Endonuclease Activity of MutL Protein of the Rhodobacter sphaeroides Mismatch Repair System
M. V. Monakhova1, A. I. Penkina2, A. V. Pavlova2, A. M. Lyaschuk3, V. V. Kucherenko4, A. V. Alexeevski1,5, V. G. Lunin3, P. Friedhoff6, G. Klug7, T. S. Oretskaya2, and E. A. Kubareva1*
1Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; E-mail: kubareva@belozersky.msu.ru2Lomonosov Moscow State University, Chemistry Department, 119991 Moscow, Russia
3Gamaleya Research Institute of Epidemiology and Microbiology, 123098 Moscow, Russia
4Lomonosov Moscow State University, Bioengineering and Bioinformatics Department, 119991 Moscow, Russia
5Research Institute of System Development, 117218 Moscow, Russia
6Institut für Biochemie, Justus-Liebig-Universität, 35392, Gießen, Germany; E-mail: Sekretariat.Biochemie@chemie.bio.uni-giessen.de
7Institut für Mikrobiologie und Molekularbiologie, Justus-Liebig-Universität, 35392, Gießen, Germany; E-mail: gabriele.klug@mikro.bio.uni-giessen.de
* To whom correspondence should be addressed.
Received September 12, 2017; Revision received November 11, 2017
We have purified the MutL protein from Rhodobacter sphaeroides mismatch repair system (rsMutL) for the first time. rsMutL demonstrated endonuclease activity in vitro, as predicted by bioinformatics analysis. Based on the alignment of 1483 sequences of bacterial MutL homologs with presumed endonuclease activity, conserved functional motifs and amino acid residues in the rsMutL sequence were identified: five motifs comprising the catalytic site responsible for DNA cleavage were found in the C-terminal domain; seven conserved motifs involved in ATP binding and hydrolysis and specific to the GHKL family of ATPases were found in the N-terminal domain. rsMutL demonstrated the highest activity in the presence of Mn2+. The extent of plasmid DNA hydrolysis declined in the row Mn2+ > Co2+ > Mg2+ > Cd2+; Ni2+ and Ca2+ did not activate rsMutL. Divalent zinc ions inhibited rsMutL endonuclease activity in the presence of Mn2+ excess. ATP also suppressed plasmid DNA hydrolysis by rsMutL. Analysis of amino acid sequences and biochemical properties of five studied bacterial MutL homologs with endonuclease activity revealed that rsMutL resembles the MutL proteins from Neisseria gonorrhoeae and Pseudomonas aeruginosa.
KEY WORDS: DNA repair, mismatch, MutL proteinDOI: 10.1134/S0006297918030082
Abbreviations: a.a., amino acid; bp, base pair; DTT, 1,4-dithiothreitol; ecMutL, MutL from Escherichia coli; GHKL, Gyrase, Hsp90, Histidine Kinase, MutL family of ATPases; IPTG, isopropyl β-D-1-thiogalactopyranoside; LB, lysogeny broth; MMR, mismatch repair system; PMSF, phenylmethylsulfonyl fluoride; rsMutL, mutL from Rhodobacter sphaeroides.
All living organisms, from bacteria to humans, possess a system for
repair of noncanonical nucleotide pairs (mismatches) and small loops,
which was named the DNA mismatch repair (MMR) system [1-3]. Impairments in MMR at any
stage of DNA repair cause accumulation of mutations, resulting in the
development of various types of tumors [4, 5]. Because of the essential role of MMR in
functioning of any living organism, studies of MMR are of extreme
importance.
MMR corrects DNA damage by excising an extended single-stranded fragment of the newly synthesized DNA and then filling the resulting gap. The signal for the MMR is hydrolysis of the phosphodiester bond in one of the DNA strands [6-8]. In cells of γ-proteobacteria, including Escherichia coli, this break is introduced by the MutH protein that recognizes the monomethylated 5′-Gm6ATC-3′/3′-CTAG↓-5′ sequence (m6A is 6-methyl-2′ deoxyadenosine; ↓ is the site of hydrolysis). The catalytic activity of MutH is stimulated by the complex of the MutS and MutL proteins [9]. MutH homologs are absent in eukaryotes and other types of bacteria that do not use methylation as a marker of the nascent DNA strand. According to the existing hypothesis, the key role in the strand discrimination in these organisms belongs to the DNA polymerase III β-subunit that determines the strand polarity and directly interacts with the MMR proteins [1]. However, this hypothesis is still debated.
MutL is a weak dimeric ATPase from the GHKL family [10]. For a long time, it had been assumed that the major role of MutL is coordination of protein–protein interactions during recognition and removal of one of the DNA strands; hence, MutL was classified as a molecular mediator [11]. However, discovery of the endonuclease activity of eukaryotic MutLα and then of bacterial MutL homologs in organisms that lack the MutH gene led to the suggestion of a key role of MutL in repair initiation [12, 13]. It was found that in these organisms, MutL has a conserved endonuclease motif that is supposedly required for the introduction of a single break in the nascent DNA strand [14]. The mechanism of this process is virtually unexplored; bacterial MMR systems with the endonuclease activity of MutL are considerably less studied than the MMR in E. coli and humans. At the same time, the MMR system of bacteria is particularly interesting because of high functional and species diversity of prokaryotes.
MutL proteins possessing the endonuclease motifs have been purified from only five bacterial species: Pseudomonas aeruginosa, Neisseria gonorrhoeae, Thermus thermophilus, Aquifex aeolicus, and Bacillus subtilis [15-19]. All of them are homodimers, which are simpler and easier to study than their heterodimer analogs in eukaryotes. Therefore, the data obtained for bacterial MutL proteins can be used for the reconstruction of the major stages of the MMR mechanism at the molecular level.
In this work, we developed a method for MutL (rsMutL) purification from Rhodobacter sphaeroides. Interest in this rod-like Gram-negative purple nonsulfur photoheterotrophic α-proteobacteria is due to the variability of its metabolic and life cycles, photosynthetic activity, and ability to survive under diverse environmental conditions [20, 21]. We analyzed the primary structure of rsMutL, predicted its secondary structure, and identified conserved amino acid residues and motifs by comparing amino acid sequences of many rsMutL bacterial analogs with supposed endonuclease activity. We also demonstrated that rsMutL hydrolyzes DNA, and its activity is affected by nonprotein factors such as divalent metal cations and ATP. We compared our results with the data of a few studies of the mechanism of action of bacterial MutL proteins with endonuclease activity.
MATERIALS AND METHODS
Bioinformatics analysis of the rsMutL sequence. The sequence of MutL protein from R. sphaeroides strain 2.4.1 was obtained from the UniProt database (accession number Q3IYV3). The sequence of MutL from E. coli strain K12 (ecMutL; UniProt accession number P06722) was also used for searching for MutL homologs. We searched 4379 representative bacterial and archaeal genomes (representative microbial genomes sample in the Refseq database, NCBI) for rsMutL and ecMutL homologs using the TBLASN program (e-value, 0.001; word length, 3).
The sequences were aligned with the Muscle program using default settings. All rsMutL and ecMutL homologs were divided into three groups based on the presence of the endonuclease motif XII (Table 1). To distinguish groups among all full-size MutL homologs, we chose the rsMutL fragment 449-462 (14 a.a.) that included motif XII. All the obtained sequence fragments were clustered according to the percentage of functionally similar amino acid residues using the Protdist program from the PHYLIP software package. The largest cluster included fragments from proteins with proven endonuclease activity. Proteins of this cluster were assigned to group 1 with presumable endonuclease activity. Proteins that lacked sequences reliably similar to motif XII and, therefore, had no endonuclease activity, were assigned to group 2. Group 3 included proteins whose fragments had low but significant similarity to motif XII. In most of these proteins, at least one of the essential residues in motif XII [14] was substituted by a functionally different amino acid.
Table 1. Conserved motifs in bacterial MutL
proteins and their functions
* The consensus motifs were identified in [22] by
alignment of 208 amino acid sequences of bacterial MutL proteins;
endonuclease motifs were identified by alignment of 192 sequences. Bold
letters in gray boxes, identical residues; bold letters, conserved
(over 90%) residues. Amino acid residues comprising the consensus
motifs in MutL are underlined.
Then we aligned sequences of rsMutL and ecMutL homologs from groups 1 and 2, respectively, except sequences that lacked the N-terminal fragments (including motif I, at least partially) or C-terminal fragments (including motif XVI, at least partially) (Fig. 1). Only one sequence from each group with 95% identity was retained in the final alignment. Conserved residues were identified with a specially written program.
Fig. 1. Comparison of amino acid sequences of MutL proteins and their homologs from various organisms. Amino acid sequences of ecMutL and human MLH1 lacking the endonuclease activity (group 2) are framed. Numbers of the first amino acid residues in the rows are indicated to the right of the sequences. The numbers of omitted residues in non-conserved fragments are shown in parentheses. Conserved residues in the selected sequences are shown in bold letters on gray background. Secondary structure elements (α-helices and β-strands) were determined based on the structures of the ecMutL N-terminal domain (PDB 1B63) and C-terminal domain of N. gonorrhoeae MutL (PDB 3NCV) and are shown under the rsMutL sequence. Conserved motifs are indicated under the alignment according to Table 1 and [22]. Conserved amino acids found in group 1 (1483 sequences) and group 2 (390 sequences) of prokaryotic MutL proteins with endonuclease activity are shown: X, 99% conserved amino acids; +, 95% or more conserved amino acids; –, 90% or more conserved amino acids. Functionally conserved positions K/R, D/E, F/Y, and A/G are indicated with the letter corresponding to the consensus residue; H denotes the MILVFY group of hydrophobic residues. Capital letter, residue is present in 95% or more sequences; small letter, residue is present in 90-95% sequences.

Supplemental materials can be found on the website http://mouse.belozersky.msu.ru/NPIDB/MutL/ and include:
1) Alignment 1 of 1483 amino acid sequences of proteins with predicted endonuclease activity and ecMutL sequence (for comparison) in the FASTA format. A subgroup of 93 sequences lacking endonuclease motifs XIV and XV occupies the lowest rows in the alignment.
2) Alignment 2 of 309 amino acid sequences of proteins presumably lacking endonuclease activity and rsMutL sequence (for comparison) in the FASTA format.
3) The names and taxonomic groups of bacteria and archaea whose sequences were used for alignment.
Construction of plasmids containing the rsMutL gene. The plasmid encoding rsMutL with an additional N-terminal sequence (36 a.a.) including the His6 tag (His6-linker-rsMutL) was obtained as described in [16]. The rsMutL gene was amplified by PCR using R. sphaeroides (strain 2.4.1.) genomic DNA as a template. DNA was isolated from R. sphaeroides cells by the alkaline lysis method [27]. The primers 5′-AAGGGGCCATATGGGAAGCCGGTGGCGGAG-3′ and 5′-AAGGGGAAGACGAGATCTTCATCGCCGGCCGAAGAGCC-3′ contained the recognition sites of BglII and NdeI restriction endonucleases, respectively. The resulting PCR product was treated with BglII/NdeI and cloned into the pET15b plasmid vector that contained the sequence encoding an additional 36-a.a. fragment including the His6-tag.
Plasmid constructs for expression of rsMutL tagged with His6 directly at the N- or C-ends (His6-rsMutL and rsMutL-His6, respectively) were obtained using the pVax1-MutL plasmid (derivative of the pVax1 plasmid) that contained the rsMutL gene optimized for expression in E. coli (Evrogen, Russia). DNA sequences coding for His6-rsMutL and rsMutL-His6 were cloned into the pET-45b(+) (Novagen, Germany) and pQE13 (Qiagen, Germany) vectors by the BamHI/AgeI or BamHI/Kpn2I sites, respectively. Cloning was verified by restriction analysis and sequencing.
Purification of rsMutL protein. Escherichia coli BL21(DE3) cells were transformed with the His6-linker-rsMutL and His6-rsMutL plasmids, and M15(pREP4) cells were transformed with the rsMutL-His6 by the heat-shock method. The cells were grown in 3 liters of LB medium containing 100 μg/ml ampicillin on a shaker at 37°C to the optical density A600 = 0.5. Protein expression was induced by adding 0.1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG), and the cells were incubated for another 16-18 h at 18°C. The cells were collected by centrifugation; the pellet was frozen and stored at –20°C. Protein expression was monitored by withdrawing aliquots before and after induction and analyzing them by electrophoresis in 8% SDS-polyacrylamide gel. The gels were stained with PageBlue™ solution (Thermo Fisher Scientific, USA) containing Coomassie Brilliant Blue G-250.
Frozen cells were thawed on ice for 30 min, resuspended in buffer A (20 mM Tris-HCl, pH 7.9, 1 M NaCl, 1 mM 1,4-dithiotreitol (DTT), 5% glycerol (v/v), 0.05% Tween 20 (v/v), and 1 mM phenylmethylsulfonyl fluoride (PMSF)) containing 5 mM imidazole, and disintegrated by sonication. The obtained cell lysate was centrifuged at 18,000 rpm (JA-10 rotor, fixed angel; Beckman, USA) for 30 min at 4°C to remove the debris. The supernatant was added to 1 ml of Ni-NTA-agarose (Thermo Fisher Scientific) and incubated for 1 h at 4°C with gentle mixing. The sorbent with the bound protein was separated by centrifugation at 800 rpm (F-35-6-30 rotor, fixed angel; Eppendorf, Germany) for 5 min at 4°C. The supernatant was removed except the last 10 ml that were used for the resuspension of the sorbent and its transfer to a 15-ml column at 4°C. After sedimentation of the sorbent, the liquid was allowed to drain, and the sorbent was washed with 15 ml of buffer A containing 60 mM imidazole to remove unbound proteins. The rsMutL was eluted from the column with 500 μl of buffer A containing 500 mM imidazole. The collected fraction was loaded on the column again, and the whole procedure was repeated 2-3 times. Final elution was performed with 500 μl of fresh buffer; DTT was added to the eluate to the final concentration of 10 mM.
rsMutL was further purified by size-exclusion chromatography on a 10/300-mm ENrich SEC 650 column (Bio-Rad, USA). The column was equilibrated with 10 mM HEPES-KOH buffer (pH 8.0) containing 300 mM KCl, 1 mM DTT, 5% glycerol (v/v), and 0.05% Tween 20 (v/v); the same buffer was also used as an eluent. The protein was loaded on the column in a volume of 1 ml and eluted at a rate of 1 ml/min. Elution was monitored at 280 nm; protein-containing fraction were collected and analyzed by electrophoresis in 8% SDS-polyacrylamide gel. Protein concentration in the fractions was determined spectrophotometrically with a NanoDrop ND-1000 spectrophotometer (PeqLab, USA). The collected fractions were concentrated using Amicon Ultra centrifuge concentrators (Merck, Germany) with 10-kDa cut-off membrane. After concentration, protein fractions were aliquoted (5-10 μl), flash-frozen in liquid nitrogen, and thawed immediately before use.
Hydrolysis of plasmid DNA by rsMutL. The pUC-MMR plasmid (10-20 nM) used earlier for studying MMR in E. coli [28] was incubated with rsMutL in 10 mM HEPES-KOH (pH 8.0) containing 100 mM KCl and 5 mM divalent metal salt at 37°C for 0-90 min. The reaction was terminated by adding 50 mM EDTA and 10 U of proteinase K with subsequent incubation of the mixture for 30 min at 37°C. To develop the optimal reaction conditions, the rsMutL concentration was varied from 0.1 to 2 µM. The endonuclease activity of rsMutL was assayed in the presence of MgCl2, MnCl2, CaCl2, CoCl2, NiCl2, CdCl2, or ZnCl2 (5 mM). The ATP concentration varied from 0 to 5 mM. The reaction products were analyzed by electrophoresis in 1% agarose gel containing 0.5 µg/ml bromide in TAE buffer (40 mM Tris-acetate, pH 7.5, 1 mM EDTA). The gels were photographed, and the images were processed with the Image Lab software (Bio-Rad). In the control experiments, the pUC-MMR plasmid (20 nM) was incubated with BamHI or nicking endonuclease Bpu10I for 60 min at 37°C in 10 μl of 10 mM Tris-HCl (pH 8.0) containing 5 mM MgCl2, 100 mM KCl, 0.02% Triton X-100, and 0.1 mg/ml BSA (for BamHI) or 10 mM Tris-HCl (pH 8.5) containing 10 mM MgCl2, 100 mM KCl, and 0.1 mg/ml BSA (for Bpu10I).
RESULTS AND DISCUSSION
Characterization of the primary and secondary structures of rsMutL. The sequence of MutL from R. sphaeroides strain 2.4.1 was obtained from the UniProt database (Q3IYV3). rsMutL consists of 616 a.a. and has a molecular mass of 65.8 kDa. Its isoelectric point is 6.51 as determined with the Protein Calculator v. 3.4 software.
Proteins of the MutL family are composed of N- and C-terminal domains connected by a variable linker [1]. According to the Pfam database, the N- and C-terminal domains of rsMutL include a.a. 1-343 and 427-616, respectively. It was found that MutL sequence contains 16 conserved motifs [22]. To identify the same motifs in rsMutL, we aligned the rsMutL sequence with nine sequences of other MutL proteins. For some of these proteins, positions of the conserved motifs have been already determined (Fig. 1). We found that rsMutL contained all 16 motifs (Fig. 1 and Table 1). All of them (except VI) coincided with the consensus sequences of the earlier identified motifs [22]. Seven conserved motifs (I, II, III, V, VII, IX, and X) presumably form the ATP-binding site in rsMutL (Table 1). The sequences corresponding to motifs VIII (N-terminal domain) and XIII (C-terminal domain) might be involved in the interaction with the β-clamp.
The motif VI localized in the N-terminal domain is essential for the MutS protein binding. It is not conserved enough to predict which residues in this motif are involved in the interaction with MutS. Endonuclease activity was experimentally demonstrated for MutL proteins from bacteria belonging to five different classes: β-proteobacteria (N. gonorrhoeae), Bacilli (B. subtilis), γ-proteobacteria (P. aeruginosa), Deinococci (T. thermophilus), and Aquificae (A. aeolicus). The sequences of MutL proteins from these bacterial species were included in the alignment in Fig. 1. All of them possess a full set of endonuclease motifs (XI, XII, XIV-XVI) that form the catalytic site for DNA hydrolysis (Table 1). These motifs are absent in MutL proteins lacking endonuclease activity (E. coli MutL protein and human MLH1 in the alignment).
Previous analysis of 208 MutL sequences revealed several conserved motifs in the protein primary structure [22]. To estimate with higher reliability how conserved and, therefore, functionally significant these motifs are, we analyzed a wider range of bacterial and archaeal MutL homologs. Initially, this sample included 2544 sequences: 1483 of them we predicted to have the endonuclease activity (alignment 1); 309 sequences presumably lacked this activity (alignment 2). The sequences were selected based on the presence/absence of the endonuclease motif XII, which has the highest informational content among all endonuclease motifs.
We compared the number of conserved or functionally conserved (by 90% or higher) positions in the MutL sequences. The N-terminal domains of MutL proteins from groups 1 and 2 contained 71 and 82% of conserved residues, respectively; 67% of these residues overlapped. The content of conserved residues in the C-terminal domains of proteins from groups 1 and 2 comprised 50 and 34%, respectively; 24% of these residues overlapped. The higher content of conserved residues in the C-terminal domain in proteins with presumed endonuclease activity confirmed our expectations and indirectly proved the validity of MutL protein classification into groups – it is known that enzyme active sites, residues in close vicinity to the active sites, and ligand- and ion-binding sites are generally more conserved than the sequence fragments at the protein globule surface, including those involved in protein–protein interactions [29].
The results of analysis of conserved residues in motifs XI, XII, XIV, XV, and XVI in proteins with presumed endonuclease activity (group 1) are shown in Table 2. Because the analyzed proteins were assigned to this group based solely on the presence of motif XII (Table 1), we used consensus sequences from Table 1 to check all group 1 proteins for the presence of other endonuclease motifs. Note that the consensus sequences were deduced based on the most conserved amino acids and did not guarantee identification of all the motifs. Thus, the motif XII consensus sequence (Table 1) was found in 81% sequences (1203 out of 1483) of the analyzed group. Because of this, we lowered the criteria for the consensus sequences by including in them only residues whose substitution would significantly decrease the MutL endonuclease activity [14]. The [D/E]XHXXXE consensus was found in the corresponding region in 93.7% aligned sequences (1390 out of 1483); motif XIV (with Cys in the corresponding position) was found in 100% of these sequences (1390 out of 1390). Motif XV was found in 1373 out of 1390 sequences; functionally important Cys [14] of this motif was observed in 99.7% sequences (1386 out of 1390). The motif XI consensus was found in 89.4% sequences (1243 out of 1390).
Table 2. Conserved amino acid residues in
endonuclease motifs XI, XII, XIV, XV, and
XVI
Interestingly, none of 93 sequences without “weakened” motif XII consensus contained consensus sequences for motifs XI, XIV, and XV. At the same time, most of these sequences, as well as 1390 sequences with the “weakened” motif XII consensus, contained motif XVI. This corresponds well with the results of Kosinski et al. [14], who demonstrated strong correlation between the presence of all five motifs in the MutL sequence. They suggested that such correlation serves as evidence of a common functional activity that involves all these motifs. However, there exist MutL proteins that lack some of the endonuclease motifs [22].
Therefore, according to our data, group 1 MutL proteins containing motif XII in the C-terminal domain could be divided into two subgroups. The first includes 1390 proteins that contain all five endonuclease motifs; moreover, their motif XII contains all functionally characterized amino acid residues. These proteins possess endonuclease activity with a high degree of probability. The second subgroup includes 93 sequences that contain only motif XII, in which one of the functionally active residues is replaced with a residue with different properties. Proteins of this subgroup lack all other endonuclease motifs, and their properties are of particular interest.
The secondary structure of rsMutL was predicted based on the alignment of its amino acid sequence with sequences of MutL proteins with known spatial structures. To predict the secondary structure of the rsMutL N-terminal domain, we used the X-ray data obtained for ecMutL (PDB 1B63, 1NHI, 1BKN), PMS1 from Saccharomyces cerevisiae (PDB 3H4L), human PMS2 (PDB 1H7S), and human MLH1 (PDB 4P7A). To predict the secondary structure of the C-terminal domain, we used the structural data obtained for MutL from B. subtilis (PDB 3GAB, 3KDG, 3KDK), MutL from A. aeolicus (PDB 5B42), MutL from N. gonorrhoeae (PDB 3NCV), and MLH1/PMS1 heterodimer from S. cerevisiae (PDB 4E4W). In most cases, the elements of the secondary structure in these sequences coincided. After comparing the sequences of rsMutL and its homologs using the Muscle software, we identified α-helical and β-strand regions in the MutL structure (indicated under rsMutL amino acid sequence in Fig. 1).
We found that rsMutL is most evolutionarily close to its homologs from N. gonorrhoeae (40% identity) and P. aeruginosa (44% identity). Both these microorganisms are proteobacteria that belong to different classes. MutL from N. gonorrhoeae (β-proteobacteria) is a homodimer with ATPase and endonuclease activities [30]. MutL from P. aeruginosa displays similar properties, even though P. aeruginosa belongs to the γ-proteobacteria and does not possess MutH homologs [31]. We concluded that rsMutL of the R. sphaeroides DNA repair system should exhibit the DNA-hydrolyzing activity.
Purification of rsMutL. We amplified the mutL gene by PCR using R. sphaeroides genomic DNA as a template and cloned the resulting fragment into the pET15b vector containing the nucleotide sequence coding for the additional 36 a.a. including His6 tag. The resulting His6-linker-rsMutL plasmid was used for transformation of E. coli cells. The cells were grown in the presence of 100 μg/ml ampicillin, and expression of the recombinant protein was induced with IPTG. The aliquots of cells before and after induction were analyzed by Laemmli gel electrophoresis [32]. Because the recombinant protein contained an additional N-terminal sequence (MGSSHHHHHHSSGLVPRGSHMGSRWRSDSDSAKAGG) that included the His6-tag separated from the rsMutL sequence by 26 a.a., recombinant rsMutL could be purified by affinity chromatography. It is commonly believed that the presence of such additional sequence would not affect the properties of the modified protein, as it has been confirmed by protein crystallography data [33].
rsMutL was first purified by the method described for MutL from N. gonorrhoeae [16] that included cell lysis by sonication, centrifugation of the lysate, and chromatography of the resultant supernatant on Ni-NTA-agarose. However, this procedure yielded only small amounts of homogenous rsMutL protein. The main problems were low efficiency of rsMutL binding to the sorbent, contamination of protein preparations with DNA, and low solubility of rsMutL. To improve rsMutL binding to Ni-NTA-agarose, we created two new plasmids containing optimized rsMutL gene. One of them coded for the rsMutL protein with the His6-tag immediately at the N-terminus (His6-rsMutL), the other with the His6-tag immediately at the C-terminus (rsMutL-His6). However, modifications of the mutL gene codon content for expression in E. coli and changes in the position of the His6 tag failed to increase the protein yield (data not shown). Therefore, we modified the purification procedure. To promote correct protein folding, the concentration of IPTG was decreased to 0.1 mM, and protein expression was induced at decreased temperature (18°C) for 16-18 h. All solutions for rsMutL purification contained 0.05% Tween 20 to increase protein solubility and 1 M NaCl to prevent DNA binding to rsMutL.
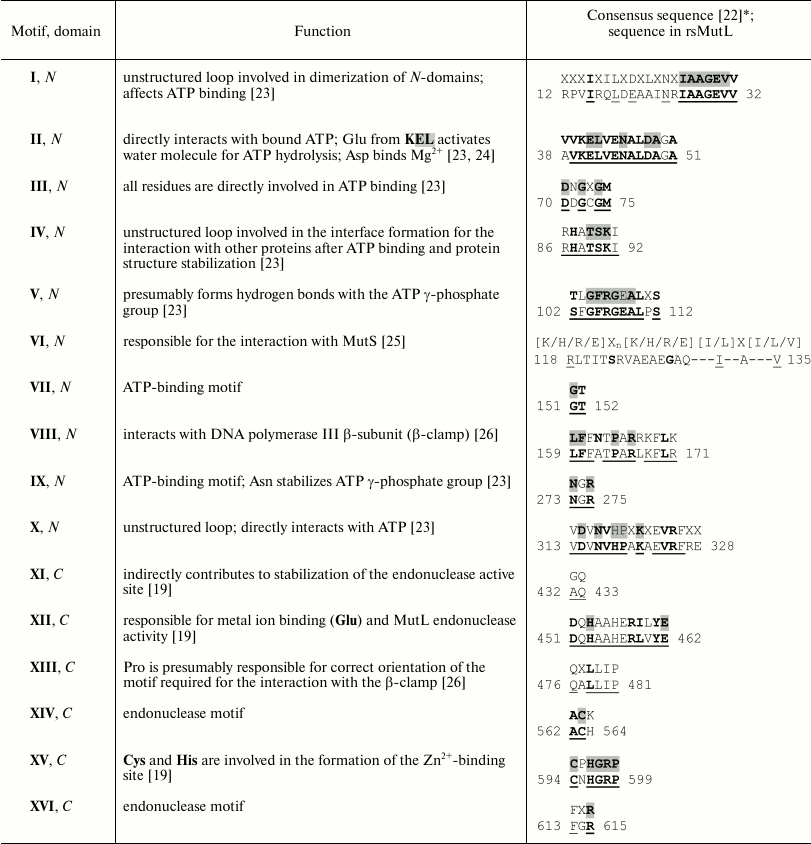
rsMutL purified by the metal-chelating chromatography was separated from the products of its proteolysis and other proteins by size-exclusion chromatography on an ENrich SEC 650 column (10 × 300 mm; Bio-Rad) using an AKTA Explorer system (GE Healthcare Life Sciences, USA). Protein-containing fractions (elution volume, 9-12 ml; first peak) were collected, analyzed by Laemmli gel electrophoresis (Fig. 2), and concentrated. The yield of rsMutL was 0.2 mg (concentration, 8 μM).
Fig. 2. rsMutL purification. a) Chromatographic separation of rsMutL from proteins with lower molecular masses. b) Analysis of fractions after size-exclusion chromatography: 1) MutL protein before chromatography; 2-5) fractions eluted with 9-10 ml; 6) 10-180 kDa molecular mass markers (Thermo Fisher Scientific).
DNA-hydrolyzing activity of rsMutL. To assay the rsMutL endonuclease activity, we used the pUC-MMR plasmid DNA. This plasmid was chosen because it can imitate the in vivo conditions, since the R. sphaeroides genome consists of two circular chromosomes and five plasmids [34]. It is also known that MutL proteins from B. subtilis, N. gonorrhoeae, A. aeolicus, T. thermophilus, and P. aeruginosa do not discriminate mismatches and hydrolyze canonical DNA in the absence of other MMR proteins [35]. All these MutL proteins display the maximum DNA-hydrolyzing activity in the presence of Mn2+ in the reaction mixtures [1].
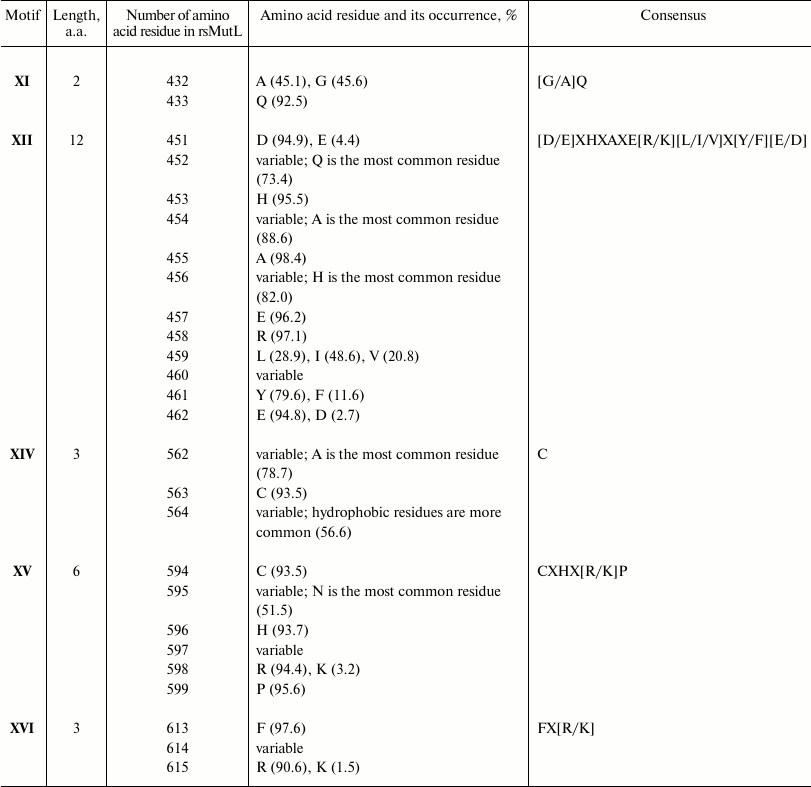
The supercoiled pUC-MMR plasmid (10 or 20 nM) was incubated with rsMutL at 37°C in the presence or absence of MnCl2. The concentration of rsMutL varied from 0 to 2 µM. The efficiency of DNA hydrolysis was estimated from the amount of formed nicked DNA (plasmid with a single-stranded break). The reaction mixture was analyzed by electrophoresis in 1% agarose gel containing ethidium bromide. pUC-MMR treated with the nicking endonuclease Bpu10I (nicked form) and restriction endonuclease BamHI (linearized form) was used as control. The nicked plasmid DNA has a lower mobility in the gel than the supercoiled DNA, while the mobility of the linearized form is between those of the supercoiled and nicked plasmid DNA (Fig. 3).
Fig. 3. Hydrolysis of the pUC-MMR plasmid by rsMutL at 37°C for 60 min. a) Electrophoresis in 1% agarose gel containing ethidium bromide: 1) DNA ladder (kb); 2) intact pUC-MMR; 3) pUC-MMR hydrolyzed by rsMutL in the absence of divalent metal cations; 4) pUC-MMR hydrolyzed by rsMutL in the presence of Mn2+; 5) pUC-MMR hydrolyzed by restriction endonuclease BamHI; 6) pUC-MMR hydrolyzed by nicking endonuclease Bpu10I; sc, supercoiled DNA; nc, nicked DNA; ln, linearized DNA. b) Dependence of the pUC-MMR (20 nM) hydrolysis on the rsMutL concentration (0-2 μM) in the presence of 5 mM MnCl2.
rsMutL did not hydrolyze pUC-MMR in the absence of divalent metal cations (Fig. 3). The presence of Mn2+ in the reaction mixture resulted in the formation of the nicked plasmid DNA. The efficiency of pUC-MMR hydrolysis increased with change in protein concentration from 0 to 0.5 μM and reached 50% at the rsMutL concentration of 1 μM. Further increase in the protein concentration did not influence on the yield of the hydrolyzed DNA. Also, the gel contained trace amounts of linearized DNA. Therefore, we demonstrated for the first time that rsMutL is an endonuclease capable of introducing single-single breaks in DNA.
Effect of divalent metal cations on DNA hydrolysis by rsMutL. The published data on the effect of divalent metals on the activity of MutL proteins from various organisms are contradictory [15-19] (Table 3).
Table 3. Effect of metal ions and ATP on the
endonuclease activity of MutL proteins from different
microorganisms
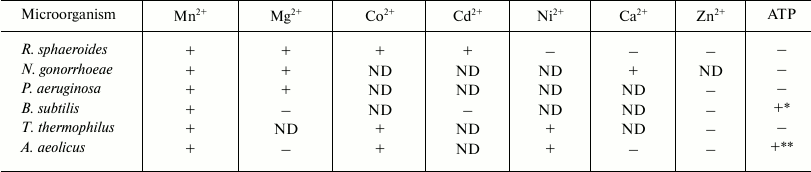
Note: +, stimulation of MutL endonuclease activity; −, no
effect or inhibition (in the case of ATP) of MutL endonuclease
activity; ND, not determined.
* Stimulation of endonuclease activity at 0.5 mM ATP; inhibition of
endonuclease activity at 5 mM ATP [19].
** Effect of ATP on the MutL endonuclease activity depended on the
concentrations of MutL, Mn2+, and ATP; stimulation was
observed at the MutL concentration of 0.16-100 nM, 1 mM ATP,
and 1 mM Mn2+ [45]; inhibition was
observed at 100 nM MutL, 5 mM ATP, and 5 mM
Mn2+ [18].
We studied the efficiency of the pUC-MMR plasmid hydrolysis by rsMutL within 60 min in the presence of divalent cations that presumably modulate the endonuclease activity: Mg2+, Mn2+, Ca2+, Co2+, Ni2+, Cd2+, Zn2+ (Fig. 4). The efficiency of DNA cleavage decreased in the order Mn2+ > Co2+ > Mg2+ > Cd2+, while Ni2+, Ca2+, and Zn2+ produced no effect on the rsMutL endonuclease activity.
Fig. 4. Efficiency of pUC-MMR plasmid (20 nM) hydrolysis by rsMutL (1 μM) in the presence of divalent metal cations (5 mM) at 37°C for 60 min.
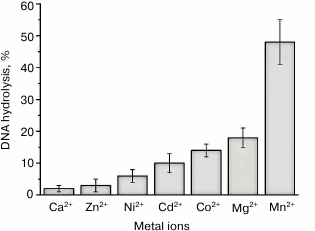
Therefore, rsMutL most efficiently hydrolyzed plasmid DNA in the presence of MnCl2, which correlates well with the data of other studies on DNA hydrolysis by MutL proteins from bacterial and human cells [1, 19]. It is known that many DNA polymerases use Mn2+ as a cofactor in DNA synthesis in vitro. Divalent manganese ion increases the enzyme activity, but it reduces fidelity and processivity of the polymerase [36, 37], which results in misincorporation of nucleotides in the DNA. Note that Co2+, like Mn2+ (but unlike other Me2+), increase the efficiency of DNA polymerases, although to a lesser extent, and promote incorporation of incorrect nucleotides into DNA [38].
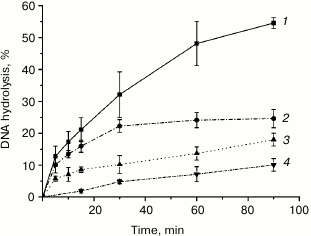
Unlike Mn2+, Mg2+ is essential for the function of MutS, the first protein in the mismatch repair system, and formation of the MutS–MutL complex [39-42]. We investigated the kinetics of plasmid DNA cleavage by rsMutL in the presence of MgCl2 and MnCl2 (Fig. 5) and found that within the time interval of 0-90 min, Mn2+ was a more efficient cofactor in the DNA hydrolysis reaction than Mg2+. Since both MutL and MutS lose their activity when incubated at 37°C (data not shown), we limited the reaction time to 90 min. Note, DNA hydrolysis in the presence of either Mn2+ and Mg2+ proceeded with preferential formation of nicked DNA; however, trace amounts of linearized DNA were also formed, as follows from the presence of the corresponding low-intensity zones in the agarose gel (Fig. 3). Similar results were observed for MutL proteins from other bacteria [15-19].
Fig. 5. Kinetics of pUC-MMR plasmid (20 nM) hydrolysis by rsMutL (1 μM) in the presence of (1) 5 mM MnCl2, (2) 5 mM MnCl2 and 0.5 mM ATP, (3) 5 mM MgCl2, and (4) 5 mM MgCl2 and 0.5 mM ATP at 37°C.
As expected, Ca2+ did not activate DNA hydrolysis by rsMutL. For many endonucleases, Ca2+ promotes formation of the enzyme–substrate complex but blocks the hydrolytic activity of the enzyme [43, 44].
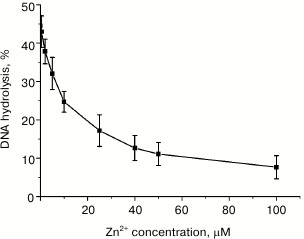
Divalent zinc ions play an important yet poorly understood role in MutL function. X-ray analysis showed [19] that MutL from B. subtilis coordinates Zn2+ not only in its active site, but also in a special Zn2+-binding domain. This coordination is provided by the side groups of Glu468, Cys604, and His606 residues, all of which are conserved in the MutL proteins with endonuclease activity. According to the alignment of the C-terminal domains from B. subtilis MutL and rsMutL (Fig. 1), these residues correspond to Glu457, Cys594, and His596 in rsMutL. In B. subtilis MutL, Zn2+ binding ensures convergence of amino acid residues in the enzyme active site. Interestingly, Zn2+ by itself does not stimulate hydrolysis of the supercoiled plasmid DNA by B. subtilis MutL; however, the amount of the nicked and linearized DNA formed in the presence of equimolar concentrations of Zn2+ and Mn2+ was two times higher than in the presence of Mn2+ alone under the same conditions [19]. It was suggested that Zn2+ performs a structural function, similar to iron ions in the IdeR (iron-dependent repressors) proteins, whose primary and secondary structures are very similar to the structure of the C-terminal domains in MutL proteins with endonuclease activity [19]. On the other hand, Zn2+ activates MutL from A. aeolicus [18]. We found that Zn2+ did not promote DNA hydrolysis by rsMutL. On the contrary, addition of increasing Zn2+ concentrations (0-100 µM) to the reaction mixture containing 5 mM Mn2+ inhibited DNA hydrolysis irrespectively of the Zn2+ concentration added (Fig. 6). Similar results were obtained for MutL for P. aeruginosa [15]. This suggests a high affinity of Zn2+ for the Me2+-binding sites in rsMutL and the ability of Zn2+ to replace Mn2+ in them. Apparently, Zn2+ is not involved in DNA hydrolysis by rsMutL.
Fig. 6. Dependence of pUC-MMR plasmid (10 nM) hydrolysis by rsMutL (500 nM) on the concentration of Zn2+ (0-100 μM) in the presence of 5 mM Mn2+ in the reaction mixture (37°C, 60 min).
Therefore, divalent metal cations significantly affect the ability of rsMutL to introduce single-stranded breaks in DNA and its specificity.
Effect of ATP on MutL endonuclease activity. As mentioned above, the presence of characteristic conserved motifs in the rsMutL sequence indicates that rsMutL belongs to the family of GHKL ATPases. ATP binding and hydrolysis might cause significant conformational changes in the protein molecule. Indeed, ATP was found to affect the endonuclease activity of MutL, although the results of these studies are quite contradictory (Table 3). ATP strongly activates DNA hydrolysis by human MutLα. Low ATP concentrations stimulate the activity of MutL from B. subtilis, while high ATP concentrations inhibit the enzyme [19]. Similarly, ATP can either stimulate [45] or inhibit [18] MutL from A. aeolicus depending on the ATP concentration, enzyme concentration, and the presence of Mg2+. ATP inhibits DNA hydrolysis by MutL proteins from N. gonorrhoeae, T. thermophiles, and P. aeruginosa [15-17].
In this work, we analyzed the effect of ATP on the rsMutL activity. In most studies on DNA hydrolysis by MutL, the concentration of ATP is 0.5 mM [18, 30]. We found that addition of 0.5 mM ATP to the reaction mixture containing Mn2+ or Mg2+ decreased the extent of plasmid DNA hydrolysis approximately two times when the reaction was carried out for 5 to 90 min (Fig. 5), which correlates well with the data obtained for MutL enzymes from N. gonorrhoeae, A. aeolicus, T. thermophiles, and P. aeruginosa. Inhibition of MutL endonuclease activity by ATP might be important for cell functioning. The intracellular concentration of ATP is approximately 3-5 mM [17], which is high enough to considerably decrease the ability of MutL to hydrolyze DNA, thereby preventing introduction of undesirable breaks in the canonical DNA.
In conclusion, we purified for the first time the MutL protein, a component of the mismatch repair system from R. sphaeroides, and demonstrated its endonuclease activity. Comparison of rsMutL with its analogs from A. aeolicus, B. subtilis, N. gonorrhoeae, T. thermophilus, and P. aeruginosa showed that all these proteins hydrolyze DNA in the presence of Mn2+ (Table 3). However, only rsMutL and MutL from N. gonorrhoeae introduce single-stranded breaks in DNA in the presence of Mg2+ and Co2+. Divalent zinc ions do not stimulate DNA cleavage by any of the MutL proteins studied, which indicates the necessity for ion coordination for the active protein structure formation. ATP significantly inhibits the DNA-hydrolyzing activity of rsMutL and all other enzymes presented in Table 3 (except MutL from B. subtilis). Analysis of MutL amino acid sequences revealed functionally important conserved motifs (Tables 1 and 2) in these proteins. Considering all these data, MutL from R. sphaeroides mostly resembles MutL proteins from N. gonorrhoeae and P. aeruginosa in its primary structure and biochemical properties.
Acknowledgments
This work was supported by the Russian Foundation for Fundamental Research (project No. 16-04-00575) and the Russian Science Foundation (project No. 16-14-10319; Bioinformatic analysis of MutL sequences from various organisms). This work was supported in part by Lomonosov Moscow State University Development Program (PNR 5.13).
REFERENCES
1.Guarne, A. (2012) The functions of MutL in mismatch
repair: the power of multitasking, Prog. Mol. Biol. Transl.
Sci., 110, 41-70.
2.Iyer, R. R., Pluciennik, A., Burdett, V., and
Modrich, P. L. (2006) DNA mismatch repair: functions and mechanisms,
Chem. Rev., 106, 302-323.
3.Jiricny, J. (2013) Postreplicative mismatch repair,
Cold Spring Harb. Perspect. Biol., 5, a012633.
4.Lynch, H. T., Lynch, P. M., Lanspa, S. J., Snyder,
C. L., Lynch, J. F., and Boland, C. R. (2009) Review of the Lynch
syndrome: history, molecular genetics, screening, differential
diagnosis, and medicolegal ramifications, Clin. Genet.,
76, 1-18.
5.Michailidi, C., Papavassiliou, A. G., and Troungos,
C. (2012) DNA repair mechanisms in colorectal carcinogenesis, Curr.
Mol. Med., 12, 237-246.
6.Lahue, R. S., and Modrich, P. (1988)
Methyl-directed DNA mismatch repair in Escherichia coli,
Mutat. Res., 198, 37-43.
7.Längle-Rouault, F., Maenhaut-Michel, G., and
Radman, M. (1987) GATC sequences, DNA nicks and the MutH function in
Escherichia coli mismatch repair, EMBO J., 6,
1121-1127.
8.Modrich, P. (1989) Methyl-directed DNA mismatch
correction, J. Biol. Chem., 264, 6597-6600.
9.Perevozchikova, S. A., Romanova, E. A., Oretskaya,
T. S., Friedhoff, P., and Kubareva, E. A. (2013) Modern concepts of the
structural and functional organization of the system for the DNA
noncanonical nucleotide pair repair, Acta Naturae, 5,
27-44.
10.Hall, M. C., Shcherbakova, P. V., and Kunkel, T.
A. (2002) Differential ATP binding and intrinsic ATP hydrolysis by
amino-terminal domains of the yeast Mlh1 and Pms1 proteins, J. Biol.
Chem., 277, 3673-3679.
11.Sancar, A., and Hearst, J. E. (1993) Molecular
matchmakers, Science, 259, 1415-1420.
12.Kadyrov, F. A., Dzantiev, L., Constantin, N., and
Modrich, P. (2006) Endonucleolytic function of MutLα in human
mismatch repair, Cell, 126, 297-308.
13.Kadyrov, F. A., Holmes, S. F., Arana, M. E.,
Lukianova, O. A., O'Donnell, M., Kunkel, T. A., and Modrich, P. (2007)
Saccharomyces cerevisiae MutLα is a mismatch repair
endonuclease, J. Biol. Chem., 282, 37181-37190.
14.Kosinski, J., Plotz, G., Guarne, A., Bujnicki, J.
M., and Friedhoff, P. (2008) The PMS2 subunit of human MutLα
contains a metal ion binding domain of the iron-dependent repressor
protein family, J. Mol. Biol., 382, 610-627.
15.Correa, E. M. E., Martina, M. A., De Tullio, L.,
Argaran, C. E., and Barra, J. L. (2011) Some amino acids of the
Pseudomonas aeruginosa MutL D(Q/M)HA(X)(2)E(X)(4)E conserved
motif are essential for the in vivo function of the protein but
not for the in vitro endonuclease activity, DNA Repair,
10, 1106-1113.
16.Duppatla, V., Bodda, C., Urbanke, C., Friedhoff,
P., and Rao, D. N. (2009) The carboxy-terminal domain is sufficient for
endonuclease activity of Neisseria gonorrhoeae MutL, Biochem.
J., 423, 265-277.
17.Fukui, K., Nishida, M., Nakagawa, N., Masui, R.,
and Kuramitsu, S. (2008) Bound nucleotide controls the endonuclease
activity of mismatch repair enzyme MutL, J. Biol. Chem.,
283, 12136-12145.
18.Iino, H., Kim, K., Shimada, A., Masui, R.,
Kuramitsu, S., and Fukui, K. (2011) Characterization of C- and
N-terminal domains of Aquifex aeolicus MutL endonuclease:
N-terminal domain stimulates the endonuclease activity of C-terminal
domain in a zinc-dependent manner, Biosci. Rep., 31,
309-322.
19.Pillon, M. C., Lorenowicz, J. J., Uckelmann, M.,
Klocko, A. D., Mitchell, R. R., Chung, Y. S., Modrich, P., Walker, G.
C., Simmons, L. A., Friedhoff, P., and Guarne, A. (2010) Structure of
the endonuclease domain of MutL: unlicensed to cut, Mol. Cell,
39, 145-151.
20.Bijlsma, J. J., and Groisman, E. A. (2003) Making
informed decisions: regulatory interactions between two-component
systems, Trends Microbiol., 11, 359-366.
21.Capra, E. J., and Laub, M. T. (2012) Evolution of
two-component signal transduction systems, Annu. Rev.
Microbiol., 66, 325-347.
22.Banasik, M., and Sachadyn, P. (2014) Conserved
motifs of MutL proteins, Mutat. Res., 769, 69-79.
23.Ban, C., and Yang, W. (1998) Crystal structure
and ATPase activity of MutL: implications for DNA repair and
mutagenesis, Cell, 95, 541-552.
24.Dutta, R., and Inouye, M. (2000) GHKL, an
emergent ATPase/kinase superfamily, Trends Biochem. Sci.,
25, 24-28.
25.Plotz, G., Raedle, J., Brieger, A., Trojan, J.,
and Zeuzem, S. (2003) N-terminus of hMLH1 confers interaction of
hMutLα and hMutLβ with hMutSα, Nucleic Acids
Res., 31, 3217-3226.
26.Pillon, M. C., Miller, J. H., and Guarne, A.
(2011) The endonuclease domain of MutL interacts with the beta sliding
clamp, DNA Repair (Amst.), 10, 87-93.
27.Davis, R. W., Botstein, D., and Roth, J. R.
(1980) Advanced Bacterial Genetics: a Manual for Genetic
Engineering, Cold Spring Harbor, Cold Spring Harbor Laboratory,
N.Y.
28.Winkler, I., Marx, A. D., Lariviere, D., Heinze,
R. J., Cristovao, M., Reumer, A., Curth, U., Sixma, T. K., and
Friedhoff, P. (2011) Chemical trapping of the dynamic MutS–MutL
complex formed in DNA mismatch repair in Escherichia coli, J.
Biol. Chem., 286, 17326-17337.
29.Echave, J., Spielman, S. J., and Wilke, C. O.
(2016) Causes of evolutionary rate variation among protein sites,
Nat. Rev. Genet., 17, 109-121.
30.Namadurai, S., Jain, D., Kulkarni, D. S., Tabib,
C. R., Friedhoff, P., Rao, D. N., and Nair, D. T. (2010) The C-terminal
domain of the MutL homolog from Neisseria gonorrhoeae forms an
inverted homodimer, PloS One, 5, 13726.
31.Jacquelín, D. K., Filiberti, A.,
Argaraña, C. E., and Barra, J. L. (2005) Pseudomonas
aeruginosa MutL protein functions in Escherichia coli,
Biochem. J., 388, 879-887.
32.Laemmli, U. K. (1970) Cleavage of structural
proteins during the assembly of the head of bacteriophage T4,
Nature, 227, 680-685.
33.Carson, M., Johnson, D. H., McDonald, H.,
Brouillette, C., and Delucas, L. J. (2007) His-tag impact on structure,
Acta Crystallogr. D. Biol. Crystallogr., 63,
295-301.
34.Choudhary, M., Mackenzie, C., Nereng, K. S.,
Sodergren, E., Weinstock, G. M., and Kaplan, S. (1994) Multiple
chromosomes in bacteria: structure and function of chromosome II of
Rhodobacter sphaeroides 2.4.1, J. Bacteriol., 176,
7694-7702.
35.Ban, C., Junop, M., and Yang, W. (1999)
Transformation of MutL by ATP binding and hydrolysis: a switch in DNA
mismatch repair, Cell, 97, 85-97.
36.Lakhin, A. V., Efremova, A. S., Makarova, I. V.,
Grishina, E. E., Shram, S. I., Tarantul, V. Z., and Gening, L. V.
(2013) Effect of Mn(II) on the error-prone DNA polymerase iota activity
in extracts from human normal and tumor cells, Mol. Genet.
Mikrobiol. Virusol., 1, 14-20.
37.Frank, E. G., and Woodgate, R. (2007) Increased
catalytic activity and altered fidelity of human DNA polymerase iota in
the presence of manganese, J. Biol. Chem., 282,
24689-24696.
38.Zakharcheva, K. A., Gening, L. V., Kazachenko, K.
Yu., and Tarantul, V. Z. (2017) Cells resistant to toxic concentrations
of manganese have increased ability to repair DNA, Biochemistry
(Moscow), 82, 101-110.
39.Grilley, M., Welsh, K. M., Su, S. S., and
Modrich, P. (1989) Isolation and characterization of the Escherichia
coli mutL gene product, J. Biol. Chem., 264,
1000-1004.
40.Prolla, T. A., Pang, Q., Alani, E., Kolodner, R.
D., and Liskay, R. M. (1994) MLH1, PMS1, and MSH2 interactions during
the initiation of DNA mismatch repair in yeast, Science,
265, 1091-1093.
41.Drotschmann, K., Aronshtam, A., Fritz, H. J., and
Marinus, M. G. (1998) The Escherichia coli MutL protein
stimulates binding of Vsr and MutS to heteroduplex DNA, Nucleic
Acids Res., 26, 948-953.
42.Acharya, S., Foster, P. L., Brooks, P., and
Fishel, R. (2003) The coordinated functions of the E. coli MutS
and MutL proteins in mismatch repair, Mol. Cell, 12,
233-246.
43.Mordasini, T., Curioni, A., and Andreoni, W.
(2003) Why do divalent metal ions either promote or inhibit enzymatic
reactions? The case of BamHI restriction endonuclease from combined
quantum-classical simulations, J. Biol. Chem., 278,
4381-4384.
44.Pingoud, A., Fuxreiter, M., Pingoud, V., and
Wende, W. (2005) Type II restriction endonucleases: structure and
mechanism, Cell Mol. Life Sci., 62, 685-707.
45.Mauris, J., and Evans, T. C. (2009) Adenosine
triphosphate stimulates Aquifex aeolicus MutL endonuclease
activity, PLoS One, 4, 71-75.