Recombinant Human Erythropoietin with Additional Processable Protein Domains: Purification of Protein Synthesized in Escherichia coli Heterologous Expression System
T. M. Grunina1, A. V. Demidenko1, A. M. Lyaschuk1, M. S. Poponova1, Z. M. Galushkina1, L. A. Soboleva1, S. A. Cherepushkin2, N. B. Polyakov1,3, D. A. Grumov1, A. I. Solovyev1, V. G. Zhukhovitsky1,4, I. S. Boksha1,5, M. E. Subbotina1,6, A. V. Gromov1*, V. G. Lunin1,6, and A. S. Karyagina1,6,7*
1Gamaleya National Research Center of Epidemiology and Microbiology, Ministry of Health of the Russian Federation, 123098 Moscow, Russia; E-mail: alexander.v.gromov@gmail.com, akaryagina@gmail.com2State Research Institute of Genetics and Selection of Industrial Microorganisms, 117545 Moscow, Russia
3Vernadsky Institute of Geochemistry and Analytical Chemistry, Russian Academy of Sciences, 119991 Moscow, Russia
4Sechenov Moscow State Medical University, 119991 Moscow, Russia
5Mental Health Research Center, 115522 Moscow, Russia
6All-Russia Research Institute of Agricultural Biotechnology, 127550 Moscow, Russia
7Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119992 Moscow, Russia
* To whom correspondence should be addressed.
Received July 4, 2017; Revision received August 3, 2017
Three variants of human recombinant erythropoietin (rhEPO) with additional N-terminal protein domains were obtained by synthesis in an Escherichia coli heterologous expression system. These domains included (i) maltose-binding protein (MBP), (ii) MBP with six histidine residues (6His) in N-terminal position, (iii) s-tag (15-a.a. oligopeptide derived from bovine pancreatic ribonuclease A) with N-terminal 6His. Both variants of the chimeric protein containing MBP domain were prone to aggregation under nondenaturing conditions, and further purification of EPO after the domain cleavage by enterokinase proved to be impossible. In the case of 6His-s-tag-EPO chimeric protein, the products obtained after cleavage with enterokinase were successfully separated by column chromatography, and rhEPO without additional domains was obtained. Results of MALDI-TOF mass spectrometry showed that after refolding 6His-s-tag-EPO formed a structure similar to that of one of native EPO with two disulfide bonds. Both 6His-s-tag-EPO and rhEPO without additional protein domains purified after proteolysis possessed the same biological activity in vitro in the cell culture.
KEY WORDS: erythropoietin, heterologous expression, Escherichia coliDOI: 10.1134/S0006297917110062
Abbreviations: a.a., amino acid residue; BMP-2, bone morphogenetic protein-2; DTT, dithiothreitol; EPO, erythropoietin; 6His, six histidine amino acid residues; MBP, maltose-binding protein; rhEPO, recombinant human erythropoietin; s-tag, 15-a.a. oligonucleotide derived from bovine pancreatic ribonuclease A.
Erythropoietin (EPO) is a hormone (cytokine) produced in kidney cells
that performs multiple functions in the organism primarily related to
regulation of erythropoiesis. EPO functions via its interaction with
the erythropoietin receptor located on the surface of erythroid
progenitor cells in bone marrow, which stimulates production of
erythrocytes. Injections of recombinant human EPO (rhEPO) are used in
medicine for therapy of various forms of anemia related to kidney
dysfunctions, cancer, and chemotherapy [1]. Direct
and indirect effects of EPO on bone tissue remodeling have been
actively investigated recently [2-4]. The possibility of using rhEPO separately or in
combination with other factors such as bone morphogenetic protein-2
(BMP-2) for healing bone defects with systemic or local application has
been investigated in laboratory studies [5, 6].
EPO consists of 166 amino acid residues (a.a.). It is glycosylated during its synthesis in the human body – N-linked glycans are attached to Asn24, Asn38, and Asn83 and O-linked glycan to Ser126 [7, 8]. Oligosaccharides are linked to the growing protein chain during the translation process in endoplasmic reticulum and then are “remodeled” in the Golgi apparatus [9]. The EPO molecule contains two disulfide bonds: one between residues Cys7 and Cys161 and another between residues Cys29 and Cys33. Data are available indicating that glycans are involved in folding and secretion, provide protein stabilization, define metabolic pathways [9, 10], and determine features of biological activity manifestation [10, 11]. As a rule, rhEPO (alfa and beta) are derived from genetically modified Chinese hamster ovary (CHO) cells. The preparation comprises a heterogeneous mixture of glycoforms – molecules differing in glycan structure. It was shown by competitive binding experiments that glycoforms with lower content of N-linked glycans demonstrate decrease in in vivo activity and increase in in vitro activity [10]. Elevated content of sialic acids in oligosaccharides increases biological activity in vivo, prolongs blood serum half-life, but at the same time decreases its affinity to EPO receptor [11]. Several variants of rhEPO are produced by various drug manufacturers for medical applications, including a modified second-generation EPO (darbepoetin alfa) with prolonged action. All the preparations are based on rhEPO produced by synthesis in eukaryotic cells that differ in degree and characteristics of protein glycosylation, specific activity, pharmacokinetics, and other parameters. Moreover, the commercially available preparations of EPO are used in scientific publications describing the effect of EPO on biological and physiological processes such as bone tissue remodeling (see review Wu et al. [3]), but the exact preparation used in a study is often not mentioned, even though the selection of a particular preparation can greatly define the results of an experiment.
Production of recombinant proteins in Escherichia coli seems like a good alternative to synthesis in eukaryotic cells due to lower cost and better standardization of the produced product. The obvious problems of biosynthesis of eukaryotic proteins in E. coli include the lack of glycosylation and problems with formation of disulfide bonds during protein folding, which can be detrimental for biological activity of the protein. However, these problems were solved in the case of EPO. For example, it was shown in the study of Jeong et al. that a partially purified protein isolated from E. coli cells had correct structure with two S–S-bonds and was active in cell culture experiments in vitro [12]. Only a handful of publications have been devoted to the production of recombinant EPO in E. coli and its purification [12-14]. Low solubility of the protein and tendency to aggregation is among the problems associated with this, which can be overcome either by introduction of amino acid substitution of the asparagine residues at positions subjected to glycosylation with lysine residues [14], or by constructing chimera proteins with additional protein domains increasing the solubility of the protein with subsequent cleavage of EPO via proteolytic hydrolysis [12].
The objective of this study was the production of highly purified non-glycosylated rhEPO synthesized in E. coli cells with correctly formed disulfide bonds possessing biological activity and being adequate for further investigation of the possibility of its application in regenerative medicine, in particular, for investigation of bone tissue reparation using combined application with recombinant BMP-2.
MATERIALS AND METHODS
Escherichia coli strain BL21(DE3) (E. coli B F– dcm ompT hsdS(rB–mB–) gal λ(DE3) (Agilent Technologies, USA) and plasmid vector pET30a(+) (Novagen, USA) were used in the study.
Preparation of gene construct encoding 6His-s-tag-EPO. The gene encoding the amino acid sequence of the Entero-EPO protein consisting of the sequence of the enteropeptidase-specific hydrolytic site (Entero) and EPO carrying the BamHI recognition site at the 5′-terminus and Kpn2I site at the 3′-terminis with optimized codon composition and RNA secondary structure was produced synthetically. The codon composition of the synthetic gene for expression in E. coli was optimized using the JCat program (http://www.jcat.de/), and correction of the secondary structure of the transcribed RNA was optimized using the DINAMelt web server (http://mfold.rna.albany.edu/?q=DINAMelt/Two-state-folding). Nucleotide sequences were synthesized by the Evrogen (Russia). The construct was inserted into the pET30a(+) plasmid between the BglII and AgeI sites. A producer of the 6His-s-tag-EPO protein carrying six histidine residues (6His) and s-tag (15-a.a. oligonucleotide from the bovine pancreatic ribonuclease A) at the N-terminus was obtained by cloning in E. coli BL-21 (Fig. 1).
Fig. 1. Amino acid sequence of 6His-s-tag-EPO protein. The EPO sequence is indicated with bold italic, the 6His sequence by underlining, the sequence hydrolyzed by enterokinase by underlined italic (the enterokinase hydrolysis site is located directly after the last underlined residue), cysteine residues in the EPO sequence are in bold underlined italic, and s-tag in bold underlined.

Calculated values of molecular mass and some properties of 6His-s-tag-EPO and EPO polypeptides. Information about these polypeptides was acquired using internet source http://web.expasy.org/compute_pi/. Molecular masses of the 6His-s-tag-EPO and EPO polypeptides are 23,828.05 and 18,396.14 Da, respectively. Absorption of aqueous protein solutions with concentration of 1 mg/ml at 280 nm must be 0.953 and 1.206, and the theoretical isoelectric points (pI) are 6.75 and 8.75, respectively.
Cultivation of transformed E. coli cells. The transformed E. coli BL-21 cells were cultivated in LB medium with addition of respective antibiotics on a shaker at 180 rpm and 37°C to OD600 1.0-1.2. Protein synthesis was induced by 0.5 mM IPTG, and then the culture was incubated for an additional 4 h under the same conditions and centrifuged for 30 min at 5000g and 10°C. The biomass was stored at –20°C.
Disruption of E. coli cells, preparation of fraction of readily soluble cellular proteins, and isolation of inclusion bodies. The thawed biomass of bacterial cells was resuspended in lysis buffer (20 mM Tris-HCl (pH 8.0), 100 mM NaCl, 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride by adding it at ratio no less than 1 : 10 (w/v)), 100 µg/ml of lysozyme was added, and the mixture was incubated for 20 min at room temperature followed by cell disruption using a Sonics Vibracell ultrasonic processor (40% amplitude, 2.5 min interrupting for 3 s every 2 s) on ice. The mixture was centrifuged for 30 min at 20,000g and 10°C. Soluble cellular proteins were in the supernatant. The precipitate containing inclusion bodies was washed twice with lysis buffer and once with 20 mM Tris-HCl (pH 8.0) buffer.
Column chromatography and electrophoresis. Column chromatography on WorkBeads 40 Ni and WorkBeads 40 S sorbents (Bio-Works, Sweden) was conducted using a BioLogic low-pressure chromatographic system (Bio-Rad, USA) or Akta Start device (GE Healthcare Life Sciences, USA). Protein concentration was determined from absorption of the protein solution at 280 nm using theoretical (calculated) protein absorption coefficients and with Bicinchoninic Acid Protein Assay (AppliChem, Germany).
Protein extracts and protein samples from different purification steps prior to PAGE were prepared in Laemmli sample buffer containing 2 mM EDTA, 125 mM Tris-HCl (pH 6.8), 20% glycerol, 4% SDS, and 0.015% Bromophenol blue with addition of 0.2 M dithiothreitol (DTT) reducer or without it. Sample aliquots were diluted with the sample buffer at ratio 1 : 1, heated for 5 min at 95°C, and centrifuged for 5 min at 16,000g; the supernatant was loaded onto a polyacrylamide gel. Protein electrophoresis in the gel was carried out using a SE 260 Amersham Mighty Small II instrument for vertical electrophoresis for 8 × 9 cm gels and a reagent kit from GE Healthcare (USA). Following electrophoresis, the gel was stained with Coomassie Brilliant Blue R-250 according to the standard procedure. To evaluate the apparent molecular mass, protein markers with molecular mass 14-97 kDa (Bio-Rad) were used. Gels were imaged using a Gel Doc™ XR+ system (Bio-Rad), and density of gel bands was assessed using Quantity One software (Bio-Rad).
Refolding of 6His-s-tag-EPO and protein purification on WorkBeads 40 S sorbent. Following the disruption of 1 g of wet biomass, precipitate containing washed inclusion bodies (250 mg) was dissolved in 10 ml of 6 M guanidine hydrochloride, 0.1 M DTT, and 20 mM Tris-HCl (pH 8.0), incubated at 4°C overnight, and centrifuged for 30 min at 9000g. Then the denatured protein was diluted with refolding buffer containing 0.5 M L-arginine, 20 mM Tris-HCl (pH 8.5) at ratio 1 : 15 to the final protein concentration of ~0.1 mg/ml and incubated for 24 h at 4°C. The solution of refolded protein was dialyzed against 10 volumes of buffer containing 4 M urea, 20 mM Tris-HCl (pH 6.8) for 24 h and centrifuged for 30 min at 9000g. The supernatant was subjected to column chromatography on the WorkBeads 40 S sorbent. The supernatant (200 ml) was loaded on a column with 15 ml of WorkBeads 40 S sorbent equilibrated with buffer containing 4 M urea, 30 mM Tris-HCl (pH 6.8) at a rate 1 ml/min and washed with the buffer at a rate 1.5 ml/min until absorbance at 280 nm reached a plateau, followed by elution with linear gradient from 0 to 1 M NaCl concentration in the same buffer. Four 3-ml fractions were collected during elution. Monomeric protein was in the first three fractions, while some aggregates were observed in the electrophoregram of the last fraction. Three fractions with total volume of 9 ml were combined and dialyzed against 20 mM Tris-HCl (pH 8.0) overnight followed by estimation of the protein concentration based on the solution absorbance at 280 nm, which was 0.105. The approximate protein concentration in this fraction was 0.1 mg/ml based on the theoretical extinction coefficient (0.953 for 6His-s-tag-EPO). Then the protein was subjected to proteolytic hydrolysis with enterokinase.
Proteolytic hydrolysis of proteins with enterokinase. Proteolytic hydrolysis of the proteins was performed according to the method of Gasparyan et al. [15] with modifications.
Treatment of the 6His-s-tag-EPO protein with enterokinase was conducted in buffer containing 10 mM Tris-HCl (pH 7.5-8.0) and 200 mM NaCl. An aliquot (10 µl) of enterokinase solution with specific activity of 0.1 unit/µl was added to 9 ml of the protein solution with concentration of 0.1 mg/ml and incubated for 3 h at room temperature (25°C). Hydrolysis efficiency was evaluated with PAGE.
Following the hydrolysis of 6His-s-tag-EPO with enterokinase, the cleaved rhEPO was purified by affinity chromatography on WorkBeads 40 Ni sorbent. A column with 3 ml of the sorbent was equilibrated with 20 mM solution of Tris-HCl (pH 8.0), protein binding and column washing with five column volumes was conducted in the same buffer, and all operations were carried out with flow rate 1 ml/min. The target protein purified from the products of hydrolysis was eluted in the first washing fractions. Absorption of the protein solution at 280 nm was 0.08, which approximately corresponded to protein concentration of 0.066 mg/ml considering the theoretical extinction coefficient (1.206 for EPO). Hence, considering the volume of combined fraction (8 ml), the total amount of purified protein was 0.53 mg. The protein preparation was homogenous in SDS-PAGE.
The combined fractions following chromatography on WorkBeads 40 Ni were dialyzed against 25 mM ammonium acetate buffer (pH 4.5) for 24 h at 4°C. The ratio of dialyzed sample and dialysis buffer was 1 : 10. The produced protein preparations were lyophilized and used for further investigations.
Evaluation of protein biological activity in vitro. Biological activity of erythropoietin preparations was evaluated with the in vitro proliferation test with human erythroleukemia cell line TF-1 (ATCC CRL-2003). Cells were cultivated at 37°C in an atmosphere of 5% CO2 in RPMI 1640 medium (PanEko, Russia) supplemented with 10% fetal bovine serum (HyClone, USA), gentamycin (PanEko) to concentration of 10 µg/ml, and granulocyte macrophage colony-stimulating factor (GM-CSF) (PanEko) at final concentration of 2 ng/ml. Prior to the test for determination of specific activity, the cells were subjected to the so-called “starvation stage”: incubation for 22-26 h in medium with minimum serum content (0.5%) without added GM-CSF and other growth factors at 37°C and 5% CO2 atmosphere. Following the starvation, staged cells were pelleted from the culture medium by centrifugation at 800 rpm, discarding supernatant and resuspending the cells in cold RPMI 1640 medium. The procedure was repeated three times. After the third centrifugation, the cells were resuspended in a volume of the test medium (RPMI 1640 supplemented with 5% fetal bovine serum and 10 µg/ml gentamycin, without GM-CSF) to adjust cell concentration to 600-800 thousand cells per ml. Protein samples (EPO variants) were filter-sterilized though a 0.2-µm filter unit (Corning, Germany) and diluted in the test medium. Samples with different dilutions in the test medium were prepared in 96-well plates (volume in each well 50 µl, 2-fold dilutions from 1000 to 4 ng/ml and from 10 to 0.04 ng/ml). The medium without EPO was used as a negative control, and recombinant EPO from mammalian cells – Epostim (epoetin beta; Farmapark, Russia) – was used as a reference sample. Aliquots (50 µl, 30-40 thousand cells) of the prepared cells were introduced into the wells containing samples. The plate was incubated for 68-72 h at 37°C in 5% CO2. On completion of incubation, 10 µl of WST-1 substrate mixture (Roche, Switzerland) was added to each well. Then the plate was incubated at 37°C in 5% CO2 for 5-6 h with visual control of color development. Optical density in the wells was recorded at wavelength 450 nm with a Tecan Infinite M 200 Pro plate reader (Tecan Group Ltd., Switzerland). The specific activity was calculated by the regression model using the PLA 3.0 program (Stegmann Systems GmbH, Germany).
Trypsinolysis of proteins prior to mass spectrometry. The proteins separated with PAGE were hydrolyzed within the gel fragments according to the protocol suggested by Shevchenko et al. [16] with and without addition of DTT and iodoacetamide.
Tryptic peptides were extracted from the gel using sequentially 50 and 90% aqueous acetonitrile solutions in 5% formic acid by adding 50 µl of each solution to the cut gel fragment. An aliquot (2 µl) of the combined extract was mixed with 0.5 µl saturated solution of 2,5-dihydroxybenzoic acid in 30% acetonitrile solution containing 0.5% (vol) glacial acetic acid on a steel target, which was then air-dried at room temperature.
To investigate formation of disulfide bonds in the recombinant protein, the protein was trypsinized in solution. The reaction was carried out as follows: 100 µl of protein solution (approximately 100 µg) in 20 mM ammonium acetate buffer (pH 8.0) was mixed with 100 µl of 100 mM ammonium bicarbonate (pH 8.3). The reaction mixture was divided in two portions. DTT and iodoacetamide were added to one portion using the procedure described above for trypsinolysis in gel [16]. Only trypsin was added to another portion. In both cases, the protein/enzyme ratio was 20 : 1. The reaction was conducted at 37°C for 17 h.
The reaction mixture was dried using a Savant SPD121P vacuum concentrator (Thermo Scientific, USA) and dissolved in 20 µl 0.1% formic acid. The sample was applied onto the target according to the procedure described above for trypsinolysis in gel.
MALDI mass spectrometry. The peptides were analyzed using an UltrafleXtreme MALDI time-of-flight mass spectrometer (Bruker Daltonics, Germany). Positive ions were detected in reflectron mode with voltage on ion source IS1 of 20.12 kV and IS2 of 17.82 kV, on lenses 7.47 kV, and on reflectron Ref1 21.07 kV and Ref2 10.80 kV.
Ions were detected in the m/z range 700-5000 Th (Thompson = Da/e). The peaks of autolytic fragments of trypsin and keratin, which were excluded from the final lists of detected masses, were used as internal standards.
RESULTS AND DISCUSSION
In the beginning, production of recombinant EPO via biosynthesis in E. coli producers was performed via two variants of chimeric constructs of EPO with maltose-binding protein (MBP). Except for a few details, this approach was similar to the one suggested in the work of Jeong et al. [12], where they produced active rhEPO. The synthetic gene was designed encoding the amino acid sequence of the MBP-EPO protein, which included amino acid sequences of MBP from E. coli and human erythropoietin with the human enterokinase recognition site between them. A similar chimeric construct was designed including N-terminal 6His-tag (its use for improving efficiency of two functional domains has been described for numerous other biotechnologically important recombinant proteins [17]). The hybrid proteins MBP-EPO and 6His-MBP-EPO were isolated and purified to homogenous state both from the fraction of readily soluble cytoplasmic proteins as well as from inclusion bodies, and then the chimeric proteins were effectively hydrolyzed with enterokinase (data not shown). However, all further attempts to separate the products of proteolytic hydrolysis – MBP and rhEPO – by column chromatography were unsuccessful. We suggest that even after proteolysis with enterokinase, MBP and EPO remain in a stable complex due to the opposite charges of these protein domains and, probably, other types of interactions.
Hence, despite the advantageous properties of the MBP domain facilitating solubility of the construct as a whole, in this case it demonstrated a significant drawback manifested by substantial aggregation with the protein linked to it (EPO); the observed phenomenon of “sticking” to the target (so-called “passenger”) protein was reported for other constructs [18]. The significant size of the maltose-binding domain (40 kDa) represents another disadvantage; thus, when this affinity domain is located at the N-terminal of the chimeric protein, it frequently interrupts synthesis (at least in our case), which results in production of incomplete recombinant products.
We decided to use another approach for production of rhEPO – introduction of an additional domain at the N-terminus of the chimeric protein including 6His, s-tag, and enterokinase recognition site (Fig. 1). This approach was successfully used in the work of Karyagina et al. in 2017 [19] for production of recombinant morphogenetic protein 2 (rhBMP-2) via synthesis in E. coli. This protein was characterized by the presence of disulfide bonds and tendency to aggregation as in the case of EPO.
The results of 6His-s-tag-EPO isolation via protein chromatography on WorkBeads 40 S following dissolution of inclusion bodies and refolding using the method of Wang et al. [20] are presented in Fig. 2a. In the first three fractions eluted from WorkBeads 40 S, we found using PAGE (after sample treatment with buffer without reducer) practically homogenous protein corresponding to ~26 kDa (based on mobility), and the following fraction contained high molecular weight aggregates (Fig. 2b, 1-4). An electrophoretically homogenous protein band with slightly lesser mobility than in the samples without reducer (~28 kDa) was observed in all fractions after treatment of the protein samples with sample buffer containing reducer (DTT) (Fig. 2b, 5-8).
Fig. 2. Electrophoregrams of separation in 12% polyacrylamide gel of protein samples produced during purification of 6His-s-tag-EPO protein preparations from cells of E. coli BL-21(DE3) producer at the stages of inclusion bodies dissolution and refolding (a), chromatography on WorkBeads 40 S sorbent (b) and enterokinase treatment followed by chromatography on WorkBeads 40 Ni sorbent (c). a) Lanes: 1) extract of strain producer cells prior to protein synthesis induction with IPTG; 2) extract of strain producer cells after protein synthesis induction with IPTG; 3) inclusion bodies of strain producer after washing; 4) 6His-s-tag-EPO protein prior to refolding; 5) 6His-s-tag-EPO after refolding. b) Fractions of 6His-s-tag-EPO protein after refolding eluted during chromatography of 6His-s-tag-EPO on WorkBeads 40 S sorbent (panel (a), lane 5). c) Lanes: 1, 5 and 2, 6) protein of the first three fractions of 6His-s-tag-EPO eluted from WorkBeads 40 S, which were not treated (1 and 2) and treated (5 and 6) with enterokinase; 3, 7) protein fractions not bound to the sorbent; 4, 8) fractions of enterokinase-treated 6His-s-tag-EPO protein eluted after chromatography on WorkBeads 40 Ni; M) molecular mass protein markers 14-97 kDa (Bio-Rad, USA). Presence or absence of reducer (DTT) in sample buffer is marked under corresponding lanes.
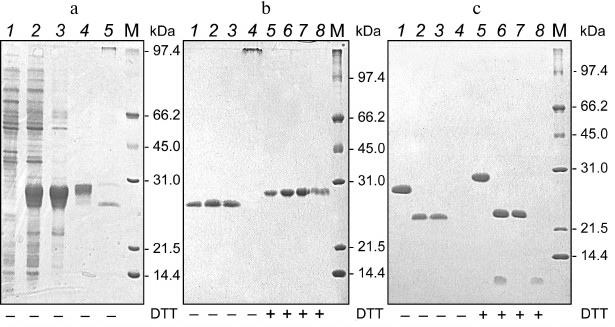
In total, the fractions collected after purification of refolded 6His-s-tag-EPO protein on WorkBeads 40 Ni contained ~0.9 mg of protein.
The data of purification efficiency and protein yield at different stages are presented in the table.
Yield of target protein during purification of 6His-s-tag-EPO and rhEPO
without additional domains
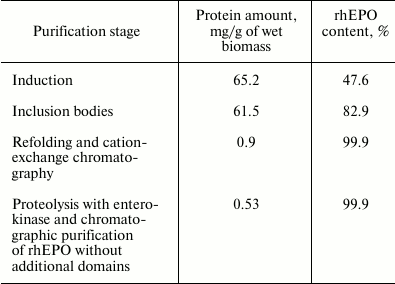
Despite the good yields of the MBP-EPO and 6His-MBP-EPO chimeric forms following the renaturation stage and affinity purification (data not shown), it was impossible to separate the rhEPO without additional domains from the carrier protein (MBP or 6His-MBP). So, the variant of rhEPO purification after enterokinase-mediated proteolysis of the 6His-s-tag-EPO construct was the only one acceptable even if the large losses during refolding and following cation-exchange chromatography were taken into account. The losses and low yield of rhEPO can be explained by the fact that only a small portion of the protein folds correctly, while the largest portion exhibits a tendency to aggregate due to absence or incorrect formation of disulfide bonds.
To investigate the structure of 6His-s-tag-EPO purified after refolding on WorkBeads 40 S sorbent, the respective band from polyacrylamide gel (Fig. 2b, lane 6) was subjected to proteolysis in the gel with sequential addition of DTT and iodoacetamide to the gel (see “Materials and Methods” for details). The MALDI mass spectrum of the resulting tryptic peptides is presented in Fig. 3a, and their sequences as well as modification are given in Table S1 (see Supplement to this paper on the site of the journal (http://protein.bio.msu.ru/biokhimiya) and Springer site (https://link.springer.com/journal/10541)). The mass-spectrometric analysis revealed several peaks corresponding to cysteine-containing peptides. These are peptides: T3+IAA (peak with m/z 763.446), T5+2IAA (peak with m/z 2803.1903) – this peptide contained two cysteine residues that formed one of the disulfide bonds in the native protein; T18+IAA (peak with m/z 1210.5994); and T19+IAA (peak with m/z 969.4527). The cysteine residues in these peptides are present in the form of carbamidomethyl cysteines. As presented in Fig. 3b, the identified tryptic peptides cover 83% of the sequence, which is considered as a good result. The absence of a number of peptides can be explained by their small mass (<700 Da), thus they are located outside the selected detection range, and the absence of the peak in the mass spectrum corresponding to a peptide (29-54) can be explained by high content of aspartic acid in it (it contains seven aspartic acid residues), which likely makes it difficult to detect it in the form of a positive ion and/or makes it impossible to extract from the gel.
Fig. 3. MALDI mass spectra of tryptic peptides of 6His-s-tag-EPO protein (a, c) and diagrams illustrating coverage of the 6His-s-tag-EPO sequence with tryptic peptides (b, d). a) MALDI mass spectrum of tryptic peptides produced during analysis of the protein band with apparent molecular mass of ~28 kDa (Fig. 2b, lane 6). Before the trypsinolysis the protein in the gel was first treated with DTT, and cysteine residues reduced with iodoacetamide; b) diagram illustrating the coverage of the protein sequence with tryptic peptides which spectrum is presented in panel (a); c) MALDI mass spectrum of tryptic peptides produced during analysis of the protein band with apparent molecular mass of ~26 kDa (Fig. 2b, lane 2). The protein in gel was not treated with DTT and iodoacetamide prior to trypsinolysis; d) diagram illustrating the coverage of the sequence of the protein with tryptic peptides whose mass spectrum is presented in panel (c). In panels (b) and (d), the peptides identified in the mass spectrum are labeled with rectangles with the peptide name inside (T-peptide No.), and the corresponding amino acid sequence is presented above. Cysteine-containing peptides are designated as gray rectangles; disulfide bonds between the identified peptides are shown with dashed line (peptides are highlighted with gray color).
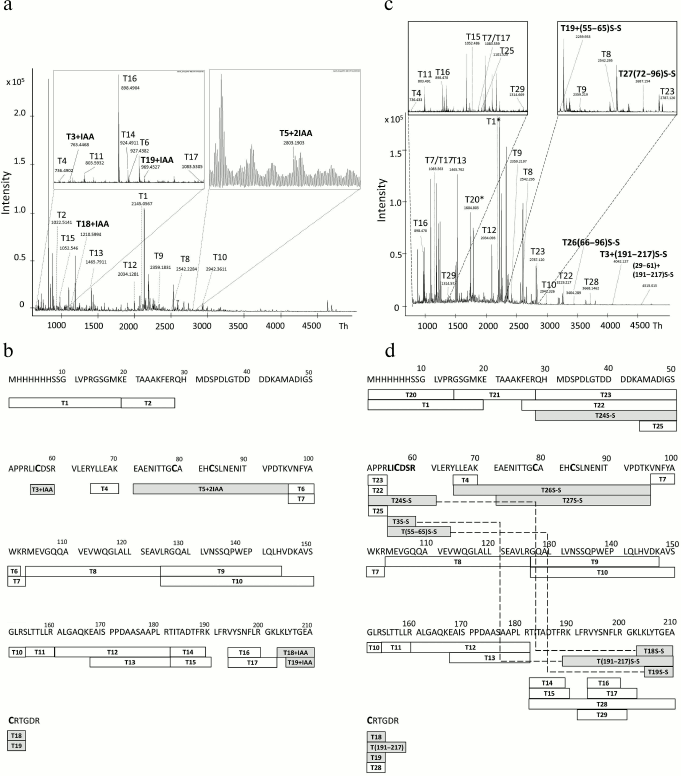
To investigate the soluble form of 6His-s-tag-EPO after refolding and purification using MALDI TOF spectrometry, the forms were first transferred into 25 mM ammonium acetate (pH 7.5) via dialysis and treated with trypsin also in the presence of DTT and iodoacetamide (mass spectra not presented). Analysis of the mass spectra showed that the set of peptides produced by trypsinolysis in the gel and in solution was practically identical, except that the mass spectrum peaks corresponding to peptides (26-54) and (127-153) were absent among the peptides produced from the solution. In addition, we note that the peaks corresponding to all cysteine-containing peptides are present in the spectrum similarly to the case of the experiment based on in-gel trypsinolysis.
To evaluate the efficiency of refolding of the protein, the respective band in polyacrylamide gel (Fig. 2b, lane 2) was subjected to trypsinolysis in the gel, but DTT and iodoacetamide were not added during the reaction. The buffer without reducer was used for gel loading. The MALDI mass spectrum of the resulting tryptic peptides is presented in Fig. 3c, and their sequences and modifications are given in Table S2 (see Supplement). The peaks corresponding to carbamidocysteine peptides are absent in the mass spectrum (Fig. 3, a and b), and the peaks confirming the presence of disulfide bonds in the protein are present. In particular, the peak with m/z 3404.029, designated in Fig. 3c as T26S-S, and the peak with m/z 2687.154 (T27S-S) confirm the presence of a disulfide bond between the Cys79 and Cys83 residues. The presence of a second disulfide bond between the Cys57 and Cys211 residues is corroborated by the presence of peaks in the mass spectrum with m/z 4515.0148 (T24+T18S-S), 4042.127 (T3+(191-217)S-S), and 2259.933 (T19+T(55-65)S-S). Hence, the mass-spectrometric analysis reliably established the presence of two disulfide bonds in the protein.
The analysis of mass spectra of the tryptic peptides produced in solution (mass spectra not shown) also confirms the presence of two disulfide bonds in the protein. In particular, the peak with m/z 2687.144 (T27S-S) is present in the spectrum, which confirms the presence of a disulfide bond between the Cys79 and Cys83 residues, as reported for the “in-gel” method. The second disulfide bond between the Cys57 and Cys111 residues is confirmed by the presence of peaks with m/z 2433.129 (T(55-61)+T(204-217)S-S), m/z 4042.127 (T3+(191-217)S-S), m/z 2845.3242 (T(44-61)+T(206-213)S-S) and m/z 3086.2388 (T(44-61)+T(204-213)S-S).
Preparation of rhEPO without additional protein domains was conducted using proteolytic hydrolysis of the 6His-s-tag-EPO with enterokinase at its specific recognition site with subsequent purification of the cleaved fragment using chromatography on the WorkBeads 40 Ni sorbent.
The combined fractions 1-3 (Fig. 2b) containing 6His-s-tag-EPO after refolding, chromatography on WorkBeads 40 S, and dialysis against 20 mM Tris-HCl (pH 8.0) with volume of 9 ml were treated with enterokinase for 3 h and purified from the cleaved fragments using the WorkBeads 40 Ni sorbent. EPO was not retained on the column and was eluted in the first washing fractions, and the 6His-s-tag fragment was captured by the sorbent and eluted with 2 M urea solution in 20 mM Tris-HCl (pH 8.0) supplemented with 400 mM imidazole. Aliquots of the fractions were treated with sample buffer with and without DTT, and then subjected to electrophoresis in 15% polyacrylamide gel (Fig. 2c). The volume of washing fractions was 4 ml, and in total two fractions were combined (total volume 8 ml). The resulting protein solution had concentration of 0.066 mg/ml and the protein was electrophoretically homogenous in the presence of reducing agent (DTT). It is worth mentioning that the cleaved fragment 6His-s-tag was not detected in the gel under nonreducing conditions (Fig. 2c, lanes 2 and 4), and it is present in the gel as a band of the corresponding mobility under reducing conditions (Fig. 2c, lanes 6 and 8). This is probably because the 6His-s-tag fragment aggregates after cleavage, although no aggregates were identified in the gel.
The refolded 6His-s-tag-EPO before and after hydrolysis with enterokinase and purification was used for measuring in vitro activity with the TF-1 cell line. The specific activity of the proteins calculated by the regression model was the same in both cases and was 13.4% of the activity of epoetin beta for 6His-s-tag-EPO before enterokinase hydrolysis, 12.9% for the 6His-s-tag-EPO treated with enterokinase and purified. The results of activity measurements are presented in Fig. 4. It is likely that the reduced activity of the recombinant EPO without additional domains produced by bacterial biosynthesis in comparison with the recombinant epoetin beta of eukaryotic origin can be explained by the lack of glycosylation of the former, differences in the secondary (conformational) structure of the EPO synthesized in bacterial and eukaryotic cells, or by the introduction of the additional seven amino acid residues at the N-terminal of the rhEPO produced in E. coli following treatment with enterokinase (see Fig. 1).
Fig. 4. Dependence of optical density in the well on the sample concentration during determination of in vitro specific activity of 6His-s-tag-EPO before and after cleavage with enterokinase: 1) epoetin beta; 2) 6-His-s-tag-EPO; 3) recombinant EPO produced following the treatment of 6-His-s-tag-EPO with enterokinase and purification.
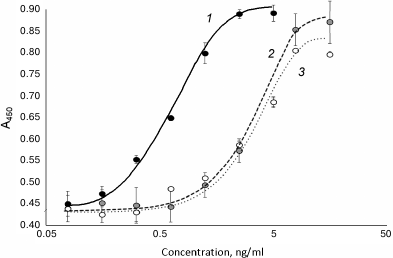
The effect of almost one order of magnitude reduction of the activity of EPO of bacterial origin on introduction of the 20-a.a. sequence including 10 histidines at the N-terminal was reported by Boissel et al. [13], who measured activity in the proliferation test with splenocytes; moreover, proteolytic hydrolysis of the additional N-terminal sequence resulted in significant (6-fold) increase in the activity of erythropoietin of bacterial origin (we did not observe this in our case). Almost identical in vitro activity of both EPO variants produced in this work – 6His-s-tag-EPO and rhEPO after cleavage of the additional domain – makes both variants worth further investigation with regard of the possibility of their application in vivo, in particular for local administration in an effort to improve osseointegration. The availability of the additional domain in the case of 6His-s-tag-EPO could play a favorable role due to increase in the protein molecular mass and, hence, decrease in the rate of protein clearance from the organism. The s-tag additional domain facilitates better solubility of eukaryotic proteins synthesized in E. coli, such as rhBMP-2 cytokines in comparison with the forms of these proteins without the additional domain [19, 21]. We have demonstrated encouraging results of application of rhBMP-2 recombinant protein with the additional s-tag domain [19] previously in several model studies on reparation of bone tissue and increasing the strength of the cornea (for application in keratoprosthesis) with small and large animals [22-26]. The work on production of the form of rhEPO for regenerative medicine and osteoplastic treatment could be further advanced in the direction of production of recombinant form of protein with additional domains facilitating concentration of the target protein in the area of injury. In this case, the difference in activity in comparison with eukaryotic rhEPO would be compensated by the increase in local concentration of the cytokine.
Hence, in this investigation the 6His-s-tag-EPO chimeric protein and the product of its proteolytic hydrolysis rhEPO, produced via bacterial synthesis in E. coli, demonstrated biological activity in vitro. It was found that the presence of the additional protein domain at the N-terminal position did not affect the cytokine activity. The presence of two disulfide bonds in the refolded form of the protein corresponding to the native folding of the protein chain was demonstrated using MALDI TOF mass spectrometry.
Acknowledgments
This work was financially supported by the Russian Science Foundation (project No. 16-15-00133).
REFERENCES
1.Cazzola, M., Mercuriali, F., and Brugnara, C.
(1997) Use of recombinant human erythropoietin outside the setting of
uremia, Blood, 89, 4248-4267.
2.Shiozawa, Y., Jung, Y., Ziegler, A. M., Pedersen,
E. A., Wang, J., Wang, Z., Song, J., Wang, J., Lee, C. H., Sud, S.,
Pienta, K. J., Krebsbach, P. H., and Taichman, R. S. (2010)
Erythropoietin couples hematopoiesis with bone formation, PLoS
One, 5, e10853.
3.Wu, C., Giaccia, A. J., and Rankin, E. B. (2014)
Osteoblasts: a novel source of erythropoietin, Curr. Osteoporos.
Rep., 4, 428-432.
4.Li, C., Shi, C., Kim, J., Chen, Y., Ni, S., Jiang,
L., Zheng, C., Li, D., Hou, J., Taichman, R. S., and Sun, H. (2015)
Erythropoietin promotes bone formation through EphrinB2/EphB4
signaling, J. Dent. Res., 94, 455-463.
5.Sun, H., Jung, Y., Shiozawa, Y., Taichman, R. S.,
and Krebsbach, P. H. (2012) Erythropoietin modulates the structure of
bone morphogenetic protein 2-engineered cranial bone, Tissue Eng.
Part A, 18, 2095-2105.
6.Rolfing, J. H., Jensen, J., Jensen, J. N., Greve,
A. S., Lysdahl, H., Chen, M., Rejnmark, L., and Bunger, C. (2014) A
single topical dose of erythropoietin applied on a collagen carrier
enhances calvarial bone healing in pigs, Acta Orthop.,
85, 201-209.
7.Lai, P. H., Everett, R., Wang, F. F., Arakawa, T.,
and Goldwasser, E. (1986) Structural characterization of human
erythropoietin, J. Biol. Chem., 261, 3116-3121.
8.Recny, M. A., Scoble, H. A., and Kim, Y. (1987)
Structural characterization of natural human urinary and recombinant
DNA-derived erythropoietin. Identification of des-arginine 166
erythropoietin, J. Biol. Chem., 262, 17156-17163.
9.Helenius, A., and Aebi, M. (2001) Intracellular
functions of N-linked glycans, Science, 291,
2364-2369.
10.Higuchi, M., Oh-eda, M., Kuboniwa, H., Tomonoh,
K., Shimonaka, Y., and Ochi, N. (1992) Role of sugar chains in the
expression of the biological activity of human erythropoietin, J.
Biol. Chem., 267, 7703-7709.
11.Egrie, J. C., and Browne, J. K. (2001)
Development and characterization of novel erythropoiesis stimulating
protein (NESP), Br. J. Cancer, 84, 3-10.
12.Jeong, T. H., Son, Y. J., Ryu, H. B., Koo, B. K.,
Jeong, S. M., Hoang, P., Do, B. H., Song, J. A., Chong, S. H.,
Robinson, R. C., and Choe, H. (2014) Soluble expression and partial
purification of recombinant human erythropoietin from E. coli,
Protein Express. Purif., 95, 211-218.
13.Boissel, J. P., Lee, W. R., Presnell, S. R.,
Cohen, F. E., and Bunn, H. F. (1993) Erythropoietin structure-function
relationships. Mutant proteins that test a model of tertiary structure,
J. Biol. Chem., 268, 5983-5993.
14.Narhi, L. O., Arakawa, T., Aoki, K., Wen, J.,
Elliott, S., Boone, T., and Cheetham, J. (2001) Asn to Lys mutations at
three sites which are N-glycosylated in the mammalian protein decrease
the aggregation of Escherichia coli-derived erythropoietin,
Protein Eng., 14, 135-140.
15.Gasparian, M. E., Ostapchenko, V. G., Schulga, A.
A., Dolgikh, D. A., and Kirpichnikov, M. P. (2003) Expression,
purification, and characterization of human enteropeptidase catalytic
subunit in Escherichia coli, Protein Express. Purif.,
31, 133-139.
16.Shevchenko, A., Tomas, H., Havlis, J., Olsen, J.
V., and Mann, M. (2006) In-gel digestion for mass spectrometric
characterization of proteins and proteomes, Nat. Protoc.,
1, 2856-2860.
17.Sun, P., Tropea, J. E., and Waugh, D. S. (2011
Enhancing the solubility of recombinant proteins in Escherichia
coli by using hexahistidine-tagged maltose-binding protein as a
fusion partner, Methods Mol. Biol., 705, 259-274.
18.Raran-Kurussi, S., and Waugh, D. S. (2016) A dual
protease approach for expression and affinity purification of
recombinant proteins, Anal. Biochem., 504, 30-37.
19.Karyagina, A. S., Boksha, I. S., Grunina, T. M.,
Demidenko, A. V., Poponova, M. S., Sergienko, O. V., Lyashchuk, A. M.,
Galushkina, Z. M., Soboleva, L. A., Osidak, E. O., Bartov, M. S.,
Gromov, A. V., and Lunin, V. G. (2017) Two variants of recombinant
human bone morphogenetic protein 2 (rhBMP-2) with additional protein
domains: synthesis in an Escherichia coli heterologous
expression system, Biochemistry (Moscow), 82,
613-624.
20.Wang, Y. J., Liu, Y. D., Chen, J., Hao, S. J.,
Hu, T., Ma, G. H., and Su, Z. G. (2010) Efficient preparation and
PEGylation of recombinant human non-glycosylated erythropoietin
expressed as inclusion body in E. coli, Int. J. Pharm.,
386, 156-164.
21.Karyagina, A. S., Boksha, I. S., Grunina, T. M.,
Demidenko, A. V., Poponova, M. S., Sergienko, O. V., Lyashchuk, A. M.,
Galushkina, Z. M., Soboleva, L. A., Osidak, E. O., Semikhin, A. S.,
Gromov, A. V., and Lunin, V. G. (2016) Optimization of rhBMP 2 active
form production in a heterologous expression system using
microbiological and molecular genetic approaches, Mol. Genet.
Microbiol. Virol., 31, 208-213.
22.Bartov, M. S., Gromov, A. V., Poponova, M. S.,
Savina, D. M., Nikitin, K. E., Grunina, T. M., Manskikh, V. N., Gra, O.
A., Lunin, V. G., Karyagina, A. S., and Gintsburg, A. L. (2016) Modern
approaches to studies of new osteogenic biomaterials on the model of
regeneration of critical-size cranial defects in rats, Bull. Exp.
Biol. Med., 162, 273-276.
23.Gaifullin, N. M., Karyagina, A. S., Gromov, A.
V., Terpilovsky, A. A., Malanin, D. A., Demeshchenko, M. V., and
Novochadov, V. V. (2016) Morphological characteristics of
osteointegration after application of titanium implants with bioactive
coating and recombinant bone morphogenetic protein rhBMP-2,
Morfologiya, 149, 77-84.
24.Zakharov, V. D., Zairat’yants, O. V.,
Andreev, A. Yu., Osidak, E. O., Borzenok, S. A., Krasheninnikov, S. V.,
Karyagina, A. S., and Domogatsky, S. P. (2016) Effect of rhBMP-2 growth
factor in composition with collagen carrier on morphological and
mechanical properties of cornea, Oftal’mokhirurgiya,
4, 20-28.
25.Zakharov, V. D., Andreev, A. Yu.,
Zairat’yants, O. V., Osidak, E. O., Borzenok, S. A.,
Krasheninnikov, S. V., Karyagina, A. S., and Domogatsky, S. P. (2016)
Morphological changes in rabbit cornea caused by the bone and cartilage
growth factor rhBMP-2 used as a component of intracorneal implant,
Klin. Eksp. Morfol., 4, 36-42.
26.Bartov, M. S., Gromov, A. V., Manskikh, V. N.,
Makarova, E. B., Rubshteyn, A. P., Poponova, M. S., Savina, D. M.,
Savin, K. S., Nikitin, K. E., Grunina, T. M., Boksha, I. S., Orlova, P.
A., Krivozubov, M. S., Subbotina, M. E., Lunin, V. G., Karyagina, A.
S., and Gintsburg, A. L. (2018) Recombinant human Bone Morphogenetic
Protein-2 (rhBMP-2) with additional protein domain produced by
synthesis in Escherichia coli: in vivo activity in models
on small and large laboratory animals, Bull. Exp. Biol. Med., in
press.
Supplementary Tables S1 and S2 (PDF)