Structural Insight into Interaction between C20 Phenylalanyl Derivative of Tylosin and Ribosomal Tunnel
G. I. Makarov1,2, N. V. Sumbatyan2, and A. A. Bogdanov1,3*
1Lomonosov Moscow State University, Faculty of Chemistry, 119991 Moscow, Russia2South Ural State University, 454080 Chelyabinsk, Russia
3Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119992 Moscow, Russia; E-mail: bogdanov@belozersky.msu.ru
* To whom correspondence should be addressed.
Received April 21, 2017; Revision received May 10, 2017
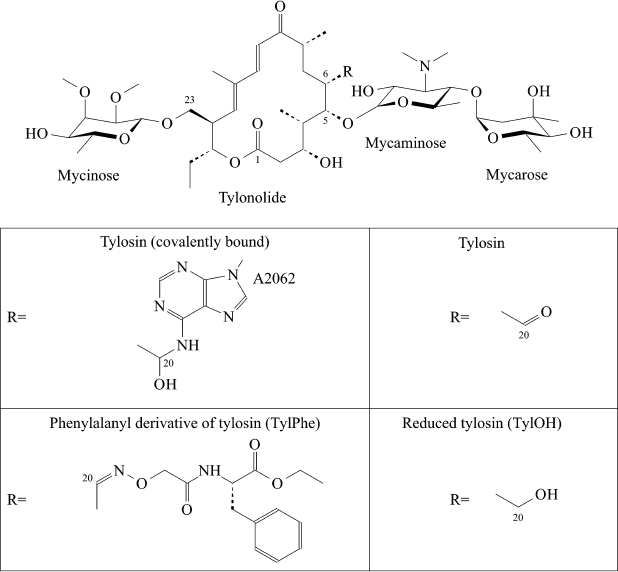
Macrolides are clinically important antibiotics that inhibit protein biosynthesis on ribosomes by binding to ribosomal tunnel. Tylosin belongs to the group of 16-membered macrolides. It is a potent inhibitor of translation whose activity is largely due to reversible covalent binding of its aldehyde group with the base of A2062 in 23S ribosomal RNA. It is known that the conversion of the aldehyde group of tylosin to methyl or carbinol groups dramatically reduces its inhibitory activity. However, earlier we obtained several derivatives of tylosin having comparable activity in spite of the fact that the aldehyde group of tylosin in these compounds was substituted with an amino acid or a peptide residue. Details of the interaction of these compounds with the ribosome that underlies their high inhibitory activity were not known. In the present work, the structure of the complex of tylosin derivative containing in position 20 the residue of ethyl ester of 2-imino(oxy)acetylphenylalanine with the tunnel of the E. coli ribosome was identified by means of molecular dynamics simulations, which could explain high biological activity of this compound.
KEY WORDS: ribosome, simulation, antibiotics, tylosin, molecular dynamicsDOI: 10.1134/S0006297917080077
Abbreviations: MD, molecular dynamics; NPET, nascent peptide exit tunnel; OMT, 5-O-mycaminosyltylonolide; PTC, peptidyltransferase center; TylPhe, phenylalanyl derivative of tylosin.
Macrolides comprise a large family of natural and semisynthetic
antibiotics that suppress the translation process in bacterial cells.
Their target is the exit tunnel incorporated into large subunit of the
ribosome (nascent peptide exit tunnel, NPET) whose function consists of
transportation of a nascent polypeptide chain from the
peptidyltransferase center (PTC) to the ribosome surface where first
processes of posttranslational modification and folding of protein
molecules are realized. Macrolides contain 12-, 14-, or 16-membered
macrolactone rings connected with one or several carbohydrate residues
[1]. X-ray analysis of complexes of the ribosome
with macrolides reveals that their lactone ring forms numerous
hydrophobic contacts with nucleotide residues of 23S rRNA that form the
NPET walls, while the carbohydrate residues are oriented along the main
axis of the NPET [2, 3]. It is
generally accepted that the mechanism of protein synthesis suppression
by macrolides consists either in creation of steric obstruction for
polypeptide chain advancing through the NPET or in allosteric
inhibition of the PTC due to formation specific complexes of macrolides
with NPET walls and/or a nascent peptide [4].
Some macrolides have been used in humans (for example, erythromycin) or in veterinary (for example, tylosin) for many years. However, like for all other classes of antibiotics, the number of macrolide-resistant pathogens grows year by year. Numerous efforts to chemically modify existing macrolides are constantly attempted aiming to overcome bacterial resistance to them. Development of ketolides, a new class of promising semisynthetic antibiotics obtained by modification of 14-membered macrolides, can be mentioned here as a positive example [5]. A series of amino acid and peptide derivatives of 16-membered macrolides – tylosin, desmycosin, and 5-O-mycaminosyltylonolide (OMT) – was synthesized in our laboratory [6, 7]. Some 16-membered macrolides containing an ethylaldehyde group in C6 position of the lactone ring have the unique ability to covalently bind with A2062 nucleotide residue of 23S rRNA (here and elsewhere numeration accepted for E. coli rRNA is used) (Fig. 1). Many compounds obtained by us efficiently bind to ribosomes and suppress protein synthesis in cell-free systems to roughly the same extent as the parental antibiotics [8]. With that a paradoxical situation arose – amino acid and peptide residues substituting the aldehyde group of antibiotics only slightly changed their activity, but it was known that reduction of this group of Tyl to carbinol or a methyl group entirely canceled the ability of the compound to suppress growth of bacteria and inhibit protein synthesis in vitro, and it decreases efficiency of its binding to the ribosome by orders of magnitude [8]. It was clear that in the case of compounds studied by us, the absence of their ability to form covalent bond with A2062 is compensated by interaction between amino acid or peptide residues with ribosome components. However, apart from the simplest dipeptide derivative of tylosin, atomic structures of complexes of ribosomes with these compounds are not available. Therefore, in the present work we used the molecular dynamics (MD) simulation approach for investigation of details of the interaction of the ribosome with the phenylalanyl derivative of tylosin (TylPhe; Fig. 1), one of the C20 macrolide derivatives that binds to the ribosome and suppresses its activity with greatest efficiency [8]. MD simulations of the complexes of reduced (TylOH; Fig. 1) and unmodified tylosin with the ribosome were also performed.
It should be noted that the MD simulation method is now arbitrarily widely applied for investigation of conformational mobility of complicated structures such as the ribosome and its interaction with ligands [9]. Formerly, using this approach we successfully found a rational explanation for the anomalous inhibition activity of amino acid derivatives of one of the members of the tylosin family [10].
Fig. 1. Structures of tylosin derivatives modeled in present work.
MATERIALS AND METHODS
Simulated systems. The structure of the E. coli ribosome was derived from the 3.1 Å resolution X-ray structure (PDB ID: 4V7U) [2] with addition of modified bases to 23S rRNA in compliance with the database [11]. Positions of modified bases were optimized by energy minimization by combination of the steepest descent method with the L-BFGS algorithm [12], followed by computation of 2-ns molecular dynamics controlled by a velocity-rescaling thermostat with stochastic correction. Incidentally, positions of all unmodified bases were fixed, whereas modified bases, ions, and water moved, and erythromycin contained in the source X-ray structure was removed. After optimization, regions having at least one atom within a cubic area with 7-nm edge that included the entire NPET and PTC in a way that the center of this region was located at the ribosomal tunnel aligned along an imaginary Z-axis were separated. A similar approach was applied in published works [13, 14]. Tylosin was placed in the selection by superimposition of the structure of the Haloarcula marismortui ribosome complex with tylosin having 3.0 Å resolution (PDB ID: 1K9M) [3]. Conformations of mycinose and mycarose residues in the binding site were optimized by docking with rDock software [15] retaining position, orientation, and conformation of the lactone ring and mycaminose residue unchanged. Other derivatives of tylosin were placed in the manner described above, whereby the phenylalanyl substituent of TylPhe was placed by docking similarly to mycarose and mycinose residues. During simulations, the mycaminose residue was protonated at the 3′-dimethylamine group in all compounds.
MD conditions. All simulations of molecular dynamics and analysis of trajectories obtained were done using GROMACS [16, 17] software version 5.0.4 and 5.1.4. Canonical and modified amino acid and nucleotide residues were modeled with the parm99sb force field [18], while tylosin and its derivatives were modeled with the GAFF force field [19]. Optimized three-dimensional structures and molecular electrostatic potentials of newly parameterized residues and compounds were prepared by quantum-chemical Hartree–Fock calculations with the 6-31G* basis set. Partial charges were evaluated with the RESP model [20].
All simulations were run at T = 300 K with 0.1-ps coupling time controlled by a velocity-rescaling thermostat with additional stochastic correction [21] and isotropic constant-pressure boundary conditions controlled by a Berendsen barostat [22] with 5-ps coupling time. Electrostatic interactions were computed using the particle-mesh Ewald method (PME) [23] with 0.125-nm grid step and fourth-order interpolation. The simulated system was centered in a cubic box with 8.8-nm edge filled with TIP4PEW [24] water molecules so that system edges was covered by a 0.9 nm thick solvent layer. Negative charge of the system was neutralized by adding potassium ions with optimized parameters [25], whereby added ions were positioned close to negatively charged group [26] (program kindly provided by A. Zalevsky). Besides that, some of the water molecules were in a random manner replaced by potassium, magnesium, and chlorine ions to prevent elution of potassium and magnesium counterions to the water phase. In such manner, 24 potassium ions, 2 magnesium ions and, 28 chloride ions were added to reach 7 mM MgCl2 and 100 mM KCl concentration in the water phase. In total, 444 potassium ions, 28 chloride ions, and 82 magnesium ions were contained in the system during simulations. Residues having at least one atom located within 0.1 nm from the edge of the simulated regions of the ribosome were positionally restrained, whereas the other atoms could move freely. A similar approach, as referenced above, was applied for modeling of the ribosome tunnel so far and does not affect movability of rRNA residues composing the NPET walls. In all simulations, the integration time step was 2 fs and coordinates were recorded into the trajectory file every 25 ps. To limit lengths of covalent bonds with hydrogen atoms, the LINCS algorithm was applied [27]. The number and duration of trajectories are given in the table.
Duration and number of trajectories obtained during simulation of
tylosin and its derivatives

Computation of well-tempered metadynamics of TylPhe phenylalanyl substituent was performed with the PLUMED software version 2.3 [28] working in conjunction with GROMACS software version 5.1.4. Cartesian coordinates of the phenyl ring in the substituent were chosen as collective variables, and biasing potential was adjusted by addition of a three-dimensional Gaussians with 2 kJ/mol height and 1 Å width every 4000 steps (8 ps) until the Gaussian width reached approximately 0.1 kJ/mol. The bias factor was 10.
Methods of trajectory analysis. Analysis of position and probability of hydrogen bonds was based on purely geometrical considerations: a hydrogen bond was considered to exist if an atom-donor of hydrogen and an atom-acceptor was spaced less than 3.5 Å apart and the angle between the line connecting these two atoms and bond between atom-donor and hydrogen covalently bound to it was less than 30°. Probability of a hydrogen bond was calculated as the ratio of number of trajectory frames containing this hydrogen bond to total number of frames in the trajectory.
Also, as part of the analysis a specifically developed program was used to detect the occurrence of stacking interactions between bases in a defined area. Detection was based on purely geometrical considerations being guided by three values calculated for every residue pair analyzed: angle between base planes, length of segment intercepted on normal originating from the center of the first base by the second base plane – an estimation of interplanar space, and distance between second base center and point in which the mentioned normal is cut by the second base plane – spacing of centers. In so doing, three cases were discerned:1) if interplanar angle was less than 30°, interplanar distance was between 1.8 and 5.2 Å and spacing of centers was less than 4 Å, then it was assumed that bases formed stacking interaction;2) if interplanar angle was less than 30°, interplanar distance was less than 1.8 Å, and spacing of centers was between 5.5 and 8 Å, then it was assumed that bases lay in one plane (and can form a complementary pair that is necessary to test by hydrogen bond analysis); 3) otherwise, it is thought that interaction is absent.
Probability of stacking interaction formation was estimated as the ratio of the number of frames in which this stacking interaction was found to the total number of frames in the trajectory.
RESULTS
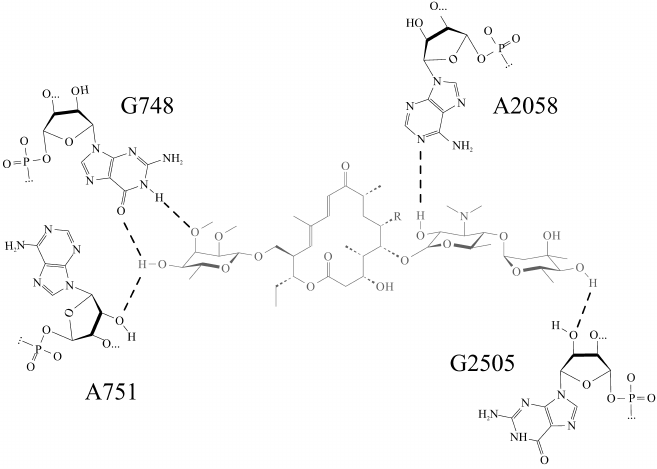
Molecular dynamics simulations of a complex of the ribosome with tylosin derivatives (TylPhe and TylOH) undertaken in present work clarify principles of their interactions with the NPET walls. The “tylosin core” of all simulated in the present work derivatives of tylosin (Fig. 1) retains almost identical conformation and position in the NPET, so that every compound forms five hydrogen bonds with 23S rRNA nucleotide residues located on the NPET walls (Fig. 2). Besides that, the aldehyde group of tylosin not bound covalently with nucleotide residue A2062 forms an unstable hydrogen bond with the exocyclic amino group of the nucleotide base of A2062. It is worth noting that the carbinolamino group that forms as a result of covalent binding of the tylosin aldehyde group to the exocyclic amino group of A2062 base includes an asymmetric center, which must have S-configuration because it assists preservation of noncovalent interactions between the “tylosin core” and the NPET. In contrast, when the R-configuration of the carbinolamine carbon atom is present, this hydrogen bond is destabilized.
Fig. 2. Most stable hydrogen bonds of the “tylosin core” (shown in gray) in tylosin (Tyl) and reduced tylosin (TylOH). Hydrogen bonds are shown by dotted lines.
Using classical docking approaches, we were not able to find a conformation of TylPhe in which the phenylalanyl substituent forms stable contacts with the NPET walls and is retained during MD simulations. All structures of the TylPhe complex predicted by this method dissipated, and its phenylalanyl substituent lost contact with the NPET components and transferred into its lumen, even though its “tylosin core” retained its position and conformation. Hence, we resorted to a metadynamics method as an instrument to search for possible conformations of the phenylalanyl substituent using the coordinates of the geometrical center of its benzene ring as collective variables. In this manner, conformations of linker group allowing various positions of the benzene ring in the tunnel were implicitly assumed. During metadynamics, a set of phenylalanyl substituent conformations was obtained, and one of them corresponded to the formation of stacking interaction between the C2610 base and the phenyl ring. It is important to note that the “tylosin core” did not change its conformation and did not shift from the tylosin-binding site in all variations of phenylalanyl substituent conformation, which additionally attests to strength of its interaction with the NPET walls.
Using the GROMOS method [29], we clustered states of the system obtained during metadynamics on phenylalanyl substituent conformation to reveal conformations in which the system stayed during metadynamics and which, therefore, correspond to free energy minima and select states being closest to centers of the ten most occupied clusters. Each of these was used as the source state for equilibrium MD simulations. States obtained in this manner were clustered using the same method. The phenylalanyl substituent takes up such conformation that its aromatic ring forms stacking interaction with the C2610 base, and the N3H of the U1782 base forms a hydrogen bond with the ester oxygen of the phenylalanyl residue in first, third, and seventh by occupancy clusters containing respectively 50, 9, and 2% of all frames. With that, the stacking interaction between the C2610 base and the benzene ring was found by geometrical criterion in only 19% of frames referring to first cluster, in 42% of frames referring to third cluster, and in 77% of frames referring to seventh cluster. Foregoing a hydrogen bond with U1782 residue stabilizes in the same series: from 11% occurrence in the frames of first cluster to 72% in the frames of third and seventh clusters. Intramolecular hydrogen bonds between oxygen and nitrogen of the oxime fragment and the 3-hydroxyl group of tylonolide were also slightly stabilized in the same series. In general, the conformation of the phenylalanyl substituent in which it interacts with the C2610 and U1782 bases occurs in 61% of all frames, so that the most frequently encountered conformations obtained by metadynamics converted into three highly similar conformations favoring these interactions.
Stabilization of TylPhe noncovalent interactions in the third and especially in the seventh cluster relative to the first suggests that the first cluster may represent a certain “early” state of TylPhe binding with still not steady interactions, but the seventh cluster may represent a “late” state in which noncovalent interactions are finally steady. It counts in favor of this suggestion that states referred to third and seventh cluster occur near the ends of trajectories, but states referred to first cluster occur near the sources of trajectories.
We obtain 200 ns MD trajectories of this system originating from its states that are closest to centers of the first, the third, and the seventh cluster to test this suggestion. With that, it was recognized that the phenylalanyl substituent conformation found in the first cluster changes during simulation with disruption of stacking interactions, but its conformation found in the seventh cluster is retained and, as could be expected, the conformation of the substituent found in the third cluster transfers into it. This stable conformation is shown in Fig. 3.
Fig. 3. Conformation of TylPhe phenylalanyl substituent modeled by MD methods. TylPhe is shown as dark gray, residues of 23S rRNA are shown as light gray, and hydrogen bond is shown by black dashes.
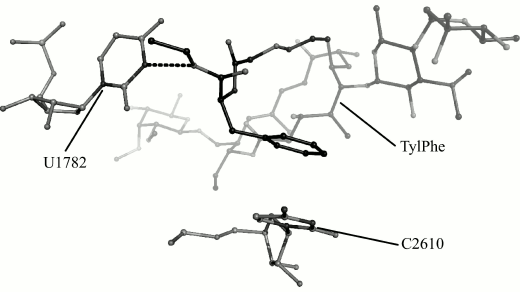
In the TylPhe conformation found in this manner, its phenylalanyl substituent forms two stable contacts with 23S rRNA provided by stacking interactions between the C2610 base and the phenyl ring of TylPhe as well as a hydrogen bond between N3H of the U1782 base and the ester oxygen of the phenylalanyl residue. Besides, the nature of one of three hydrogen bonds of the mycinose residue being characteristic for tylosin derivatives changes: the 2′-hydroxyl group of the ribose residue of A751 forms a hydrogen bond with the 4′-hydroxyl group of the mycinose residue serving as hydrogen donor rather than acceptor as with other modeled derivatives.
The role of the phenylalanyl residue that offers the configuration described above in the interaction between TylPhe and the NPET can be evaluated by comparison of TylPhe with the tylosin derivative that is unable to covalently bind with rRNA and protein residues too due to reduction of the Tyl aldehyde group at position 20 (Fig. 1). For this purpose, fifteen 40-ns trajectories of the system containing reduced tylosin and sixteen 40-ns trajectories of the system containing TylPhe in position and conformation corresponding to the seventh cluster were simulated. With that, the initial velocities of atoms for any trajectory were generated over again to additionally inject an element of randomness into the behavior of the simulated systems. In seven of fifteen trajectories of the system, containing reduced tylosin obtained in this way, this derivative began to leave the macrolide-binding site as observed by variation of root-mean-square deviation (RMSD) value of reduced tylosin and by means of visual analysis. It is important to emphasize that in none of sixteen systems containing TylPhe its displacement from the tylosin major binding site was observed. At the same time, in four of sixteen trajectories the conformation of the phenylalanyl substituent was changed, followed by disruption of stacking interactions and the hydrogen bond described above. Hence, the phenylalanyl substituent taking the conformation determined by us significantly strengthens interaction of TylPhe with the ribosome tunnel in comparison to reduced tylosin, which is in excellent agreement with experimental data [8].
DISCUSSION
As mentioned above, the main goal of the present work was to find an answer to the question of why TylPhe features high and comparable with unmodified tylosin ability to inhibit ribosome translational activity, whereas TylOH is essentially inactive. It was evident that the explanation of unexpected behavior of TylPhe lies in special properties of the phenylalanyl moiety (Fig. 1) and peculiarities of its interaction with 23S rRNA or proteins in the ribosome tunnel area. In addition, TylPhe very strongly binds to the ribosome, and what is more, the phenylalanyl substituent and “tylosin core” of TylPhe synergistically strengthen interactions of each other with ribosome components [8]. It is important that, as shown in this work, the “tylosin core” of both TylPhe and unmodified tylosin binds with the ribosome in the same manner. The same conclusion was made when the macrolactone position of tylosin and its alanylalanyl derivative was compared by X-ray analysis [30].
From metadynamics calculations and equilibrium MD simulations, we propose the conformation of the TylPhe phenylalanyl residue that is stabilized by strong noncovalent interactions and contributes to stable TylPhe binding. We have found that the complex of TylPhe with the bacterial ribosome should be even more stable than that of tylosin itself due to formation of the more stable hydrogen bond between the 4′-hydroxyl group of the mycinose residue of TylPhe and the O6 atom of nucleotide residue G748. Conversely, the 2′-hydroxyl group of nucleotide residue A751 forms a hydrogen bond with the same 4′-hydroxyl group of mycinose acting as a donor, so that one hydrogen bond in the cases of tylosin bound and unbound covalently to residue A2062 and in reduced tylosin is replaced by two H-bonds in the case of TylPhe. This redistribution requires a minimal shift of the “tylosin core” in the tylosin-binding site and it is not prohibited for tylosin and all its derivatives. The reason why it was not observed during the present simulations probably stems from the fact that the “tylosin core” of all derivatives except TylPhe falls into local energetic minima, which it could not leave during equilibrium MD simulations. In contrast, metadynamics simulations have shown that the mycinose residue in TylPhe was in a more favorable position.
We have found that the highly flexible nucleotide residue A2062 retains its mobility, since its conformation is not stabilized by any interactions with TylPhe. Only in an “early” state, the exocyclic amino group of adenine in A2062 can still form weak hydrogen bonds with oxygen and nitrogen atoms of the oxime fragment of the phenylalanyl substituent, and the amide nitrogen of the phenylalanyl residue can form an equally weak hydrogen bond with the N7 atom of the A2062 adenine. It is likely that formation of highly stable stacking interactions with cytosine in C2610 and a hydrogen bond with the uridine base in U1782 by phenylalanine residue is more favorable. It should be emphasized that in the E. coli ribosome the nucleotide residue C2610 is bound by strong stacking interactions with nucleotide residue G2581. It is worth noting that the conformation of the phenylalanyl substituent proposed earlier [8], in which its phenyl group forms stacking interaction with the base of the nonconservative residue 2586, occurs among conformations found by us with metadynamics. However, this conformation is less favorable, since in the conformation of the suggested earlier phenylalanyl substituent is not able to form stable hydrogen bonds.
Previously, it was shown that tylosin binds the E. coli ribosome more strongly than TylPhe, although the latter appears to be more active as a translation inhibitor [8]. From the data presented here, it is clear that even very strong interactions of the phenylalanyl group of TylPhe with the NPET walls cannot compensate the lack of the covalent bond formed by tylosin with the A2062 base. However, in the conformation of the phenylalanyl substituent found by us, TylPhe shifts into the NPET lumen more profoundly than unmodified tylosin and, as one can suggest, should block more efficiently the promotion of a nascent polypeptide chain through the tunnel. There are numerous evidences that antibiotics that bind in the NPET interact with the nascent polypeptide chain, and their activity varies depending on the nature of the amino acid sequence of the growing polypeptide chain [31, 32]. This circumstance may also be important for the increase in inhibiting activity of TylPhe relatively to tylosin observed in experiment.
We believe that the ability to answer questions such as those discussed in this work will help to develop new low-toxicity antibacterial agents that will overcome pathogen resistance to antibiotics.
Acknowledgments
We thank the Research Computing Center of Lomonosov Moscow State University for the opportunity to compute molecular dynamics using the “Lomonosov” supercomputer.
This research was supported by the Russian Science Foundation (project No. 14-24-00061-P, tylosin derivative simulations), by the Russian Foundation for Basic Research (project No. 16-04-00709-a, tylosin simulations), and by the Government of the Russian Federation (Resolution No. 211 from 16.03.2013, contract No. 02.A03.21.0011).
REFERENCES
1.Omura, S. (ed.) (2003) Macrolide Antibiotics:
Chemistry, Biology and Practice, 2nd Edn., Academic Press, N.Y.
2.Dunkle, J. A., Xiong, L., Mankin, A. S., and Cate,
J. H. D. (2010) Structures of the Escherichia coli ribosome with
antibiotics bound near the peptidyl transferase center explain spectra
of drug action, Proc. Natl. Acad. Sci. USA, 107,
17152-17157.
3.Hansen, J., Ippolito, J., Ban, N., Nissen P.,
Moore, P., and Steitz, T. (2002) The structures of four macrolide
antibiotics bound to the large ribosomal subunit, Mol. Cell,
10, 117-128.
4.Wilson, D. N. (2009) The A-Z of bacterial
translation inhibitors, Crit. Rev. Biochem. Mol. Biol.,
44, 393-433.
5.Ruan, Z.-X., Huangfu, D.-S., Xu, X.-J., Sun, P.-H.,
and Chen, W.-M. (2013) 3D-QSAR and molecular docking for the discovery
of ketolide derivatives, Exp. Opin. Drug Discov.,
8, 427-444.
6.Korshunova, G. A., Sumbatyan, N. V., Fedorova, N.
V., Kuznetsova, I. V., Shishkina, A. V., and Bogdanov, A. A. (2007)
Peptide derivatives of tylosin-related macrolides, Russ. J. Bioorg.
Chem., 33, 218-226.
7.Sumbatyan, N. V., Kuznetsova, I. V., Karpenko, V.
V., Fedorova, N. V., Chertkov, V. A., Korshunova, G. A., and Bogdanov,
A. A. (2010) Amino acid and peptide derivatives of the tylosin family
of antibiotics modified by aldehyde function, Russ. J. Bioorg.
Chem., 36, 245-256.
8.Starosta, A. L., Karpenko, V. V., Shishkina, A. V.,
Mikolajka, A., Sumbatyan, N. V., Schluenzen, F., Korshunova, G. A.,
Bogdanov, A. A., and Wilson, D. N. (2010) Interplay between the
ribosomal tunnel, nascent chain, and macrolides influences drug
inhibition, Chem. Biol., 17, 504-514.
9.Makarov, G. I., Makarova, T. M., Sumbatyan, N. V.,
and Bogdanov, A. A. (2016) Investigation of ribosomes using molecular
dynamics simulation methods, Biochemistry (Moscow), 81,
1579-1588.
10.Shishkina, A., Makarov, G., Tereshchenkov, A.,
Korshunova, G., Sumbatyan, N., Golovin, A., Svetlov, M., and Bogdanov,
A. (2013) Conjugates of amino acids and peptides with
5-O-mycaminosyltylonolide and their interaction with the ribosomal exit
tunnel, Bioconjug. Chem., 24, 1861-1869.
11.Cannone, J. J., Subramanian, S., Schnare, M. N.,
Collett, J. R., D’Souza, L. M., Du, Y., Feng, B., Lin, N.,
Madabusi, L. V., Muller, K. M., Pande, N., Shang, Z., Yu, N., and
Gutell, R. R. (2002) The comparative RNA web (CRW) site: an online
database of comparative sequence and structure information for
ribosomal, intron, and other RNAs, BMC Bioinformatics, 3,
1-31.
12.Byrd, R., Lu, P., and Nocedal, J. (1995) A
limited memory algorithm for bound constrained optimization, SIAM J.
Sci. Statist. Comput., 16, 1190-1208.
13.Petrone, P., Snow, C., Lucent, D., and Pande, V.
(2008) Side-chain recognition and gating in the ribosome exit tunnel,
Proc. Natl. Acad. Sci. USA, 105, 16549-16554.
14.Lucent, D., Snow, C., Aitken, C., and Pande, V.
(2010) Non-bulk-like solvent behavior in the ribosome exit tunnel,
PLoS Comput. Biol., 6, e1000963.
15.Ruiz-Carmona, S., Alvarez-Garcia, D., Foloppe,
N., Garmendia-Doval, A. B., Juhos, S., Schmidtke, P., Barril, X.,
Hubbard, R. E., and Morley, S. D. (2014) rDock: a fast, versatile and
open source program for docking ligands to proteins and nucleic acids,
PLoS Comput. Biol., 10, e1003571.
16.Van der Spoel, D., Lindahl, E., Hess, B.,
Groenhof, G., Mark, A., and Berendsen, H. (2005) GROMACS: fast,
flexible, free, J. Comput. Chem., 26, 1701-1718.
17.Van der Spoel, D., Lindahl, E., Hess, B., and
Kutzner, C. (2008) GROMACS 4: algorithms for highly efficient,
load-balanced, and scalable molecular simulation, J. Chem. Theory
Comp., 4, 435-447.
18.Hornak, V., Abel, R., Okur, A., Strockbine, B.,
Roitberg, A., and Simmerling, C. (2006) Comparison of multiple Amber
force fields and development of improved protein backbone parameters,
Prot. Struct. Funct. Bioinform., 65, 712-725.
19.Wang, J., Wolf, R. M., Caldwell, J. W., Kollman,
P. A., and Case, D. A. (2004) Development and testing of a general
Amber force field, J. Comput. Chem., 25, 1157-1174.
20.Bayly, C. I., Cieplak, P., Cornell, W., and
Kollman, P. A. (1993) A well-behaved electrostatic potential based
method using charge restraints for deriving atomic charges: the RESP
model, J. Phys. Chem., 97, 10269-10280.
21.Bussi, G., Donadio, D., and Parrinello, M. (2007)
Canonical sampling through velocity rescaling, J. Chem. Phys.,
126, 014107-014106.
22.Berendsen, H., Postma, J., van Gunsteren, W.,
DiNola, A., and Haak, J. (1984) Molecular dynamics with coupling to an
external bath, J. Chem. Phys., 81, 3684-3690.
23.Darden, T., York, D., and Pedersen, L. (1993)
Particle mesh Ewald: an Nlog(N) method for Ewald sums in
large systems, J. Chem. Phys., 98, 10089-10092.
24.Horn, H. W., Swope, W. C., Pitera, J. W., Madura,
J. D., Dick, T. J., Hura, G. L., and Head-Gordon, T. (2004) Development
of an improved four-site water model for biomolecular simulations:
TIP4P-EW, J. Chem. Phys., 120, 9665-9678.
25.Joung, I. S., and Cheatham, T. E. (2008)
Determination of alkali and halide monovalent ion parameters for use in
explicitly solvated biomolecular simulations, J. Phys. Chem. B,
112, 9020-9041.
26.Athavale, S., Petrov, A., Hsiao, C., Watkins, D.,
Prickett, C., Gossett, J., Lie, L., Bowman, J., O’Neill, E., Hud,
C. B. N., Wartell, R., Harvey, S., and Williams, L. (2012) RNA folding
and catalysis mediated by iron (II), PLoS One, 7,
1-7.
27.Hess, B., Bekker, H., Berendsen, H. J., and
Fraaije, J. G. (1997) LINCS: a linear constraint solver for molecular
simulations, J. Comput. Chem., 18, 1463-1472.
28.Tribello, G. A., Bonomi, M., Branduardi, D.,
Camilloni, C., and Bussi, G. (2014) PLUMED 2: new feathers for an old
bird, Comp. Phys. Commun., 185, 604-613.
29.Daura, X., Gademann, K., Jaun, B., Seebach, D.,
Gunsteren, W. F., and Mark, A. E. (1999) Peptide folding: when
simulation meets experiment, Ang. Chem. Inter. Ed., 38,
236-240.
30.Wilson, D., Harms, J., Nierhaus, K., Schlunzen,
F., and Fucini, P. (2005) Species-specific antibiotic–ribosome
interactions: implications for drug development, Biol. Chem.,
386, 1239-1252.
31.Marks, J., Kannan, K., Roncase, E. J., Klepacki,
D., Kefi, A., Orelle, C., Vázquez-Laslop, N., and Mankin, A. S.
(2016) Context-specific inhibition of translation by ribosomal
antibiotics targeting the peptidyl transferase center, Proc. Natl.
Acad. Sci. USA, 113, 12150-12155.
32.Sothiselvam, S., Neuner, S., Rigger, L.,
Klepacki, D., Micura, R., Vázquez-Laslop, N., and Mankin, A. S.
(2017) Binding of macrolide antibiotics leads to ribosomal selection
against specific substrates based on their charge and size, Cell
Rep., 16, 1789-1799.