Attenuation of Focal Adhesion Kinase Reduces Lipopolysaccharide-Induced Inflammation Injury through Inactivation of the Wnt and NF-κB Pathways in A549 Cells
D. Bai#, S. Cong#, and L. P. Zhu*
Jining No. 1 People’s Hospital, Department of Pediatrics, 272011 Jining, China; E-mail: zhuliping5280@126.com# These authors contributed equally to this work.
* To whom correspondence should be addressed.
Received September 14, 2016; Revision received October 25, 2016
Overall analysis and understanding of mechanisms are of great importance for treatment of infantile pneumonia due to its high morbidity and mortality worldwide. In this study, we preliminarily explored the function and mechanism of focal adhesion kinase (FAK) in regulation of inflammatory response induced by lipopolysaccharides in A549 cells. Flow cytometry, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, quantitative reverse transcription polymerase chain reaction, and Western blot analysis were used to explore the correlation of FAK expression with cell apoptosis, viability, and the inflammatory cytokine activity in A549 cells. The results showed that knockdown of FAK enhanced cell viability, suppressed apoptosis, and decreased inflammatory cytokine activity. In addition, downregulation of FAK could activate the Wnt and nuclear factor κB signaling pathways. These findings suggest that FAK might be involved in progression of infantile pneumonia and could be a new therapeutic target for this disease.
KEY WORDS: infantile pneumonia, lipopolysaccharides, focal adhesion kinase, Wnt and NF-κB pathways, new therapeutic targetDOI: 10.1134/S0006297917040058
Abbreviations: FAK, focal adhesion kinase; IP, infantile pneumonia; LPS, lipopolysaccharide; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (assay); NF-κB, nuclear factor κB.
Being an important cause of mortality and morbidity, infantile pneumonia
(IP) is one of the most common and severe respiratory diseases of
childhood [1]. IP is always accompanied by fever,
cough, and shortness of breath, and attacks of breathlessness can lead
to respiratory failure and heart failure, sepsis, and even death [2-5]. IP can include aspiration
pneumonia and infectious pneumonia, and the incidence and severity are
highest in the first year of life, especially in the first six months
[6-8].
Focal adhesion kinase (FAK) is focal adhesion protein that is encoded by the FAK gene. It is associated with bad prognosis of many kinds of diseases [9, 10]. To control the process of cell proliferation and metabolism, FAK, which is a non-receptor protein tyrosine kinase, senses a variety of extracellular signals [11, 12]. FAK has been shown to closely associate with cell adhesion and movement, and it can promote cell apoptosis of different tumor types [13, 14]. Feng et al. showed that silencing of FAK inhibits malignant cell proliferation and invasion in gastric cancer [15]. Moreover, by suppression of macrophage infiltration, Ninjurin-1 has been reported to inhibit tumor growth through repression of FAK signaling [16]. FAK has become an attractive selective and validated target for cancer therapy. However, there are few studies focused on the functional role of FAK in treatment of IP.
For evaluation of inflammatory response, many cytokines have been detected in previous studies. Buck et al. reported that levels of tumor necrosis factor α (TNF-α) and interleukin 1β (IL-1β) were both elevated in newborns with pneumonia [17]. During inflammatory cascade, TNF-α and IL-1β are the first cytokines that stimulate production of interleukin 6 (IL-6) [18]. Thus, in the present study, we focused on expression levels of TNF-α, IL-1β, and IL-6 to evaluate inflammatory response. Additionally, a recent study suggested that inactivation of the FAK pathway inhibits the phosphatidylinositol-3-kinase (PI3K)/AKT/mTOR pathway [19]. Moreover, FAK has been reported to promote cell proliferation by enhancing the Wnt signaling pathway [20]. Also, nuclear factor κB (NF-κB) was shown to be activated through the PI3K/AKT pathway [21]. Therefore, we hypothesized the FAK might be involved in the regulation of the Wnt and NF-κB pathways.
Pulmonary delivery is a common therapeutic routine for pulmonary disease. Type II cells, a kind of pulmonary epithelium, possess multiple functions that make them a potential target for therapy of pneumonia [22]. The A549 cell line is widely used having characteristic features of type II cells [23]. The great majority of studies reported in the literature have explored potential therapeutic targets for pneumonia utilizing A549 cells [24, 25]. Thus, we selected A549 cells to construct an inflammation model through stimulation with lipopolysaccharides (LPS) and then further studied the effect of FAK on cell viability, apoptosis, and inflammatory response as well as on related signaling pathways.
MATERIALS AND METHODS
Cell culture and LPS treatment. The A549 cell line was purchased from the American Type Culture Collection (Manassas, USA). The cells were suspended in Dulbecco’s modified Eagle’s medium (Gibco, Thermo Fisher Scientific, USA) with 4 mM glutamine, 10% fetal bovine serum, 100 U/ml penicillin, and 100 µg/ml streptomycin. The cells were cultured in the medium until they achieved 80-90% confluence. Then, the cells were treated with 0.2% trypsin and 0.02% EDTA for 5 min and then collected by centrifugation for 2 min. The cells were plated at 2·104 cells/cm2 on 24-well plates and maintained for 48 h until they reached optimal confluence [26].
SiRNA transfection. FAK-specific siRNA (si-FAK) and negative control (siNC) were designed and synthesized by GenePharma (China). Cell transfection was performed using Lipofectamine 3000 (Invitrogen Life Technologies, USA) according to the manufacturer’s instructions.
MTT assay. The viability of cells attached to the substrate was determined by a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay according to a standard procedure.
Apoptosis assay. Apoptosis was assayed to identify and quantify apoptotic cells using an Annexin V-FITC/propidium iodide (PI) apoptosis detection kit (Beijing Biosea Biotechnology, China). The A549 cells were seeded in 6-well plates. Treated cells were washed twice with cold phosphate-buffered saline (PBS) and resuspended in binding buffer. The adherent and floating cells were combined and treated following the manufacturer’s instruction and measured with flow cytometer (Beckman Coulter, USA) to differentiate apoptotic cells (Annexin-V positive and PI negative) from necrotic cells (Annexin-V and PI positive).
qRT-PCR. Total RNA was isolated from cells attached to the substrate using TRIzol reagent (Invitrogen, USA) and treated with DNase I (Promega, USA). Reverse transcription was performed utilizing the Multiscribe RT kit (Applied Biosystems, Switzerland). The reverse transcription conditions were 10 min at 25°C, 30 min at 48°C, and a final step of 5 min at 95°C. The quantitative PCR was performed using Power SYBR Green PCR master mix (Applied Biosystems) following the manufacturer’s protocol. The sequences of the synthesized primers (GenePharma) are given in the table. The fold changes in gene expression were estimated with the 2–ΔΔCt method described previously [27]. GAPDH (glyceraldehyde-3-hposphate dehydrogenase) was used as the reference gene.
Sequence of primers used in this study
Western blot analysis. The protein for Western blot analysis was extracted from cells attached to the substrate using RIPA lysis buffer (Beyotime Biotechnology, China) supplemented with protease inhibitors (Roche, China). The proteins were quantified using the BCA™ Protein Assay Kit (Pierce, USA). Equal amounts of proteins were loaded on a 12% gel and isolated using a Bis-Tris Gel system Bio-Rad (USA) following the manufacturer’s instructions. The proteins were transferred to polyvinylidene fluoride (PVDF) membranes. Then the membranes carrying the proteins were incubated with primary antibodies against FAK (sc-271195), IL-1β (sc-12742), IL-6 (sc-65989) (all from Santa Cruz, USA); against TNF-α (ab6671), Wnt3a (ab81614), Wnt5a (ab159811), β-catenin (ab27798) (all from Abcam, UK); against inhibitor of nuclear factor κBα (t-IκBα, 9247), phosphorylated IκBα (p-IκBα, 2859), p65 (t-p65, 6956), phosphorylated p65 (p-p65, 3033) (all from Cell Signaling Technology, USA), and against GAPDH (Sigma, USA). Primary antibodies were prepared in 5% blocking buffer at dilution 1 : 1000 and respectively incubated with membranes at 4°C overnight, which was followed by washing and incubation with secondary antibodies labeled with horseradish peroxidase (HRP) for 1 h at room temperature. After rinsing, the membranes carrying blots and antibodies were transferred into a Bio-Rad ChemiDoc™ XRS system with addition of 200 µl Immobilon Western Chemiluminescent HRP Substrate (Millipore, USA) to cover the membrane surface. Signals were captured and the intensity of the bands was quantified using Image Lab™ Software (Bio-Rad, China).
Statistical analysis. The results of repeated experiments are presented as mean ± SD. Statistical analyses were performed using SPSS 19.0 statistical software. The p-values were calculated by one-way analysis of variance (ANOVA); p < 0.05 was considered as statistically significant difference.
RESULTS
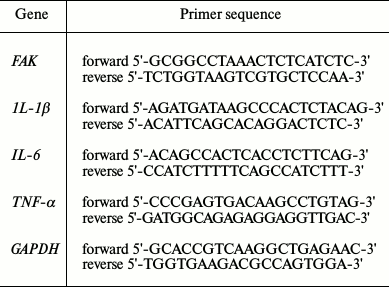
LPS induced A549 cell inflammation and decreased cell viability in a concentration-dependent manner. The injury model was constructed with stimulation by LPS of A549 cells. Cell viability of A549 cells was assayed under various concentrations (0, 5, 10, and 20 µg/ml) of LPS. Both the mRNA and protein expression levels of inflammatory cytokines were evaluated after treatment with LPS. The results showed that cell viability significantly decreased while the expression levels of inflammatory cytokines were markedly increased by LPS in comparison with the control in a concentration-dependent manner (p < 0.05 or p < 0.01) (Fig. 1).
Fig. 1. Correlation of focal adhesion kinase (FAK) with cell viability and inflammatory response. A549 cells were stimulated with LPS at 0, 5, 10, or 20 µg/ml. a) Cell viability; b) expression levels of inflammatory cytokines. Bars indicate means ± SD. * p < 0.05, ** p < 0.01 – significant difference compared with cells stimulated with 0 µg/ml LPS.
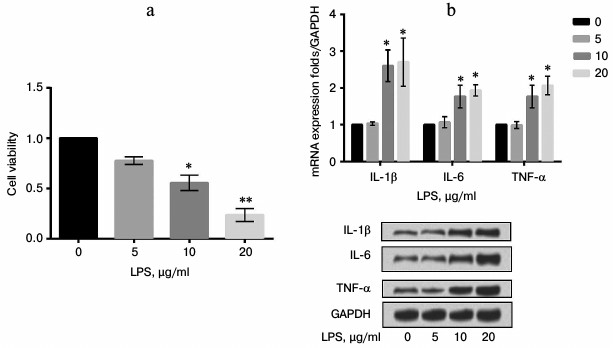
LPS upregulated expression of FAK in a concentration-dependent manner. To explore the effect of LPS on expression of FAK, mRNA and protein expression levels of FAK were estimated in cells treated with LPS at various concentrations (0, 5, 10, and 20 µg/ml). As shown in Fig. 2, the mRNA and protein expression levels of FAK were both upregulated by LPS in a concentration-dependent manner in comparison with the control (p < 0.01).
Fig. 2. mRNA and protein expression levels of focal adhesion kinase (FAK) in LPS-induced cells. The A549 cells were stimulated with LPS at 0, 5, 10, or 20 µg/ml. Bars indicate means ± SD; ** p < 0.01 – significant difference compared with cells stimulated with 0 µg/ml LPS.
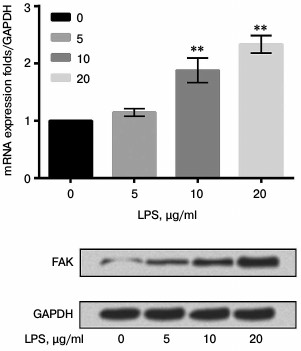
siFAK downregulated expression of FAK. A549 cells were respectively transfected with siFAK or siNC. After transfection, the mRNA and protein expression level of FAK were analyzed by qRT-PCR and Western blot assay, respectively. The results shown in Fig. 3 confirmed that siFAK downregulated the expression of FAK in both mRNA and protein levels compared with that in cells transfected with siNC (p < 0.01).
Fig. 3. Effect of small interfering RNA targeting focal adhesion kinase (siFAK) on expression of FAK. The A549 cells were respectively transfected with siFAK or its negative control (siNC). Bars indicate means ± SD; ** p < 0.01 – significant difference compared with cells transfected with siNC.
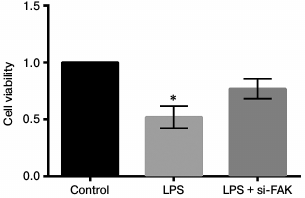
Knockdown of FAK enhanced cell viability. The A549 cells were divided into three groups – control, LPS, and LPS + siFAK groups. Cells in the LPS group were treated with 10 µg/ml LPS. Cells in LPS + si-FAK group were transfected with siFAK and treated with 10 µg/ml LPS. Cells in the control group were without any treatment. The MTT assay was employed to explore the effects of FAK knockdown on cell viability. Cell viability was remarkably decreased by LPS treatment in comparison with the control (p < 0.05). The cell viability was significantly enhanced by interference of siFAK, resulting in no significant difference in comparison with the control (Fig. 4). Thus, from this part of the work we drew the conclusion that knockdown of FAK upregulated cell viability in LPS-treated A549 cells.
Fig. 4. Effect of focal adhesion kinase (FAK) on cell viability. A549 cells were divided into three groups – control, LPS, and LPS + si-FAK groups. Cells in the LPS group were treated with 10 µg/ml LPS. Cells in LPS + si-FAK group were transfected with small interfering RNA targeting FAK (siFAK) and treated with 10 µg/ml LPS. Cells in the control group were without any treatment. Bars indicate means ± SD; * p < 0.05 – significant difference compared with control.
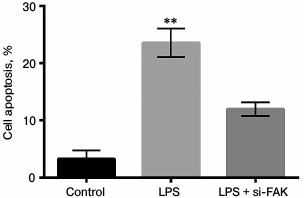
Knockdown of FAK inhibited cell apoptosis. The A549 cells were divided into three groups as described above. Cell apoptosis was assessed by flow cytometry. In Fig. 5, cell apoptosis was significantly promoted by LPS in comparison with that in control (p < 0.01). When FAK expression was inhibited, the promotion of cell apoptosis induced by LPS was reversed, resulting in no significant difference in comparison with that in control. We concluded that FAK knockdown protected cells from apoptosis.
Fig. 5. Effect of focal adhesion kinase (FAK) on cell apoptosis. A549 cells were divided into three groups – control, LPS, and LPS + si-FAK groups. Cells in the LPS group were treated with 10 µg/ml LPS. Cells in the LPS + siFAK group were transfected with small interfering RNA targeting FAK (siFAK) and treated with 10 µg/ml LPS. Cells in control group were without any treatment. Bars indicate means ± SD; ** p < 0.01 – significant difference compared with control.
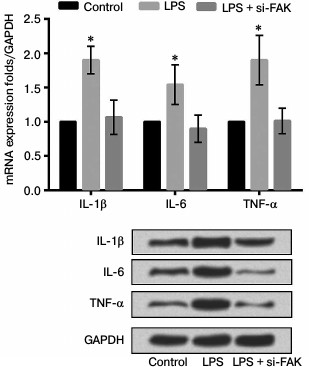
Knockdown of FAK downregulated expression levels of inflammatory cytokines. The A549 cells were divided into three groups as described above. The mRNA and protein expression levels of various inflammatory cytokines were assessed to investigate the effect of FAK on the production of LPS-induced IL-1β, IL-6, and TNF-α. As shown in Fig. 6, the mRNA and protein levels of these inflammatory cytokines were both upregulated by stimulation of LPS compared with that in control (p < 0.05). Interference by siFAK significantly suppressed the expression of inflammatory cytokines induced by LPS, leading to no prominent difference in comparison with that in control. We concluded from these results that LPS promoted inflammatory cytokine expression while FAK knockdown could alleviate the inflammatory response induced by LPS.
Fig. 6. Effect of focal adhesion kinase (FAK) on expression of inflammatory cytokines. A549 cells were divided into three groups – control, LPS, and LPS + si-FAK groups. Cells in the LPS group were treated with 10 µg/ml LPS. Cells in LPS + si-FAK group were transfected with small interfering RNA targeting FAK (siFAK) and treated with 10 µg/ml LPS. Cells in the control group were without any treatment. Bars indicate means ± SD; * p < 0.05 – significant difference compared with control.
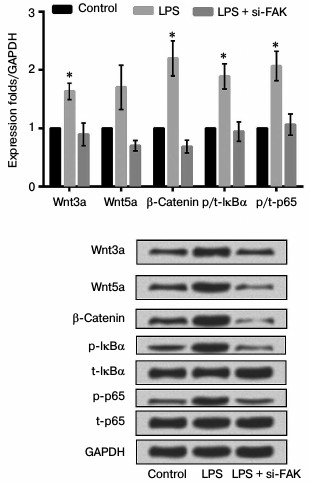
Knockdown of FAK suppressed inflammatory response through the Wnt and NF-κB pathways. To explore how the inhibition of FAK affects the production of NF-κB-mediated inflammatory cytokines, the mRNA and protein expression levels of Wnt3a, Wnt5a, β-catenin, p/t-IκBα, and p/t-p65 were analyzed by qRT-PCR and Western blot analysis. The cells were divided into three groups as described above. According to the results shown in Fig. 7, the mRNA and protein expression levels of key kinases were both markedly upregulated by stimulation of LPS in comparison with that in control (p < 0.05). While in cells transfected with siFAK the increased expressions were all reversed, leading to no obvious difference in comparison with that in control. All these results suggested that knockdown of FAK inhibited the Wnt and NF-κB signaling pathways.
Fig. 7. Effect of focal adhesion kinase (FAK) knockdown on Wnt/nuclear factor κB (NF-κB) signaling pathway. A549 cells were divided into three groups – control, LPS, and LPS + siFAK groups. Cells in the LPS group were treated with 10 µg/ml LPS. Cells in LPS + siFAK group were transfected with small interfering RNA targeting FAK (siFAK) and treated with 10 µg/ml LPS. Cells in the control group were without any treatment. Bars indicate means ± SD; * p < 0.05 – significant difference compared with control. t-IκBα, inhibitor of NF-κBα; p-IκBα, phosphorylated IκBα; p-p65, phosphorylated p65.
DISCUSSION
Virus is considered the most common cause of pneumonia in children under five years old [28, 29]. Child pneumonia has always been considered to relate to tachypnea and/or chest recession, and aspiration pneumonia is a frequent cause of morbidity and mortality in children [29]. The clinical characteristics of IP have also been widely studied. In the study of Wang et al., the levels of pulmonary surfactant proteins A and D (SP-A, SP-D) in children with pneumonia were observed, and SP-D was shown to play an important role in regulation of the immune and inflammatory responses [30]. It is considered a common respiratory infectious disease in infancy, but the mechanism is far from sufficiently clear [31].
FAK is an integrated component of cell–cell adhesion and regulates the focal adhesion complex in the basal part of endothelial cells, which has been shown to act as a transcriptional regulator in the nucleus [32-34]. As a non-receptor protein tyrosine kinase, FAK plays a vital role in cell signaling pathways such as cell proliferation, survival, and migration [33, 35-37]. The findings of Serrels et al. confirmed a potential new purpose for FAK kinase inhibitors, which can cause immune-mediated tumor regression [33].
In the present study, we preliminarily explored the function and mechanism of FAK in regulation of inflammatory response induced by LPS in A549 cells. We first used LPS to construct an inflammatory model in A549 cells, then tested FAK expression in this model and found that LPS could upregulate the expression of FAK. Determined by MTT, the cell survival rate showed that knockdown of FAK could significantly increase viability of cells treated with LPS. Flow cytometry results showed that the knockdown of FAK reduced LPS-induced apoptosis. Then we tested the expression of inflammatory factors and found that knockdown of FAK could also reduce the expression levels of inflammatory markers. Additionally, Western blot results showed that the inhibition of FAK decreased activation of the Wnt and NF-κB signaling pathways, accompanied by reduction in cell inflammation.
Wnt3a is a canonical Wnt protein that functions via a β-catenin-dependent signaling pathway [38]. In contrast, Wnt5a is a noncanonical Wnt protein that has been reported to inhibit the canonical Wnt pathway via enhancement of β-catenin degradation [39]. In the present study, the expression levels of Wnt3a and Wnt5a were both enhanced by simulation with LPS, resulting in upregulated β-catenin despite the opposite effects of Wnt3a and Wnt5a. When cells were subjected to siFAK, the effect of LPS on expression levels of Wnt3a, Wnt5a, and β-catenin were all reversed. The results suggest that knockdown of FAK activates the Wnt/β-catenin pathway.
On the other hand, NF-κB dimers consist of five members – p65/RelA, p50, RelB, cRel, and p52 [40]. Under resting conditions, IκB proteins bind to NF-κB dimers and sequester them in the cytoplasm. When cells are stimulated, IκB is phosphorylated, ubiquitinated, and degraded, leading to release of NF-κB factors [41]. In our study, upon stimulation of LPS the phosphorylation of IκBα was upregulated, which might cause release of NF-κB factors by degradation of IκB proteins. Perhaps the phosphorylated level of p65 was upregulated by stimulation with LPS. Meanwhile, knockdown of FAK reversed the increase in IκBα phosphorylation, resulting in reversed regulation of p65 phosphorylation. Thus, knockdown of FAK might inhibit inflammatory response through the NF-κB signaling pathway.
In conclusion, this article provides a preliminary exploration of FAK in regulation of cell inflammatory response. The findings provide a solid foundation for further study of FAK function, providing a new strategy and a possible target for clinical treatment of inflammation in infants.
REFERENCES
1.Wysocki, J., Sluzewski, W., Gutterman, E., Jouve,
S., Moscariello, M., and Balter, I. (2016) Active hospital-based
surveillance of invasive pneumococcal disease and clinical pneumonia in
infants and young children in two Polish counties, Arch. Med.
Sci., 3, 629-638.
2.Shu, L. H., Jiang-Jiang, X. U., Wang, S., Zhong, H.
Q., Dong, X. Y., Jiang, K., Zhang, H. Y., Xiong, Q., Wang, C., and Sun,
T. (2015) Distribution of pathogenic microorganisms and its
relationship with clinical features in children with community-acquired
pneumonia, Chinese J. Contemp. Pediatr., 17,
1056-1061.
3.Chisti, M. J., Salam, M. A., Bardhan, P. K.,
Faruque, A. S., Shahid, A. S., Shahunja, K. M., Das, S. K., Hossain, M.
I., and Ahmed, T. (2015) Treatment failure and mortality amongst
children with severe acute malnutrition presenting with cough or
respiratory difficulty and radiological pneumonia, PLoS One,
10.
4.Leung, D. T., Das, S. K., Malek, M. A., Qadri, F.,
Faruque, A. S., Chisti, M. J., and Ryan, E. T. (2015) Concurrent
pneumonia in children under 5 years of age presenting to a diarrheal
hospital in Dhaka, Bangladesh, Am. J. Trop. Med. Hyg.,
93, 895-903.
5.Yu, Z. W., Qian, J., Gu, X. H., Zhang, X. J., Pan,
J. R., and Ju, H. L. (2015) Changes in serum inflammatory factors in
wheezing infants with community-acquired pneumonia, Chinese J.
Contemp. Pediatr., 17, 815-818.
6.Zhang, X., Wu, J., Zhang, B., and Dong, L. (2015)
Potassium dehydroandrographolide succinate injection for treatment of
infantile pneumonia: a systematic review and Meta-analysis, J. Trad.
Chinese Med., 35, 125-133.
7.Zhu, R., Lei, L., Zhao, L., Jie, D., Fang, W., Yu,
S., Song, Q., Ding, Y., and Yuan, Q. (2015) Characteristics of the
mosaic genome of a human parechovirus type 1 strain isolated from an
infant with pneumonia in China, Infect. Genet. Evol., 29,
91-98.
8.Gomez Del Pulgar, T., Cebrian, A.,
Fernandez-Acenero, M. J., Borrero-Palacios, A., Puerto-Nevado, L. D.,
Martinez-Useros, J., Marin-Arango, J. P., Carames, C., Vega-Bravo, R.,
and Rodriguez-Remirez, M. (2016) Focal adhesion kinase: predictor of
tumour response and risk factor for recurrence after neoadjuvant
chemoradiation in rectal cancer, J. Cell. Mol. Med., 20,
1729-1736.
9.Uehara, K., and Uehara, A. (2016) Differentiated
localizations of phosphorylated focal adhesion kinase in endothelial
cells of rat splenic sinus, Cell Tissue Res., 83,
1-12.
10.Zheng, X., Bao, W., Yang, J., Zhang, T., Sun, D.,
Liang, Y., Li, S., Wang, Y., Feng, X., and Hao, H. (2016) Focal
adhesion kinase directly interacts with TSC2 through its FAT domain and
regulates cell proliferation in cashmere goat fetal fibroblasts, DNA
Cell Biol., 9, 480-488.
11.Troutman, S., Moleirinho, S., Kota, S., Nettles,
K., Fallahi, M., Johnson, G. L., and Kissil, J. L. (2016) Crizotinib
inhibits NF2-associated schwannoma through inhibition of focal adhesion
kinase 1, Oncotarget, doi: 10.18632/oncotarget.10248.
12.Hao, Z., Shao, H., Golubovskaya, V. M., Chen, H.,
Cance, W., Adjei, A. A., and Dy, G. K. (2016) Efficacy of focal
adhesion kinase inhibition in non-small cell lung cancer with
oncogenically activated MAPK pathways, Br. J. Cancer, 2,
203-211.
13.Heffler, M., Golubovskaya, V. M., Dunn, K. M. B.,
and Cance, W. (2013) Focal adhesion kinase autophosphorylation
inhibition decreases colon cancer cell growth and enhances the efficacy
of chemotherapy, Cancer Biol. Ther., 14, 761-772.
14.Woo, J. K., Jang, Y. S., Kang, J. H., Hwang, J.
I., Seong, J. K., Lee, S. J., Jeon, S., Oh, G. T., Lee, H. Y., and Oh,
S. H. (2016) Ninjurin1 inhibits colitis-mediated colon cancer
development and growth by suppression of macrophage infiltration
through repression of FAK signaling, Oncotarget, 20,
29592-29604.
15.Feng, R., and Yang, S. (2016) Effects of
combining erlotinib and RNA-interfered downregulation of focal adhesion
kinase expression on gastric cancer, J. Int. Med. Res.,
44.
16.Shi, R., Wang, Q., Ouyang, Y., Wang, Q., and
Xiong, X. (2016) Picfeltarraenin IA inhibits lipopolysaccharide-induced
inflammatory cytokine production by the nuclear factor-κB pathway
in human pulmonary epithelial A549 cells, Oncol. Lett.,
11, 1195-1200.
17.Buck, C., Gallati, H., Pohlandt, F., and
Bartmann, P. (1994) Increased levels of tumor necrosis factor α
(TNF-α) and interleukin 1α (IL-1α) in tracheal
aspirates of newborns with pneumonia, Infection, 22,
238-241.
18.Jodal, M., Fihn, B. M., and Sun, Y. (1990)
Correlations and interactions in the production of interleukin-6
(IL-6), IL-1, and tumor necrosis factor (TNF) in human blood
mononuclear cells: IL-6 suppresses IL-1 and TNF, Blood,
75, 40-47.
19.You, D., Xin, J., Volk, A., Wei, W., Schmidt, R.,
Scurti, G., Nand, S., Breuer, E. K., Kuo, P., and Breslin, P. (2015)
FAK mediates a compensatory survival signal parallel to PI3K-AKT in
PTEN-null T-ALL cells, Cell Rep., 10, 2055-2068.
20.Sun, C., Yuan, H., Wang, L., Wei, X., Williams,
L., Krebsbach, P. H., Guan, J. L., and Liu, F. (2016) FAK promotes
osteoblast progenitor cell proliferation and differentiation by
enhancing Wnt signaling, J. Bone Min. Res., doi:
10.1002/jbmr.2908.
21.Harvey, R. D., Silberman, J., and Lonial, S.
(2015) The PI3 kinase/Akt pathway as a therapeutic target in multiple
myeloma, Fut. Oncol., 3, 639-647.
22.Foster, K. A., Oster, C. G., Mayer, M. M., Avery,
M. L., and Audus, K. L. (1998) Characterization of the A549 cell line
as a type II pulmonary epithelial cell model for drug metabolism,
Exp. Cell Res., 243, 359-366.
23.Uboldi, C., Bonacchi, D., Lorenzi, G., Hermanns,
M. I., Pohl, C., Baldi, G., Unger, R. E., and Kirkpatrick, C. J. (2009)
Gold nanoparticles induce cytotoxicity in the alveolar type-II cell
lines A549 and NCIH441, Part. Fibre Toxicol., 6,
1-12.
24.Wu, J., Zhao, J., Yu, J., Zhang, W., and Huang,
Y. (2015) Cylindromatosis (CYLD) inhibits Streptococcus
pneumonia-induced plasminogen activator inhibitor-1 expression via
interacting with TRAF-6, Biochem. Biophys. Res. Commun.,
463, 942-947.
25.Zhao, X., Li, H., Wang, J., Guo, Y., Liu, B.,
Deng, X., and Niu, X. (2015) Verbascoside alleviates pneumococcal
pneumonia by reducing pneumolysin oligomers, Mol. Pharmacol.,
89, 376-387.
26.Cantais, A., Mory, O., Pillet, S., Verhoeven, P.
O., Bonneau, J., Patural, H., and Pozzetto, B. (2014) Epidemiology and
microbiological investigations of community-acquired pneumonia in
children admitted at the emergency department of a university hospital,
J. Clin. Virol., 60, 402-407.
27.Li, Q., Liu, L., Zhang, Q., Liu, S., Ge, D., and
You, Z. (2014) Interleukin-17 indirectly promotes M2 macrophage
differentiation through stimulation of COX-2/PGE2 pathway in the cancer
cells, Cancer Res. Treat. Offic. J. Korean Cancer Assoc.,
46, 297-306.
28.Ali, A., Akhund, T., Warraich, G. J., Aziz, F.,
Rahman, N., Urmani, F. A., Qureshi, S., Petri, W. A., Bhutta, Z.,
Zaidi, A. K., and Hughes, M. A. (2016) Respiratory viruses associated
with severe pneumonia in children under two years old in a rural
community in Pakistan, J. Med. Virol., 88, 1882-1890.
29.Dop, D., Gheonea, C., Stanescu, G. L.,
Moroşanu, A. E., Diaconu, R., Niculescu, E. C., Ognean, M. L.,
and Niculescu, D. (2015) Aspiration pneumonia in an infant with
neurological sequelae – case report, Roman. J. Morphol.
Embryol., 56, 1191-1194.
30.Wang, L. L., Zheng, S. Y., Ren, L., Xiao, Q. Y.,
Long, X. R., Luo, J., Qu-Bei, L. I., Deng, Y., Xie, X. H., and Liu, E.
M. (2016) Levels of surfactant proteins A and D in bronchoalveolar
lavage fluid of children with pneumonia and their relationships with
clinical characteristics, Chinese J. Contemp. Pediatr.,
18.
31.Kessler, B. E., Sharma, V., Zhou, Q., Jing, X.,
Pike, L. A., Kerege, A. A., Sams, S. B., and Schweppe, R. E. (2016) FAK
expression, not kinase activity, is a key mediator of thyroid
tumorigenesis and pro-tumorigenic processes, Mol. Cancer Res.,
14, 869-882.
32.Zaidi, A. (2015) FAK kinase activity is required
for the progression of c-Met/β-catenin-driven HCC, Gene
Express., 17, 79-88.
33.Serrels, A., and Frame, M. C. (2016) FAK goes
nuclear to control anti-tumor immunity – a new target in cancer
immunotherapy, Oncoimmunology, 5, e1119356.
34.Thiyagarajan, V., Lin, S. H., Chang, Y. C., and
Weng, C. F. (2016) Identification of novel FAK and S6K1 dual inhibitors
from natural compounds via ADMET screening and molecular docking,
Biomed. Pharmacother., 80, 52-62.
35.Hutchinson, L. (2016) Ovarian cancer: FAK –
new target for antiangiogenic therapy, Nature Rev. Clin. Oncol.,
13, 328.
36.Ding, L., Wang, L., Sui, L., Zhao, H., Xu, X.,
Li, T., Wang, X., Li, W., Zhou, P., and Kong, L. (2016) Claudin-7
indirectly regulates the integrin/FAK signaling pathway in human colon
cancer tissue, J. Human Genet., 61, 711-720.
37.Zhang, R., Li, L., Yuan, L., and Zhao, M. (2015)
Hypoxic preconditioning protects cardiomyocytes against
hypoxia/reoxygenation-induced cell apoptosis via sphingosine kinase 2
and FAK/AKT pathway, Exp. Mol. Pathol., 100, 51-58.
38.Liu, F., Kohlmeier, S., and Wang, C. (2008) Wnt
signaling and skeletal development, Cell. Signal., 20,
999-1009.
39.Topol, L., Jiang, X., Choi, H., Garrettbeal, L.,
Carolan, P. J., and Yang, Y. (2003) Wnt-5a inhibits the canonical Wnt
pathway by promoting GSK-3-independent β-catenin degradation,
J. Cell Biol., 162, 899-908.
40.Oeckinghaus, A., Hayden, M. S., and Ghosh, S.
(2011) Crosstalk in NF-κB signaling pathways, Nature
Immunol., 12, 695-708.
41.Mulero, M. C., Bigas, A., and Espinosa, L. (2013)
IκBα beyond the NF-κB dogma, Oncotarget,
4, 1550-1551.