Heterologous Expression and Isolation of Influenza A Virus Nuclear Export Protein NEP
A. O. Golovko1, O. N. Koroleva2*, and V. L. Drutsa3
1Lomonosov Moscow State University, Faculty of Bioengineering and Bioinformatics, 119991 Moscow, Russia; E-mail: nastiagolovko@mail.ru2Lomonosov Moscow State University, Faculty of Chemistry, 119991 Moscow, Russia; E-mail: koroleva@genebee.msu.ru
3Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 11991 Moscow, Russia; E-mail: drutsa@genebee.msu.ru
* To whom correspondence should be addressed.
Received July 19, 2017; Revision received September 5, 2017
Influenza A virus nuclear export protein NEP (NS2, 14.4 kDa) plays a key role in various steps of the virus life cycle. Highly purified protein preparations are required for structural and functional studies. In this study, we designed a series of Escherichia coli plasmid constructs for highly efficient expression of the NEP gene under control of the constitutive trp promoter. An efficient method for extraction of NEP from inclusion bodies based on dodecyl sulfate treatment was developed. Preparations of purified NEP with either N- or C-terminal (His)6-tag were obtained using Ni-NTA agarose affinity chromatography with yield of more than 20 mg per liter of culture. According to CD data, the secondary structure of the proteins matched that of natural NEP. A high propensity of NEP to aggregate over a wide range of conditions was observed.
KEY WORDS: influenza A virus, nuclear export protein (NEP), affinity chromatography, protein aggregationDOI: 10.1134/S0006297917120124
Abbreviations: a.a., amino acid residue; bp, base pairs; IPTG, isopropyl-β-D-thiogalactopyranoside; NEP, nuclear export protein; NEP-C, NEP with C-terminal (His)6-tag; NEP-N, NEP with N-terminal (His)6-tag; PCR, polymerase chain reaction.
Influenza A virus causes acute infection disease. The search for targets
in the virus structure suitable for development of therapeutic
preparations is a very important task. The viral nuclear export
protein – NEP (or NS2, 14.4 kDa, 121 a.a.), which
mediates many key processes associated with the virus propagation
cycle, can be such target [1, 2]. NEP plays an important regulating role in
transcription and replication of the viral RNA by modulating activity
of the polymerase complex [2, 3]. Moreover, NEP participates in export of viral
genetic material from the nucleus of the host cell into the cytoplasm,
thus being a mediator between viral ribonucleoprotein (vRNP) complex
with matrix protein M1 (vRNP-M1) and the receptor of the nuclear export
CRM1 [4-7]. Data are available
in the literature on the effect of NEP on other stages of the virus
life cycle such as, for example, the viral budding process. In this
case NEP interacts with the β-subunit of the FoF1-ATPase [8].
N-terminal and C-terminal domains are recognized in the NEP structure. The latter is characterized by conformational rigidity and is crystallized in the form of a dimer [9, 10]. The C-domain comprises two α-helices (C1 and C2) that form an antiparallel amphiphilic hairpin. The hydrophilic surface of the hairpin that participates in interaction with M1 protein contains Trp78 surrounded with a cluster of glutamic acid residues. The protease-sensitive N-terminus of the protein is conformationally mobile [11], which likely prevents crystallization of the full-size protein. However, molecular modeling of the 3D structure of the N-terminal fragment does not rule out the possibility of formation in the region of two antiparallel α-helices [1, 12]. The data available in the literature indicate that the N-terminal site containing two nuclear export signals (NES) ensures interaction with CRM1 [6].
The physicochemical and functional properties of NEP are not fully understood. To investigate any protein in vitro, sufficient amounts of homogenous protein preparation are required. Isolation of NEP from virions is labor consuming, requires multistep chromatography, and does not provide high yields [4]. In most studies, recombinant variants of NEP were produced in various expression systems, either bacterial or eukaryotic [6, 13, 14]. However, in many cases the nature of the study does not require protein isolation in a pure state, as interactions of NEP with other biomolecules are investigated directly in a live cell or via its immobilization on a solid support [6, 7, 15-18]. NEP has also been produced in a cell-free protein synthesis system [19]. Isolation of pure recombinant NEP has been reported only in a few studies [4, 9, 11, 14, 19]. Systems based on E. coli BL21(DE3) cells with the NEP gene under control of the T7 promoter can be considered most efficient. The expressed protein was isolated either by multistep chromatography [11] or by affinity chromatography on Ni-NTA agarose [9]. As a rule, no data on protein yields are reported, but they do not exceed 4 mg per liter of culture [20].
The objective of this study was to develop a convenient highly efficient bacterial expression system for production of recombinant NEP with N- and C-terminal (His)6-tags, as well to determine some physicochemical parameters of the protein.
MATERIALS AND METHODS
Tris (Merck, Germany), IPTG (MP Biomedicals Inc., Germany), ATP, dATP, dGTP, dCTP, and dTTP (SibEnzyme, Russia), tryptone, bacto agar, and yeast extract (Difco, USA), SDS (Sigma, USA), and Ni-NTA agarose (Invitrogen, Germany) were used.
The following enzymes were used: polynucleotide kinase, T4 DNA ligase, DNA polymerase TaqSE, reverse transcriptase AMV, and restriction endonucleases (SibEnzyme). Treatment of DNA with enzymes was conducted according to the manufacturer’s instructions.
Escherichia coli strains ER1821 and BL21(DE3) (NEB, USA) and plasmids pET30a(+) (Novagen, USA), pCYB4 (NEB), and pB1 [21] were used. Structures of synthetic oligodeoxyribonucleotides (Syntol, Russia) are presented in the table. Preparative phosphorylation of synthetic oligodeoxyribonucleotides was carried out using polynucleotide kinase [22].
Oligodeoxyribonucleotides used in this work

Escherichia coli cells were cultivated in a DYT medium (tryptone, 16 g; yeast extract, 10 g; NaCl, 5 g per liter) with ampicillin (100 mg per liter) or without it. Petri dishes with solid EHA medium (tryptone, 13 g; NaCl, 8 g; Na-citrate, 2.4 g; glucose, 1.4 g; ampicillin, 100 mg per liter) were used for cloning. Preparation of competent cells and transformation was carried out with calcium treatment; DNA isolation and other genetic engineering procedures were performed using standard protocols [23].
The main buffer solutions used: buffer A (20 mM Hepes-Na, pH 7.2, 0.5 M NaCl, 0.05% Triton X-100); TE (10 mM Tris-HCl, pH 8.0, 0.1 mM EDTA).
Polymerase chain reactions (PCR) were conducted in a TsikloTemp-107 programmable thermostat (ZAO Resurs-Pribor, Russia). PCR conditions were optimized empirically for each experiment. A minimum number of cycles (8-12) were used in each case so sufficient product was accumulated.
Primary structures of plasmid DNAs were determined with an ABI PRISM 310 automatic sequencer (Applied Biosystems, USA) using 16-sqtl primer.
Protein preparations were analyzed with denaturing SDS-PAGE according to Laemmli [24] using 12 and 15% separating gels. Gels were stained with Coomassie G-250.
CD spectra were recorded on a Chirascan CD spectrometer (Applied Photophysics, UK) in a cuvette with 0.01-cm light pathlength.
Construction of plasmids. PCR screening for clones with target plasmids. Some cells from a single colony on a Petri dish were transferred using a sterile needle to a 0.5-ml volume tube and resuspended in 8 µl of TE buffer. The suspension was covered with 10 µl of mineral oil, and the tube was heated for 5 min at 98°C. Next, its content was thoroughly mixed by shaking and separated by centrifugation. An aliquot (1 µl) of the aqueous phase was added to PCR reaction mixture with selected primers, 27-30 cycles of PCR reaction was conducted, and the products were analyzed using agarose gel electrophoresis. Colonies were selected based on the presence (or absence) of PCR products of expected size.
Production of PCR-copy of NEP gene (363 bp). AMV reverse transcriptase was used for production of cDNA from viral RNA with 20-NSr primer. The product was precipitated with ethanol and then amplified with phosphorylated primers p20-NSr and p44-NSl removing a 473-unit intron sequence in the beginning of the NEP gene encoded in the influenza virus RNA.
pB-NEP plasmid (NEP gene, 2064 bp). A linear PCR copy of pB1 plasmid (1701 bp, beta-lactamase operon, replication origin from pUC19 plasmid, linker to restriction endonuclease sites for RsrII, SexAI, and BstEII) produced using 15-ptmt and 19-ptmb primers was ligated with the 363-unit PCR fragment containing the NEP gene and used for transformation of E. coli ER1821 competent cells. PCR screening for the target clones was carried out with primers 44-NSl and 16-sqtl. Primary structure of the plasmid in the inserted region was confirmed by sequencing.
pBt-NEP plasmid (NEP gene, T7 terminator, 2123 bp). A DNA fragment with a terminator derived from the T7 phage (59 bp) was produced by PCR copying of the region in the pET30a(+) plasmid using p19-etl and p17-ttr primers. The linear PCR copy of the pB-NEP plasmid was produced using 20-NSr and 15-ptmt primers. The linearized plasmid and the insert were joined using T4 DNA ligase and used for transformation of E. coli ER1821 cells. The target clones were screened by PCR with primers 44-NSl and 17-ttr. Primary structure of the plasmid in the region of insert was confirmed by sequencing.
pBt-NEPh plasmid (NEP gene, C-terminal (His)6-tag, T7 terminator, 2156 bp). A C-terminal (His)6-tag was inserted during production of linearized pBt-NEP plasmid using phosphorylated primers p19etl and p51-chb. The PCR product was converted into a circular one by T4 DNA ligase and used for transformation of E. coli ER1821 cells. PCR screening for the target clones was carried out with primers 44-NSl and 15-sqhr. The primary structure of the plasmid in the region of the insert was confirmed by sequencing.
EpT7t-NEPh plasmid (NEP gene, C-terminal (His)6-tag, T7 terminator and promoter, 2233 bp). A DNA fragment with the bacteriophage T7 promoter (77 bp) was produced by PCR copying of the pET30a(+) plasmid region using phosphorylated primers p17-PTl and p18-PTr. A linear PCR copy of the pBt-NEPh plasmid was obtained using primers 19-ptmb and 44-NSl. The linearized plasmid and the insert were joined with T4 DNA ligase and used for transformation of E. coli BL21(DE3) cells. PCR screening for the target clones was conducted with primers 17-PTl and 16-sqtl. The primary structure of the plasmid in the region of the insert was confirmed by sequencing.
Eptat-NEPh plasmid (NEP gene, C-terminal (His)6-tag, T7 terminator, tac promoter, 2249 bp). A DNA fragment with tac promoter (93 bp) was produced by PCR copying of the region in the pCYB4 plasmid using phosphorylated primers p17-Ptal and p19-Ptar. A linear PCR copy of the pBt-NEPh plasmid was produced using primers 19-ptmb and 44-NSl. The linearized plasmid and insert were joined by T4 DNA ligase and used for transformation of E. coli ER1821 cells. PCR screening for the target clones was carried out with primers 17-Ptal and 16-sqtl. The primary structure of the plasmid in the region of the insert was confirmed by sequencing.
Eptrt-NEPh plasmid (NEP gene, C-terminal (His)6-tag, terminator, trp promoter, 2227 bp). A DNA fragment with trp promoter (71 bp) was produced by PCR copying of total genomic DNA of E. coli using phosphorylated primers p19-Ptrl and p21-Ptrr. A linear PCR copy of the pBt-NEPh plasmid was produced using primers 19-ptmb and 44-NSl. The linearized plasmid and insert were joined with T4 DNA ligase and used for transformation of E. coli ER1821 cells. PCR screening for the target clones was carried out with primers 19-ptrl and 16-sqtl. The primary structure of the plasmid in the region of the insert was confirmed by sequencing.
Eptrt-NEP plasmid (NEP gene without affinity tags, terminator, trp promoter, 2194 bp). The C-terminal (His)6-tag in the Eptrt-NEPh plasmid was deleted by preparation of the linearized copy using phosphorylated primers p20-NSr and p19-etl followed by conversion into circular form with T4 DNA ligase and transformation of E. coli ER1821 cells. PCR screening for the target clones was carried out with primers 19-Ptrl and 15-sqhr. The primary structure of the plasmid in the region of the insert was confirmed by sequencing.
Eptrt-hNEP plasmid (NEP gene, N-terminal (His)6-tag, terminator, trp promoter, 2227 bp). An N-terminal (His)6-tag was inserted during production of the linearized Eptrt-NEP plasmid using phosphorylated primers p21-Ptrr and p50-nht. The PCR product was converted into circular form with T4 DNA ligase and used for transformation of E. coli ER1821 cells. PCR screening for target clones was conducted with primers 50-nht and 16-sqtl. The primary structure of the plasmid in the region of the insert was confirmed by sequencing.
Expression and isolation of recombinant proteins. Expression of NEP-C (NEP with C-terminal (His)6-tag) in E. coli BL21(DE3) cells with EpT7t-NEPh plasmid. An aliquot (1 ml) of overnight culture of E. coli BL21(DE3) with EpT7t-NEPh plasmid was added to 100 ml of DYT medium with ampicillin (100 mg/liter), the cells were cultivated for 2 h at 37°C with intensive shaking, then IPTG was added to 2-mM final concentration followed by further cultivation for 16 h at 37°C.
Expression of NEP-C and NEP-N (NEP with N-terminal (His)6-tag) in E. coli ER1821 cells. An aliquot (0.5 ml) of overnight culture of E. coli ER1821 cells with plasmids Eptrt-NEPh or Eptrt-hNEP was added to 100 ml of DYT medium with ampicillin (100 mg/liter) and cultivated for 18 h at 37°C with intensive shaking.
Isolation of NEP-C from cytoplasm. Cells were precipitated with centrifugation, resuspended in 4 ml of buffer A, and disrupted with ultrasound at 0°C. The cellular homogenate was centrifuged (15 min, 4°C, 8000g) and (NH4)2SO4 was added to the supernatant containing NEP-C to 25% saturation at 0°C, then the precipitate was separated by centrifugation. (NH4)2SO4 was added again to the supernatant to 55% saturation at 0°C, and the formed precipitate containing no less than 95% of the target NEP-C was separated by centrifugation. The precipitate was dissolved in 0.3 ml of buffer A, the resulting solution was loaded on a 0.2-ml Ni-NTA agarose column equilibrated with buffer A, and the column was washed with buffer A (6 × 0.2 ml) followed by elution of NEP-C with buffer A supplemented with 0.3 M imidazole (4 × 0.15 ml). The resulting solution (0.6 ml) was mixed with an equal volume of saturated at room temperature solution of (NH4)2SO4, and the precipitate containing NEP-C was separated by centrifugation and dissolved in buffer for further use. An aliquot of the produced preparation was analyzed using 15% SDS-PAGE. The yield was evaluated from the intensity of staining of the NEP-C band in comparison with staining of the band of a standard protein (BSA).
Isolation of NEP-C and NEP-N from inclusion bodies. Cells were precipitated by centrifugation, resuspended in 4 ml of the buffer A, and disrupted by ultrasound at 0°C. The cellular homogenate was centrifuged (15 min, 4°C, 8000g) and the supernatant discarded. The cell debris containing inclusion bodies formed by NEP-C (NEP-N) was resuspended in 0.54 ml of 1% aqueous solution of SDS. The residue of cell components not dissolved under this treatment was removed by centrifugation (5 min, 25°C, 12,500g). A 5 M solution of potassium acetate was added to the supernatant to 1/9 of the total volume (60 µl), and the formed precipitate of potassium dodecyl sulfate was removed by centrifugation. The resulting supernatant was mixed with (NH4)2SO4 to 25% saturation at 0°C, and the resulting precipitate was removed by centrifugation. The supernatant was mixed again with (NH4)2SO4 to 55% saturation at 0°C, and the formed precipitate was separated by centrifugation. The precipitate was dissolved in 0.5 ml of buffer A, the resulting solution was loaded onto a 0.3-ml column with Ni-NTA agarose equilibrated with buffer A, and then the column was washed with buffer A (6 × 0.3 ml) followed by elution of NEP-C (NEP-N) with buffer A supplemented with 0.3 M imidazole (4 × 0.2 ml). The resulting solution (0.8 ml) was mixed with an equal volume of (NH4)2SO4 solution saturated at room temperature, and the formed precipitate of NEP-C (NEP-N) was separated by centrifugation and dissolved in buffer for further use. An aliquot of the preparation was analyzed using 15% SDS-PAGE. The yield was evaluated based on intensity of staining of the NEP-C (NEP-N) band in comparison with a standard protein (BSA).
Crosslinking with glutaraldehyde. Protein solution (6 µg of protein in 7 µl of elution buffer from the affinity column (buffer A + imidazole)) was mixed with 0.5 µl of glutaraldehyde solution in TE. The final concentration of glutaraldehyde in the sample varied in the range 0.025-1% (2.5-100 mM). The volume of the sample was adjusted to 10 µl with buffer A. The mixture was incubated at 25°C for 8-20 min and analyzed using 12% PAGE. Control crosslinking in the presence of SDS (final concentration 1%) was conducted in a similar manner.
Recording of circular dichroism spectra. Protein circular dichroism spectra were recorded in the range 190-250 nm (far UV) in a cuvette with optical pathlength of 0.01 cm in 100 mM Na-phosphate buffer, pH 7.0, with protein concentration of 0.12 mg/ml. CD is expressed in molar ellipticity units [θ] = 3300 × Δε. The molar coefficient of dichroic absorption Δε was calculated per mol of amino acid residues. The calculation was performed using the equation Δε = ΔD/(C × l), where ΔD – measured value of dichroism, C – protein concentration in M of amino acid residues, l – optical pathlength in cm. The average molecular mass of an amino acid residue was assumed to be 120.7 g/mol. Spectra were recorded at 25°C with 1-nm step and 0.5-s accumulation time for each step. The final spectrum was obtained by averaging data from five scans and subtraction of the baseline (control). The spectra were smoothed using software supplied with the instrument.
RESULTS AND DISCUSSION
To investigate NEP comprehensively, it is necessary to have a highly efficient expression system and isolation protocol for both the protein itself and its mutant forms. Based on the literature data indicating that the best results were achieved using cells of E. coli BL21(DE3) that generate RNA polymerase (RNAP) from bacteriophage T7 in combination with a plasmid containing the gene of the target protein under control of the promoter recognized by T7 RNAP [9, 11], we decided to follow the same strategy in this work. We attempted to create a similar system (Fig. 1a) that could express NEP with C-terminal (His)6-tag in the E. coli BL21(DE3) cells for its further purification [25]. The short laboratory plasmid pB1 (1701 bp, pUC19 origin of replication, β-lactamase operon) [21], which allowed rapid rearrangement (mutagenesis) of the target region via PCR copying of the entire plasmid [26], was selected as the initial one. The expression plasmid EpT7t-NEPh containing promoter and terminator of T7 RNAP as well as the NEP gene with adjacent 3′-end fragment encoding C-terminal (His)6-tag was constructed on its basis (Fig. 1a). The indicated structural elements were inserted sequentially into the plasmid as described in the “Materials and Methods” section. The required DNA fragment with the NEP gene was produced in two steps: (i) synthesis of cDNA by reverse transcription of the respective region of total influenza A virus RNA using the 20-NSr primer (complementary to the 3′-end region of the NEP gene); (ii) PCR copying of the cDNA with simultaneous removal of a 473-unit intron in the NEP gene precursor.
Fig. 1. Expression plasmids constructed in this work for production of preparations of NEP with (His)6-tag at N- or C-terminal. Designations: Ampr, ampicillin resistance gene; ori, origin of replication from the pUC19 plasmid; t, T7-terminator; PT7, bacteriophage T7 RNA polymerase promoter; Ptac, inducible tac promoter recognized by E. coli RNA polymerase; Ptrp, trp constitutive promoter recognized by E. coli RNA polymerase; His6, site encoding affinity tag DPGSHHHHHHL (a, b, c) or MHHHHHHSGGT (d).
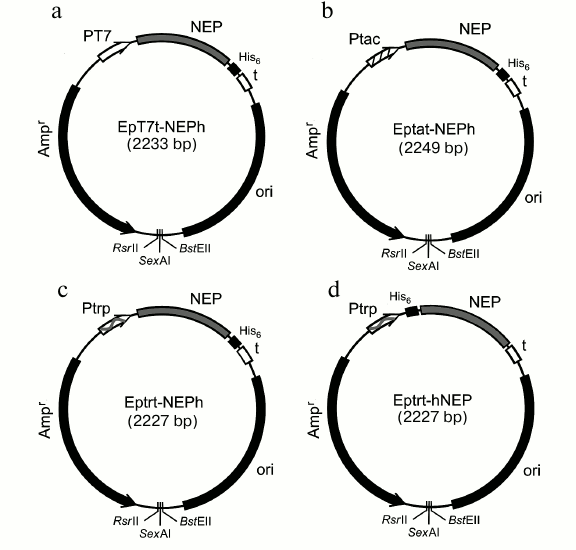
Induction of E. coli BL21(DE3) containing the pET7t-NEPh plasmid with IPTG resulted in expression of the target NEP-C (132 a.a.) containing the DPGSHHHHHHL amino acid fragment ((His)6-tag) at the C-terminal found both in cytoplasm (one third) and in inclusion bodies (two thirds). However, its total yield after affinity chromatography on Ni-NTA agarose was not as high as expected (no more than 2 mg per liter of culture). Problems of low protein expression in systems based on bacteriophage T7 RNAP were reported previously [27] and were likely related to specifics of particular proteins and difficulties with optimization of induction conditions and propagation of producer cells.
To increase yield of the target protein, we constructed another expression system where the NEP gene was transcribed with E. coli RNA polymerase. For this purpose, plasmid constructs were produced based on the EpT7t-NEPh vector, where the NEP gene with the fragment encoding N- or C-terminal (His)6-tag was under control of either the IPTG-inducible tac promoter or the constitutive trp promoter [28] (Fig. 1, b and c). The level of expression of NEP-C in E. coli ER1821 cells with Eptat-NEPh plasmid (Fig. 1b) containing tac promoter was higher than in the case with the T7 RNAP system (approximately 7 mg per liter of culture), and the protein was found both in cytoplasm and in inclusion bodies (Fig. 2a). The use of the plasmid with the strong trp promoter (Eptrt-NEPh) resulted in significant increase in the protein (NEP-C) yield (approximately 17 mg per liter of culture) (Fig. 2b); moreover, practically all the protein was found in inclusion bodies.
Fig. 2. SDS-PAGE of preparations of NEP-C (lanes 1-3) and NEP-N (lane 4) isolated from cells with Eptat-NEPh (1, 2), Eptrt-NEPh (3), and Eptrt-hNEP (4) plasmids. M, protein molecular mass markers.

The Eptrt-hNEP plasmid was constructed (Fig. 1c) allowing production of NEP-N with additional N-terminal MHHHHHHSGGT fragment to investigate the effect of (His)6-tag location on the properties of NEP. The producer with this plasmid also exhibited high yields of NEP-N (more than 20 mg per liter of culture). In this case also, all the NEP-N was found in inclusion bodies.
Special techniques were developed to isolate the protein from inclusion bodies. Following cell disruption, cell debris with inclusion bodies was precipitated by centrifugation, treated with 1% SDS, and separated from the insoluble fraction of the debris by centrifugation. Dodecyl sulfate was removed from the resulting solution as the insoluble potassium salt by adding excess potassium acetate. Then the target protein was isolated on Ni-NTA agarose. Because the protein is small and has relatively simple structure, we expected that this treatment would facilitate efficient restoration of the native protein structure. Electrophoretic analysis of the NEP-N and NEP-C in SDS-polyacrylamide gel is presented in Fig. 2. The proteins produced with this method are practically homogenous; furthermore, we found that the preparations of NEP-N and NEP-C isolated from inclusion bodies contain less admixture than NEP-C isolated from cytoplasm (Fig. 2, a and b). This phenomenon was described before for other proteins. Moreover, special techniques were developed recently allowing targeted production of protein in inclusion bodies [29].
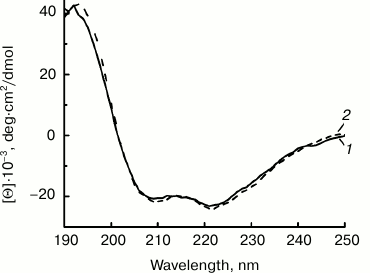
The CD spectra of NEP-C and NEP-N in the far-UV region were recorded and analyzed to confirm the native structure of the proteins isolated from inclusion bodies (Fig. 3). The spectra of the proteins with N- and C-terminal (His)6-tags are practically identical and are in good agreement with the literature data for NEP [11]. They demonstrate negative maxima at 208/209 and 221/222 nm characteristic for the spectra of a protein with significant fraction of α-helices in its secondary structure. The calculated fraction of α-helices estimated using the Greenfield–Fasman method is 58% [30]. Hence, the CD data confirm secondary structure in NEP-C and NEP-N that is practically identical to the structure of natural NEP.
Fig. 3. CD spectra of NEP-C (1) and NEP-N (2) produced by expression in E. coli cells with plasmids Eptrt-NEPh and Eptrt-hNEP and isolated from inclusion bodies.
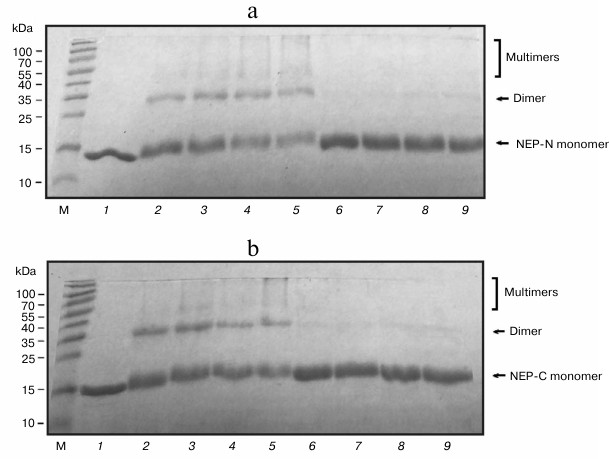
We found during work with preparations of NEP-C and NEP-N that they are prone to aggregation over a wide range of conditions (pH, concentration, buffer composition, ionic strength, temperature, and incubation time were varied). This phenomenon was observed previously. For example, formation of insoluble deposits in protein preparations was mentioned [11], although their nature was not elucidated. Crosslinking experiments were conducted to confirm the availability of aggregates in NEP-C and NEP-N solutions. Glutaraldehyde was used as a bifunctional agent as a reactant widely used for intermolecular crosslinking of proteins in complexes. This approach allows demonstrating the ability of protein for oligomerization [31, 32]. The results of electrophoretic analysis of the products of crosslinking are presented in Fig. 4. A significant amount of product is formed in the reaction mixture that corresponds to the respective NEP dimer based on its mobility (10-30%), as well as a certain amount of oligomer (Fig. 4, a and b). The crosslinking products were not formed in the presence of SDS (denaturing conditions). According to the literature, treatment of NEP with another crosslinking reagent (ethylene glycol bis-succinimidyl succinate) did not result in formation of intermolecular bonds [9]. However, a deletion variant of NEP consisting of the C-terminal domain produced significant amounts of the dimer under the same conditions. The availability of crosslinked products in our case indicate that the structure of glutaraldehyde corresponds better to the distance between the functional groups in the NEP aggregates that are linked. Hence, we have demonstrated that the full-size protein is capable of self-assembly with formation of oligomeric aggregates.
Fig. 4. SDS-PAGE of products of crosslinking of NEP-N (a) and NEP-C (b) (6 µg, 40 µM of protein) with glutaraldehyde in the presence of 1% SDS (lanes 6-9) and in its absence (lanes 2-5). Glutaraldehyde concentration: 0.025% (2.5 mM) (2, 6), 0.25% (25 mM) (3, 7), and 0.5% (50 mM) (4, 5, 8, 9), incubation time 10 min (2-4, 6-8) and 20 min (5, 9). Lane 1, protein preparations in the absence of glutaraldehyde; M, protein molecular mass markers. Locations of protein monomers, their dimers, and high molecular weight crosslinked products are marked.
In conclusion, we have developed a new and highly effective system for expression of NEP that allows production of highly purified protein preparations with (His)6-tag at the C- or N-terminal from inclusion bodies with yields higher than 20 mg/liter. The features of the produced plasmid constructs make them quite suitable for conducting fast rearrangements and production of mutant variants of the protein using the method of full-size plasmid copying.
Acknowledgments
This work was financially supported by the Russian Foundation for Basic Research (project No. 16-04-00563).
REFERENCES
1.Darapaneni, V., Prabhaker, V. K., and Kukol, A.
(2009) Large-scale analysis of influenza A virus sequences reveals
potential drug target sites of non-structural proteins, J. Gen.
Virol., 90, 2124-2133.
2.Manz, B., Schwemmle, M., and Brunotte, L. (2013)
Adaptation of avian influenza A virus polymerase in mammals to overcome
the host species barrier, J. Virol., 87, 7200-7209.
3.Odagiri, T., and Tobita, K. (1990) Mutation in NS2,
a nonstructural protein of influenza A virus, extragenically causes
aberrant replication and expression of the PA gene and leads to
generation of defective interfering particles, Proc. Natl. Acad.
Sci. USA, 87, 5988-5992.
4.Yasuda, J., Nakada, S., Kato, A., Toyoda, T., and
Ishihama, A. (1993) Molecular assembly of influenza virus: association
of the NS2 protein with virion matrix, Virology, 196,
249-255.
5.Iwatsuki-Horimoto, K., Horimoto, T., Fujii, Y., and
Kawaoka, Y. (2004) Generation of influenza A virus NS2 (NEP) mutants
with an altered nuclear export signal sequence, J. Virol.,
78, 10149-10155.
6.Brunotte, L., Flies, J., and Bolte, H. (2014) The
nuclear export protein of H5N1 influenza A viruses recruits matrix 1
(M1) protein to the viral ribonucleoprotein to mediate nuclear export,
J. Biol. Chem., 289, 20067-20077.
7.Watanabe, K., Shimizu, T., Noda, S., Tsukahara, F.,
Maru, Y., and Kobayashi, N. (2014) Nuclear export of the influenza
virus ribonucleoprotein complex: interaction of Hsc70 with viral
proteins M1 and NS2, FEBS Open Bio, 4, 683-688.
8.Gorai, T., Goto, H., Noda, T., Watanabe, T.,
Kozuka-Hata, H., Oyama, M., Takano, R., Neumann, G., Watanabe, S., and
Kawaoka, Y. (2012) F1Fo-ATPase, F-type proton-translocating ATPase, at
the plasma membrane is critical for efficient influenza virus budding,
Proc. Natl. Acad. Sci. USA, 109, 4615-4620.
9.Akarsu, H., Burmeister, W. P., Petosa, C., Petit,
I., Muller, C. W., Ruigrok, R. W., and Baudin, F. (2003) Crystal
structure of the M1 protein-binding domain of the influenza A virus
nuclear export protein (NEP/NS2), EMBO J., 22,
4646-4655.
10.Akarsu, H., Iwatsuki-Horimoto, K., Noda, T.,
Kawakami, E., Katsura, H., Baudin, F., Horimoto, T., and Kawaoka, Y.
(2011) Structure-based design of NS2 mutants for attenuated influenza A
virus vaccines, Virus Res., 155, 240-248.
11.Lommer, B. S., and Luo, M. (2002) Structural
plasticity in influenza virus protein NS2 (NEP), J. Biol. Chem.,
277, 7108-7117.
12.Salahuddin, P., and Khan, A. U. (2010) Structural
and functional analysis of NS1 and NS2 proteins of H1N1 subtype,
Genomics Prot. Bioinform., 8, 190-199.
13.Neumann, G., Watanabe, T., Ito, H., Watanabe, S.,
Goto, H., Gao, P., Hughes, M., Perez, D. R., Donis, R., Hoffmann, E.,
Hobom, G., and Kawaoka, Y. (1999) Generation of influenza A viruses
entirely from cloned cDNAs, Proc. Natl. Acad. Sci. USA,
96, 9345-9350.
14.Ward, A. C., Castelli, L. A., Lucantoni, A. C.,
White, J. F., Azad, A. A., and Macreadie, I. G. (1995) Expression and
analysis of the NS2 protein of influenza A virus, Arch. Virol.,
140, 2067-2073.
15.Hu, Y., Liu, X., Zhang, A., Zhou, H., Liu, Z.,
Chen, H., and Jin, M. (2015) CHD3 facilitates vRNP nuclear export by
interacting with NES1 of influenza A virus NS2, Cell Mol. Life
Sci., 72, 971-982.
16.Tawaratsumida, K., Phan, V., Hrincius, E. R.,
High, A. A., Webby, R., Redecke, V., and Hacker, H. (2014) Quantitative
proteomic analysis of the influenza A virus nonstructural proteins NS1
and NS2 during natural cell infection identifies PACT as an NS1 target
protein and antiviral host factor, J. Virol., 88,
9038-9048.
17.Reuther, P., Giese, S., Gotz, V., Kilb, N., Manz,
B., Brunotte, L., and Schwemmle, M. (2014) Adaptive mutations in the
nuclear export protein of human-derived H5N1 strains facilitate a
polymerase activity-enhancing conformation, J. Virol.,
88, 263-271.
18.Shimizu, T., Takizawa, N., Watanabe, K., Nagata,
K., and Kobayashi, N. (2011) Crucial role of the influenza virus NS2
(NEP) C-terminal domain in M1 binding and nuclear export of vRNP,
FEBS Lett., 585, 41-46.
19.Inglis, S. C., Barrett, T., Brown, C. M., and
Almond, J. W. (1979) The smallest genome RNA segment of influenza virus
contains two genes that may overlap, Proc. Natl. Acad. Sci. USA,
76, 3790-3794.
20.Greenspan, D., Krystal, M., Nakada, S.,
Arnheiter, H., Lyles, D. S., and Palese, P. (1985) Expression of
influenza virus NS2 nonstructural protein in bacteria and localization
of NS2 in infected eucaryotic cells, J. Virol., 54,
833-843.
21.Koroleva, O. N., Dubrovin, E. V., Yaminsky, I.
V., and Drutsa, V. L. (2016) Effect of DNA bending on transcriptional
interference in the systems of closely spaced convergent promoters,
Biochim. Biophys. Acta, 1860, 2086-2096.
22.Koroleva, O. N., Drutsa, V. L., Dolinnaya, N. G.,
Tsytovich, A. V., and Shabarova, Z. A. (1984) DNA-like duplexes
containing repetitive sequences. VII. Chemico-enzymatic synthesis of
polymers with fragment of natural promoters, Mol. Biol.
(Moscow), 18, 146-160.
23.Maniatis, T., Fritsch E., and Sambroock, J.
(1982) Molecular Cloning: A Laboratory Manual, Cold Spring
Harbor, Cold Spring Harbor Laboratory Press, N.Y.
24.Laemmli, U. (1970) Cleavage of structural
proteins during the assembly of the head of bacteriophage T4,
Nature, 227, 680-685.
25.Bornhorst, J. A., and Falke, J. J. (2000)
Purification of proteins using polyhistidine affinity tags, Methods
Enzymol., 326, 245-254.
26.Hemsley, A., Arnheim, N., Toney, M. D.,
Cortopassi, G., and Galas, D. J. (1989) A simple method for
site-directed mutagenesis using the polymerase chain reaction,
Nucleic Acids Res., 17, 6545-6551.
27.Sivashanmugam, A., Murray, V., Cui, C., Zhang,
Y., Wang, J., and Li, Q. (2009) Practical protocols for production of
very high yields of recombinant proteins using Escherichia coli,
Protein Sci., 18, 936-948.
28.Bass, S. H., and Yansura, D. G. (2000)
Application of the E. coli trp promoter, Mol.
Biotechnol., 16, 253-260.
29.Wang, X., Zhou, B., Hu, W., Zhao, Q., and Lin, Z.
(2015) Formation of active inclusion bodies induced by hydrophobic
self-assembling peptide GFIL8, Microb. Cell Fact., 14,
88.
30.Greenfield, N. J., and Fasman, G. D. (1969)
Computed circular dichroism spectra for the evaluation of protein
conformation, Biochemistry, 8, 4108-4116.
31.Richards, F. M., and Knowles, J. R. (1968)
Glutaraldehyde as a protein cross-linkage reagent, J. Mol.
Biol., 37, 231-233.
32.Kowalczyk, M., and Bardowski, J. (2003)
Overproduction and purification of the CcpA protein from Lactococcus
lactis, Acta Biochim. Pol., 50, 455-459.