, J. R. (2015) Somatic mosaicism: implications for disease
and transmission genetics, Trends Genet., 31,
382-392.
36.Zhang, N., Zhao, S., Zhang, S. H., Chen, J., Lu,
D., Shen, M., and Li, C. (2015) Intra-monozygotic twin pair discordance
and longitudinal variation of whole-genome scale DNA methylation in
adults, PLoS One, 10, e0135022.
37.Baxter, A. G., and Hodgkin, P. D. (2015) No luck
replicating the immune response in twins, Genome Med., 7,
29.
38.Greek, R., and Rice, M. J. (2013) Monozygotic
twins: identical in name only, Anesthesiology, 118,
230.
39.Van Dongen, J., Slagboom, P. E., Draisma, H. H.,
Martin, N. G., and Boomsma, D. I. (2012) The continuing value of twin
studies in the omics era, Nat. Rev. Genet., 13,
640-653.
40.McIntosh, H. (1996) 25 years ahead: will cancer
be a “background-noise kind of disease”? J. Natl. Cancer
Inst., 88, 1794-1798.
41.DeGregori, J. (2017) Connecting cancer to its
causes requires incorporation of effects on tissue microenvironments,
Cancer Res., doi: 10.1158/0008-5472.CAN-17-1207.
42.Tomasetti, C., and Vogelstein, B. (2015) Cancer
etiology. Variation in cancer risk among tissues can be explained by
the number of stem cell divisions, Science, 347,
78-81.
43.Alekseenko, I. V., Kuzmich, A. I., Pleshkan, V.
V., Tyulkina, D. V., Zinovyeva, M. V., Kostina, M. B., and Sverdlov, E.
D. (2016) The cause of cancer mutations: Improvable bad life or
inevitable stochastic replication errors? Mol. Biol. (Moscow),
50, 906-921.
44.Tomasetti, C., Li, L., and Vogelstein, B. (2017)
Stem cell divisions, somatic mutations, cancer etiology, and cancer
prevention, Science, 355, 1330-1334.
45.Waddell, N., Pajic, M., Patch, A. M., Chang, D.
K., Kassahn, K. S., Bailey, P., Johns, A. L., Miller, D., Nones, K.,
Quek, K., Quinn, M. C., Robertson, A. J., Fadlullah, M. Z., Bruxner, T.
J., Christ, A. N., Harliwong, I., Idrisoglu, S., Manning, S., Nourse,
C., Nourbakhsh, E., Wani, S., Wilson, P. J., Markham, E., Cloonan, N.,
Anderson, M. J., Fink, J. L., Holmes, O., Kazakoff, S. H., Leonard, C.,
Newell, F., Poudel, B., Song, S., Taylor, D., Waddell, N., Wood, S.,
Xu, Q., Wu, J., Pinese, M., Cowley, M. J., Lee, H. C., Jones, M. D.,
Nagrial, A. M., Humphris, J., Chantrill, L. A., Chin, V., Steinmann, A.
M., Mawson, A., Humphrey, E. S., Colvin, E. K., Chou, A., Scarlett, C.
J., Pinho, A. V., Giry-Laterriere, M., Rooman, I., Samra, J. S., Kench,
J. G., Pettitt, J. A., Merrett, N. D., Toon, C., Epari, K., Nguyen, N.
Q., Barbour, A., Zeps, N., Jamieson, N. B., Graham, J. S., Niclou, S.
P., Bjerkvig, R., Grutzmann, R., Aust, D., Hruban, R. H., Maitra, A.,
Iacobuzio-Donahue, C. A., Wolfgang, C. L., Morgan, R. A., Lawlor, R.
T., Corbo, V., Bassi, C., Falconi, M., Zamboni, G., Tortora, G.,
Tempero, M. A.; Australian Pancreatic Cancer Genome Initiative, Gill,
A. J., Eshleman, J. R., Pilarsky, C., Scarpa, A., Musgrove, E. A.,
Pearson, J. V., Biankin, A. V., and Grimmond, S. M. (2015) Whole
genomes redefine the mutational landscape of pancreatic cancer,
Nature, 518, 495-501.
46.Swanton, C. (2015) Cancer evolution constrained
by mutation order, N. Engl. J. Med., 372, 661-663.
47.Schlesner, M., and Eils, R. (2015) Hypermutation
takes the driver’s seat, Genome Med., 7, 31.
48.Salk, J. J., Fox, E. J., and Loeb, L. A. (2010)
Mutational heterogeneity in human cancers: origin and consequences,
Annu. Rev. Pathol., 5, 51-75.
49.Gatenby, R., Gillies, R., and Brown, J. (2010)
The evolutionary dynamics of cancer prevention, Nat. Rev.
Cancer, 10, 526-527.
50.Sverdlov, E. D. (2011) Genetic surgery — a
right strategy to attack cancer, Curr. Gene Ther., 11,
501-531.
51.McAloose, D., and Newton, A. L. (2009) Wildlife
cancer: a conservation perspective, Nat. Rev. Cancer, 9,
517-526.
52.Greaves, M. (2007) Darwinian medicine: a case for
cancer, Nat. Rev. Cancer, 7, 213-221.
53.Greaves, M. (2015) Evolutionary determinants of
cancer, Cancer Discov., 5, 806-820.
54.Aktipis, C. A., Boddy, A. M., Jansen, G., Hibner,
U., Hochberg, M. E., Maley, C. C., and Wilkinson, G. S. (2015) Cancer
across the tree of life: cooperation and cheating in multicellularity,
Philos. Trans. R. Soc. Lond. B. Biol. Sci., 370,
1673.
55.Breivik, J. (2016) Reframing the “Cancer
Moonshot": how experts and non-experts interpret the problem of cancer,
EMBO Rep., 17, 1685-1687.
56.DeGregori, J. (2011) Evolved tumor suppression:
why are we so good at not getting cancer? Cancer Res.,
71, 3739-3744.
57.Chalmers, Z. R., Connelly, C. F., Fabrizio, D.,
Gay, L., Ali, S. M., Ennis, R., Schrock, A., Campbell, B., Shlien, A.,
Chmielecki, J., Huang, F., He, Y., Sun, J., Tabori, U., Kennedy, M.,
Lieber, D. S., Roels, S., White, J., Otto, G. A., Ross, J. S.,
Garraway, L., Miller, V. A., Stephens, P. J., and Frampton, G. M.
(2017) Analysis of 100,000 human cancer genomes reveals the landscape
of tumor mutational burden, Genome Med., 9, 34.
58.Wood, L. D., Parsons, D. W., Jones, S., Lin, J.,
Sjoblom, T., Leary, R. J., Shen, D., Boca, S. M., Barber, T., Ptak, J.,
Silliman, N., Szabo, S., Dezso, Z., Ustyanksky, V., Nikolskaya, T.,
Nikolsky, Y., Karchin, R., Wilson, P. A., Kaminker, J. S., Zhang, Z.,
Croshaw, R., Willis, J., Dawson, D., Shipitsin, M., Willson, J. K.,
Sukumar, S., Polyak, K., Park, B. H., Pethiyagoda, C. L., Pant, P. V.,
Ballinger, D. G., Sparks, A. B., Hartigan, J., Smith, D. R., Suh, E.,
Papadopoulos, N., Buckhaults, P., Markowitz, S. D., Parmigiani, G.,
Kinzler, K. W., Velculescu, V. E., and Vogelstein, B. (2007) The
genomic landscapes of human breast and colorectal cancers,
Science, 318, 1108-1113.
59.Easwaran, H., Tsai, H. C., and Baylin, S. B.
(2014) Cancer epigenetics: tumor heterogeneity, plasticity of stem-like
states, and drug resistance, Mol. Cell., 54, 716-727.
60.Pribluda, A., de la Cruz, C. C., and Jackson, E.
L. (2015) Intratumoral heterogeneity: from diversity comes resistance,
Clin. Cancer Res., 21, 2916-2923.
61.Kaiser, J. (2009) Cancer research. Looking for a
target on every tumor, Science, 326, 218-220.
62.Mallick, P. (2015) Complexity and information:
cancer as a multi-scale complex adaptive system, in Physical
Sciences and Engineering Advances in Life Sciences and Oncology
(Janmey, P., Fletcher, D., Gerecht, S., Levine, R., Mallick, P.,
McCarty, O., Munn, L., and Reinhart-King, C., eds.) Springer
International Publishing, pp. 5-29.
63.Rickles, D., Hawe, P., and Shiell, A. (2007) A
simple guide to chaos and complexity, J. Epid. Com. Health,
61, 933-937.
64.Suki, B., Bates, J. H., and Frey, U. (2011)
Complexity and emergent phenomena, Compr. Physiol., 1,
995-1029.
65.Noble, D. (2013) A biological relativity view of
the relationships between genomes and phenotypes, Prog. Biophys.
Mol. Biol., 111, 59-65.
66.Korn, R. (2005) The emergence principle in
biological hierarchies, Biol. Phil., 20, 137-151.
67.Van Regenmortel, M. H. (2004) Reductionism and
complexity in molecular biology. Scientists now have the tools to
unravel biological and overcome the limitations of reductionism,
EMBO Rep., 5, 1016-1020.
68.Greek, R., and Hansen, L. A. (2013) Questions
regarding the predictive value of one evolved complex adaptive system
for a second: exemplified by the SOD1 mouse, Prog. Biophys. Mol.
Biol., 113, 231-253.
69.Janson, N. (2012) Non-linear dynamics of
biological systems, Contemp. Phys., 53, 137-168.
70.Greek, R., and Menache, A. (2013) Systematic
reviews of animal models: methodology versus epistemology, Int. J.
Med. Sci., 10, 206-221.
71.Merlo, L. M., Pepper, J. W., Reid, B. J., and
Maley, C. C. (2006) Cancer as an evolutionary and ecological process,
Nat. Rev. Cancer, 6, 924-935.
72.Hanahan, D., and Weinberg, R. A. (2011) Hallmarks
of cancer: the next generation, Cell, 144, 646-674.
73.Bissell, M. J., and Hines, W. C. (2011) Why
don’t we get more cancer? A proposed role of the microenvironment
in restraining cancer progression, Nat. Med., 17,
320-329.
74.Bordon, Y. (2015) Immunotherapy: checkpoint
parley, Nat. Rev. Cancer, 15, 3.
75.Smyth, M. J., Ngiow, S. F., Ribas, A., and Teng,
M. W. (2016) Combination cancer immunotherapies tailored to the tumour
microenvironment, Nat. Rev. Clin. Oncol., 13,
143-158.
76.Park, J., Kwon, M., and Shin, E. C. (2016) Immune
checkpoint inhibitors for cancer treatment, Arch. Pharm. Res.,
39, 1577-1587.
77.Postow, M. A., Callahan, M. K., and Wolchok, J.
D. (2015) Immune checkpoint blockade in cancer therapy, J. Clin.
Oncol., 33, 1974-1982.
78.Diesendruck, Y., and Benhar, I. (2017) Novel
immune check point inhibiting antibodies in cancer therapy –
opportunities and challenges, Drug Resist Updat., 30,
39-47.
79.Vreeland, T., Clifton, G., Herbert, G., Hale, D.,
Jackson, D., Berry, J., and Peoples, G. (2016) Gaining ground on a cure
through synergy: combining checkpoint inhibitors with cancer vaccines,
Exp. Rev. Clin. Immunol., 12, 1347-1357.
80.Calabrese, L., and Velcheti, V. (2017) Checkpoint
immunotherapy: good for cancer therapy, bad for rheumatic diseases,
Ann. Rheum. Dis., 76, 1-3.
81.Postow, M., and Wolchok, J. (2016) Toxicities
Associated with Checkpoint Inhibitor Immunotherapy (avilable from
http://www.uptodate/com/contents/toxicities-associated-withchckpoint-inhibitor-immunotherapy).
82.The ENCODE Project Consortium (2012) An
integrated encyclopedia of DNA elements in the human genome,
Nature, 489, 57-74.
83.Alberts, B. (2012) The end of “small
science”? Science, 337, 1583.
84.Evans, J. P., Meslin, E. M., Marteau, T. M., and
Caulfield, T. (2011) Genomics. Deflating the genomic bubble,
Science, 331, 861-862.
85.Graur, D. (2016) Rubbish DNA: the functionless
fraction of the human genome, arXiv:1601.06047v1[q-bio.GN].
86.Graur, D., Zheng, Y., and Azevedo, R. B. (2015)
An evolutionary classification of genomic function, Genome Biol.
Evol., 7, 642-645.
87.Doolittle, W. F., Brunet, T. D., Linquist, S.,
and Gregory, T. R. (2014) Distinguishing between “function”
and “effect” in genome biology, Genome Biol. Evol.,
6, 1234-1237.
88.Graur, D., Zheng, Y., Price, N., Azevedo, R. B.,
Zufall, R. A., and Elhaik, E. (2013) On the immortality of television
sets: “function” in the human genome according to the
evolution-free gospel of ENCODE, Genome Biol. Evol., 5,
578-590.
89.Graur, D. (2017) An upper limit on the functional
fraction of the human genome, Genome Biol. Evol., 9,
1880-1885.
90.Brunet, T. D., and Doolittle, W. F. (2014)
Getting “function” right, Proc. Natl. Acad. Sci.
USA, 111, E3365.
91.Nei, M. (2005) Selectionism and neutralism in
molecular evolution, Mol. Biol. Evol., 22, 2318-2342.
92.Rands, C. M., Meader, S., Ponting, C. P., and
Lunter, G. (2014) 8.2% of the human genome is constrained: variation in
rates of turnover across functional element classes in the human
lineage, PLoS Genet., 10, e1004525.
93.Brenner, S. (2010) Sequences and consequences,
Philos Trans. R. Soc. Lond. B. Biol. Sci., 365,
207-212.
94.Sverdlov, E. (2006) Biological reductionism goes
away? What is further? Vestn. Ros. Akad. Nauk, 76,
707-721.
95.Blanco-Gomez, A., Castillo-Lluva, S., Del Mar
Saez-Freire, M., Hontecillas-Prieto, L., Mao, J. H.,
Castellanos-Martin, A., and Perez-Losada, J. (2016) Missing
heritability of complex diseases: enlightenment by genetic variants
from intermediate phenotypes, Bioessays, 38, 664-673.
96.Gottesman, I., and McGue, M. (2015)
Endophenotypes, JohnWiley & Sons, Inc.
97.Te Pas, M. F., Madsen, O., Calus, M. P., and
Smits, M. A. (2017) The importance of endophenotypes to evaluate the
relationship between genotype and external phenotype, Int. J. Mol.
Sci., 18, E472.