MINI-REVIEW: Interplay between Brain BDNF and Glutamatergic Systems: A Brief State of the Evidence and Association with the Pathogenesis of Depression
N. V. Gulyaeva
Institute of Higher Nervous Activity and Neurophysiology, Russian Academy of Sciences, 117485 Moscow, Russia; E-mail: nata_gul@mail.ru
Received October 25, 2016; Revision received November 11, 2016
The excitatory neurotransmitter glutamate system and the brain-derived neurotrophic factor (BDNF) system are principally involved in phenomena of cellular and synaptic plasticity. These systems are interacting, and disclosing mechanisms of such interactions is critically important for understanding the machinery of neuroplasticity and its modulation in normal and pathological situations. The short state of evidence in this review addresses experimentally confirmed connections of these mechanisms and their potential relation to the pathogenesis of depression. The connections between the two systems are numerous and bidirectional, providing for mutual regulation of the glutamatergic and BDNF systems. The available data suggest that it is complex and well-coordinating nature of these connections that secures optimal synaptic and cellular plasticity in the normal brain. Both systems are associated with the pathogenesis of depression, and the disturbance of tight and well-balanced associations between them results in unfavorable changes in neuronal plasticity underlying depressive disorders and other mood diseases.
KEY WORDS: BDNF, glutamate, TrkB, NMDA receptors, AMPA receptors, metabotropic glutamate receptors, neuronal plasticity, synaptic plasticity, depressionDOI: 10.1134/S0006297917030087
Abbreviations: AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors; BDNF, brain-derived neurotrophic factor; LTP, long-term potentiation; NMDAR, N-methyl-D-aspartate receptors; p75 (NTR), a low-affinity nerve growth factor receptor; TrkB, type B tyrosine kinase receptor for BDNF.
In the developing nervous system, as well as in the mature brain, the
formation of synapses between neurons is dependent upon their
electrical activity, and neurotrophic factors belonging to the
neurotrophin family produced by target cells play a crucial role in
activity-dependent forming of neural networks. Interplay between
neurotransmitter and neurotrophic factor signaling pathways mediates
adaptive responses of neural networks to environmental and internal
demands [1]. The excitatory neurotransmitter
glutamate system (glutamate + receptors + transporters) and
the brain-derived neurotrophic factor (BDNF) system (BDNF +
receptors) are systems principally involved in phenomena of cellular
and synaptic plasticity. These systems are interacting, and disclosing
mechanisms of such interactions is critically important for
understanding the machinery of neuroplasticity phenomena and its
modulation under normal and pathological situations. The present short
review considers the issue of relations between glutamate and BDNF
system components and addresses experimentally confirmed connections of
these mechanisms to pathogenesis of depression.
BDNF plays several prominent roles in synaptic plasticity. BDNF signaling mediates upregulation of proteins involved in neurogenesis, learning and memory, and neuronal survival, including proteins that regulate mitochondrial biogenesis, protein quality control, and resistance of cells to oxidative, metabolic, and proteotoxic stress. BDNF signaling is negatively regulated by stress hormone glucocorticoids that impair synaptic plasticity in the brain by downregulating spine density, neurogenesis, and long-term potentiation, effects linked to glucocorticoid regulation of BDNF. Initiating signal transduction through at least two membrane receptors, TrkB (a member of a receptor family of tyrosine kinases) and p75 (NTR, a low-affinity nerve growth factor receptor), BDNF is a key player in the activity-dependent regulation of synaptic structure and function, particularly of glutamatergic synapses [2].
The neurotransmitter role of glutamate has been known for half a century, while a variety of other regulatory functions of glutamate in the brain has since been shown (control of neurogenesis, neurite outgrowth, synaptogenesis, and neuron survival). Glutamate exerts its effects by signaling through cell-surface receptors. In mammals, four families of glutamate receptors have been identified, ionotropic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors (AMPAR, subunits GluR1-GluR4), kainate receptors (subunits GluR5-GluR7, KA-1, KA-2), N-methyl-D-aspartate (NMDA) receptors (NMDAR, subunits NR1-NR3), and metabotropic G protein-coupled glutamate receptors 1 (subunits GluR1, GluR5), -2 (subunits GluR2, GluR3), and -3 (subunits GluR4, GluR6, GluR7, GluR8). Glutamate transporters EAAT and VGLUT in neuronal and glial membranes remove glutamate from the extracellular space. Synaptic glutamate activates ionotropic receptors on the membrane of dendrites coupled with the influx of Na+ (via AMPAR) and Ca2+ (via NMDAR and voltage-dependent Ca2+ channels as well as Ca2+ release from endoplasmic reticulum stores) resulting in responses changing the structure and function of neurons. Calcium ion activates kinases (e.g. Ca2+-calmodulin-dependent protein kinases and protein kinase C) that in turn activate transcription factors including AP-1, cyclic AMP response element-binding protein (CREB) and nuclear factor κB (NF-κB), and translational regulators such as Arc (Activity-Regulated Cytoskeletal-associated protein) and FMRP (Fragile X Mental Retardation Protein). Both rapid kinase- and protease-mediated and delayed transcription-dependent glutamate receptor-mediated responses underlay different forms of neuronal plasticity, and BDNF and glutamate interact to regulate developmental and adult neuroplasticity [2].
BDNF modulates several distinct aspects of synaptic transmission. It can modify glutamate signaling directly, by changing the expression of glutamate receptor subunits and Ca2+-regulating proteins, or indirectly by inducing the production of antioxidant enzymes, energy-regulating proteins, and antiapoptotic Bcl-2 family members. Importantly, glutamate stimulates the production of BDNF, which in turn modifies neuronal glutamate sensitivity, Ca2+ homeostasis, and plasticity [2]. BDNF can be released in the mature form, which activates preferentially TrkB receptors, or as proBDNF, which is coupled to the stimulation of p75 (NTR). The mature form (cleaved proBDNF) induces rapid effects on glutamate release and may induce short- and long-term effects on the postsynaptic response to the neurotransmitter. BDNF can affect glutamate receptor activity by inducing the phosphorylation of the receptor subunits, which may also affect the interaction with intracellular proteins and, consequently, their recycling and localization to defined postsynaptic sites. BDNF-induced stimulation of local protein synthesis and transcriptional activity account for the delayed effects of BDNF on glutamatergic synaptic strength.
The first experiments linking the glutamatergic and BDNF systems were published two decades ago. Jarvis et al. [3] suggested that BDNF can serve as glycine-like ligand for the NMDAR receptor. Song et al. [4] demonstrated that BDNF rapidly enhanced synaptic transmission among hippocampal neurons, suggesting that BDNF acutely activated synaptic transmission via NMDAR. Using standard whole-cell patch-clamp recordings, Lessmann et al. [5] investigated the acute effects of BDNF on single-cell-activated glutamatergic synaptic connections in microcultures of postnatal rat hippocampal neurons. In approximately 30% of the cells, glutamatergic synaptic transmission was enhanced, this enhancement being abolished in the presence of the specific Trk inhibitor k252a. These data suggested that the enhancement of unitary glutamatergic synaptic transmission was mediated predominantly by presynaptic modifications, and this modulation could participate in BDNF-dependent modification of glutamatergic synaptic transmission in the hippocampus in situ.
Physiological and biochemical evidence implicates NMDAR as a target for BDNF modulation. Acute exposure to BDNF rapidly and reversibly enhanced the magnitude of NMDAR-mediated synaptic currents, specifically enhancing the activity of NMDAR containing the NR2B subunit, this effect of BDNF being dependent on activation of TrkB receptors [6]. These results provided a potential mechanism for the proposed role for BDNF in activity-dependent synaptic plasticity and, perhaps learning and memory processes. Neuronal activity induces transcription of the BDNF gene, which modulates the function of synapses. Sensory experience is transduced into changes in gene transcription via the activation of Ca2+-signaling pathways downstream of both L-type voltage-gated calcium channels (L-VGCCs) and NMDAR, these signaling pathways converging on the regulation of transcription factors including calcium-response factor (CaRF). CaRF limits NMDAR-dependent BDNF induction by regulating expression of NMDAR subunit GluN3A [7]. BDNF increases phosphorylation of NMDAR subunits NR1 and NR2B in the postsynaptic density, linking NMDAR phosphorylation to synaptic plasticity.
AMPAR mediate most of the excitatory neurotransmission in the mammalian central nervous system and participate in forms of synaptic plasticity thought to underlie memory and learning. BDNF-mediated GluR1 subunit of AMPAR tyrosine phosphorylation suggested to regulate synaptic plasticity postsynaptically through NR2B subunits of the NMDAR [8]. Several independent studies have suggested that AMPAR can increase BDNF expression by both Ca2+-dependent and Ca2+-independent pathways (see [9] for review). AMPAR were shown to interact with the protein tyrosine kinase Lyn, recruiting the mitogen-activated protein kinase (MAPK) signaling pathway and increasing the expression of BDNF. Thus, in addition to directly enhancing glutamatergic synaptic transmission, AMPAR activation upregulates in vitro and in vivo BDNF expression. Long-term encoding of synaptic events, as in long-term memory formation, requires AMPAR stabilization and maintenance. Jourdi et al. [10] revealed a new role for BDNF in the long-term maintenance of AMPAR subunits and associated scaffolding proteins at synapses and further supported the role of BDNF as a key regulator of synaptic consolidation. Changes in BDNF and AMPAR expression appear to be dissociable, and upregulation of the former leads to enhanced trophic signaling at excitatory synapses [11]. A novel mechanism by which estrogen and BDNF regulate hippocampal synaptic plasticity in the adult brain has been suggested [12]: both estradiol and G-protein-coupled estrogen receptor 1-specific agonist G1 rapidly induce BDNF release, leading to transient stimulation of activity-regulated cytoskeleton-associated (Arc) protein translation and GluR1(GluA1)-containing AMPAR internalization in field CA3 of the hippocampus.
Numerous studies have firmly established a critical role of BDNF in hippocampal long-term potentiation (LTP), a long-term enhancement of synaptic efficacy thought to constitute cellular substrates of learning and memory. Well known synaptic plasticity phenomena, LTP and long-term depression (LTD), can be elicited by NMDAR, typically by the coincident activity of pre- and postsynaptic neurons. The early phases of LTP expression are mediated by a redistribution of AMPAR, late phases by NMDAR [13]. In acute hippocampal slices, the presence of extracellular BDNF is essential for the induction of spike-timing-dependent long-term potentiation (tLTP). By monitoring changes in green fluorescent protein (GFP) fluorescence at the dendrite of hippocampal neurons expressing GFP-tagged BDNF, Lu et al. [14] found that pairing of iontophoretic glutamate pulses with neuronal spiking resulted in BDNF secretion from the postsynaptic dendrite at the iontophoretic site. Leal et al. [15] summarized evidence concerning a role for BDNF in generating functional and structural changes at synapses required for both early- and late phases of LTP in hippocampus and information regarding pre- and/or postsynaptic release of BDNF and its action during LTP. They also discussed the effects of BDNF on the synaptic proteome, either by acting on the protein synthesis machinery and/or by regulating protein degradation. They suggest that fine-tuned control of the synaptic proteome rather than a simple upregulation of protein synthesis may play a key role in BDNF-mediated synaptic potentiation.
Studies with human carriers of BDNF Met-allele polymorphism linking stress vulnerability and risk for depression revealed an additional link between the glutamate and BDNF systems. The Val66Met polymorphism in the BDNF gene (“a BDNF loss-of-function allele”) affects episodic memory and affective behaviors and is associated with a defect in activity-dependent regulated release of BDNF. The BDNF Val66Met polymorphism has a direct effect on synaptic plasticity in the hippocampus, as well as in the medial prefrontal cortex, and the central amygdala, impairing NMDAR transmission [16]. Recently, Jing et al. [17] showed that the BDNF Val66Met polymorphism enhanced glutamatergic transmission but diminished activity-dependent synaptic plasticity in the dorsolateral striatum. Studies in BDNF Val66Met mice provided information about the relation of a glutamate receptor with BDNF [18]. In wild-type mice after chronic-restraint stress and in unstressed mice with BDNF Val66Met allele, an epigenetic activator of histone acetylation, P300, exerted the dynamic up- and downregulation of mGlu2 in hippocampus via histone-3-lysine-27-acetylation when acute stressors were applied.
Converging evidence strongly suggests that deficits in BDNF signaling contribute to the pathogenesis of several major diseases and disorders such as Huntington’s disease, Alzheimer’s disease, and depression [19]. Abnormal BDNF, trkB-TK+, and GAD67 mRNA expression was shown in the hippocampus of individuals with schizophrenia and mood disorders, indicating that fundamental properties of hippocampal signaling transmission, plasticity, and circuitry may be affected in individuals with these major mental illnesses [20]. Numerous studies show impaired synaptic plasticity of glutamatergic synapses in diseases where compromised BDNF function has been observed, such as Huntington’s disease, depression, anxiety, and the BDNF polymorphism Val66Met, suggesting that upregulating BDNF-activated pathways may be therapeutically relevant (see [21] for review).
Both glutamatergic and BDNF systems are among the most important players in the consequences of chronic stress including depression. Meta-clustering of gene coexpression links in 11 transcriptome studies from postmortem brains of human major depression subjects and non-psychiatric control subjects revealed significant differences related to genes in the identified module encoding proteins implicated in neuronal signaling and structure, including glutamate metabotropic receptors (GRM1, GRM7) and BDNF [22]. Increased expression of BDNF appears to be involved in the mechanism of action of antidepressant drugs. Treatment with most antidepressants induces expression of the gene encoding BDNF in the hippocampus and cerebral cortex. However, a relative specificity exists regarding upregulation of BDNF by antidepressants. For example, the antidepressant paroxetine, but not desipramine, enhanced synaptic plasticity in the hippocampus by increasing BDNF mRNA expression, determining later AMPAR-subunit trafficking to synaptic membranes [23]. Studies of the last decade highlight that the brain glutamate system is involved in the etiology of depression, and glutamatergic-targeting drugs are currently being explored as novel antidepressant medications. For example, recent data indicate antidepressant-like activity of group I mGlu receptor (mGluR1 and mGluR5) antagonists in animal tests and models. Chronic treatment with 2-methyl-6-(phenylethynyl)-pyridine (MPEP), a selective mGlu5 receptor antagonist, increased hippocampal but reduced cortical BDNF mRNA level (Northern blot). Thus, an antagonist of mGlu5 receptors, like most well-established antidepressants, induces hippocampal BDNF gene expression [24].
As mentioned above, several lines of evidence indicate that chronic stress and downregulation of BDNF are key components of depression pathology. Study of stress effects on major glutamate receptor-mediated BDNF-dependent synaptic plasticity phenomena, including LTP, in rodent hippocampus is a useful tool to learn more about the pathogenesis of depression [25]. Optimization of glutamatergic signaling by neuroprotective drugs (e.g. antidepressants), or neuroprotective factors such as physical exercise or calorie restriction, is at least partially due to enhancing of BDNF signaling, though there are a few studies not confirming that BDNF signaling plays a pivotal role in the antidepressant effects of glutamate-based compounds [26]. The mechanism by which chronic stress downregulates BDNF and promotes depressive-like responses is not fully understood. Evidence from animal models of depression demonstrates that chronic stress impairs hippocampal BDNF expression and that antidepressant drug effects correlate with increased BDNF synthesis and activity in the hippocampus. Growing evidence suggests that downregulated clearance of glutamate and signaling pathways involving BDNF and its receptor TrkB are intimately involved in morphological changes in the hippocampus of patients with depression. BDNF-TrkB signaling regulates glutamate transporter 1 (GLT-1) on astrocytes, which are responsible for most glutamate reuptake from the synapse [27]. Oxidative stress excessively activates glutamate receptors inducing so-called excitotoxicity and contributing to neuronal dysfunction and degeneration in acute and chronic neurological and psychiatric disorders. Alterations of the mGluR1, mGluR5, and BDNF mRNA have been shown to contribute to depression-like and anxiety-like behaviors of prenatally stressed offspring rats [28]. An increase in glucocorticoid levels associated with downregulation of BDNF is supposed to be involved in the pathophysiology of depressive disorders. Using cultured cortical neurons, Numakawa et al. [29] showed that glucocorticoid receptor (GR) interacts with BDNF TrkB receptor. TrkB–GR interaction may play a critical role in the BDNF-stimulated PLC-gamma pathway, which is required for glutamate release, and the decrease in TrkB-GR interaction caused by chronic exposure to glucocorticoids results in the suppression of BDNF-mediated neurotransmitter release via a glutamate transporter. The available data suggest that chronic stress mediates changes in several Ca2+-related components involved in BDNF synthesis, including glutamatergic neurotransmission through NMDAR. Chronic-stress-induced NMDAR stimulation could lead to dysregulated calcium signaling and decreased BDNF activity, and in this situation, neurons become vulnerable to the effects of stress, leading to dysfunctional neurotransmission and behavioral disturbances. This suggests that treatment with NMDAR antagonists may help to restore calcium signaling, promote appropriate BDNF signaling, and correct depressive symptoms [30].
Reduced BDNF levels and altered BDNF signaling have been reported in several brain diseases and behavioral disorders that also exhibit reduced levels of AMPAR subunits. BDNF treatment acutely regulates AMPAR expression and function, including synaptic AMPAR subunit trafficking. Positive allosteric modulators of AMPAR increase brain levels of BDNF, improving the viability and generation of neurons in key brain structures. AMPAR potentiators are effective in rodent models predictive of antidepressant efficacy [31]. Recent data show that positive modulators of AMPAR (ampakines, drugs structurally derived from aniracetam that potentiate currents mediated by AMPAR) increase neuronal BDNF expression, though AMPAR and BDNFR was downregulated by prolonged activation. Several groups showed using rodent models of depression that BDNF/TrkB signaling may be involved in the sustained antidepressant-like effects of LY341495, a mGlu2/3 receptor antagonist [32], and 2-methyl-6-(phenylethynyl)pyridine, a selective metabotropic glutamate receptor 5 antagonist [33].
Recent discoveries related to antidepressant properties of ketamine, a classical antagonist of NMDAR and a dissociative anesthetic in use since 1970, contributed to understanding of tight regulatory relations between glutamate and BDNF. Ketamine has emerged as an effective treatment for refractory depression. Rapid antidepressant effects of NMDAR antagonists, specifically ketamine, may be associated with disinhibition of glutamate transmission, resulting in a rapid transient burst of glutamate, followed by an increase in BDNF release and activation of downstream signaling pathways that stimulate synapse formation. Neurodegeneration is an important component in the pathogenesis of major depression associated with cognitive complaints. Patients with depression demonstrate decreased brain volumes in areas implicated in emotional regulation and cognition (e.g. hippocampus), activation of various pathways contributing to cell death paradigms, and neuronal and glial cell death. Depression is associated with reduced synapse numbers and dearborization of dendrites, and ketamine appears to potently induce mechanisms that reverse these neurodegenerative processes (see [34] for review). The available data suggest that lower BDNF levels associated with mitochondrial dysfunction, oxidative stress, inflammation, and excitotoxicity may be involved in neuronal and glial cell death in depression, leading to decreased brain volume and cognitive dysfunction with multiple recurrent episodes of dementia [35]. The major mechanisms of ketamine action induce synaptogenesis in the BDNF pathway [36]. Preclinical work has shown that the antidepressant actions of ketamine (and scopolamine) in rodent models are caused by an influx of extracellular glutamate, elevated BDNF, activation of the mammalian target of rapamycin complex 1 (mTORC1) cascade, and increased number and function of spine synapses in the prefrontal cortex [37]. In addition to blocking NMDAR, ketamine coupled to a mature AMPAR, activates eukaryotic elongation factor 2 (eEF2), and this in turn activates BDNF protein synthesis, particularly in neuronal dendrites [38-40]. The associated depolarization of AMPAR initiates calcium-dependent exocytosis of BDNF, which can bind to postsynaptic TrkB receptors and induce BDNF signaling [38, 41]. BDNF signaling activated by ketamine seems to induce transcriptional changes, since inhibition of the BDNF receptor does not block the immediate effects of ketamine, but it does prevent the delayed effects. Activation of AMPAR in the medial prefrontal cortex by ketamine rapidly stimulates BDNF release via activation of L-type voltage-dependent calcium channels (VDCC) [42]. Potential involvement of the AMPAR(NMDAR)/BDNF pathway in antidepressant-like activity has been confirmed in several animal models of depression [43, 44].
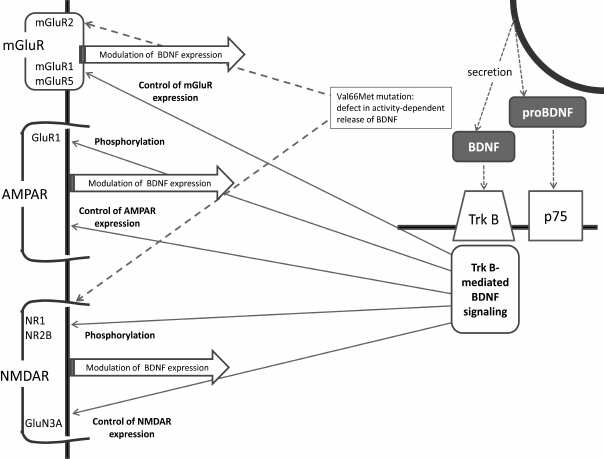
This brief review has summarized major facts relating brain BDNF and glutamatergic systems, and the main links between BDNF and glutamate receptors are shown in the figure. Indeed, the connections between the two systems are numerous and bidirectional, providing for mutual regulation of the glutamatergic and BDNF systems. The available data suggest that it is the complex and well-coordinating nature of these connections that secures optimal synaptic and cellular plasticity in the normal brain. Both systems are associated with the pathogenesis of depression, and the recent data imply that disturbance of tight and well-balanced associations between the glutamatergic and BDNF systems results in unfavorable changes in neuronal plasticity underlying depressive disorders and other mood diseases.
Main links between BDNF and glutamate receptors. AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; BDNF, brain-derived neurotrophic factor; NMDAR, N-methyl-D-aspartate receptors; p75, NTR, a low-affinity nerve growth factor receptor; TrkB, BDNF tyrosine kinase B receptor
Acknowledgements
This work was supported by the Russian Science Foundation (project No. 14-25-00136).
REFERENCES
1.Rothman, S. M., and Mattson, M. P. (2013)
Activity-dependent, stress-responsive BDNF signaling and the quest for
optimal brain health and resilience throughout the lifespan,
Neuroscience, 239, 228-240.
2.Mattson, M. P. (2008) Glutamate and neurotrophic
factors in neuronal plasticity and disease, Ann. N. Y. Acad.
Sci., 1144, 97-112.
3.Jarvis, C. R., Xiong, Z. G., Plant, J. R.,
Churchill, D., Lu, W. Y., MacVicar, B. A., and MacDonald, J. F. (1997)
Neurotrophin modulation of NMDA receptors in cultured murine and
isolated rat neurons, J. Neurophysiol., 78,
2363-2371.
4.Song, D. K., Choe, B., Bae, J. H., Park, W. K.,
Han, I. S., Ho, W. K., and Earm, Y. E. (1998) Brain-derived
neurotrophic factor rapidly potentiates synaptic transmission through
NMDA, but suppresses it through non-NMDA receptors in rat hippocampal
neuron, Brain Res., 799, 176-179.
5.Lessmann, V., and Heumann, R. (1998) Modulation of
unitary glutamatergic synapses by neurotrophin-4/5 or brain-derived
neurotrophic factor in hippocampal microcultures: presynaptic
enhancement depends on pre-established paired-pulse facilitation,
Neuroscience, 86, 399-413.
6.Kolb, J. E., Trettel, J., and Levine, E. S. (2005)
BDNF enhancement of postsynaptic NMDA receptors is blocked by ethanol,
Synapse, 55, 52-57.
7.Lyons, M. R., Chen, L. F., Deng, J. V., Finn, C.,
Pfenning, A., Sabhlok, A., Wilson, K., and West, A. E. (2016) The
transcription factor calcium-response factor limits NMDA
receptor-dependent transcription in the developing brain, J.
Neurochem., 137, 164-176.
8.Wu, K., Len, G. W., McAuliffe, G., Ma, C., Tai, J.
P., Xu, F., and Black, I. B. (2004) Brain-derived neurotrophic factor
acutely enhances tyrosine phosphorylation of the AMPA receptor subunit
GluR1 via NMDA receptor-dependent mechanisms, Brain. Res. Mol. Brain
Res., 130, 178-186.
9.O’Neill, M. J., Bleakman, D., Zimmerman, D.
M., and Nisenbaum, E. S. (2004) AMPA receptor potentiators for the
treatment of CNS disorders, Curr. Drug Targets CNS Neurol.
Disord., 3, 181-194.
10.Jourdi, H., Iwakura, Y., Narisawa-Saito, M.,
Ibaraki, K., Xiong, H., Watanabe, M., Hayashi, Y., Takei, N., and Nawa,
H. (2003) Brain-derived neurotrophic factor signal enhances and
maintains the expression of AMPA receptor-associated PDZ proteins in
developing cortical neurons, Dev. Biol., 263,
216-230.
11.Lauterborn, J. C., Pineda, E., Chen, L. Y.,
Ramirez, E. A., Lynch, G., and Gall, C. M. (2009) Ampakines cause
sustained increases in brain-derived neurotrophic factor signaling at
excitatory synapses without changes in AMPA receptor subunit
expression, Neuroscience, 159, 283-295.
12.Briz, V., Liu, Y., Zhu, G., Bi, X., and Baudry,
M. (2015) A novel form of synaptic plasticity in field CA3 of
hippocampus requires GPER1 activation and BDNF release, J. Cell.
Biol., 210, 1225-1237.
13.Luscher, C., and Malenka, R. C. (2012) NMDA
receptor-dependent long-term potentiation and long-term depression
(LTP/LTD), Cold Spring Harb. Perspect. Biol., 4, pii:
a005710.
14.Lu, H., Park, H., and Poo, M. M. (2013)
Spike-timing-dependent BDNF secretion and synaptic plasticity,
Philos. Trans. R. Soc. Lond. B Biol. Sci., 369,
20130132.
15.Leal, G., Afonso, P. M., Salazar, I. L., and
Duarte, C. B. (2015) Regulation of hippocampal synaptic plasticity by
BDNF, Brain Res., 1621, 82-101.
16.Ninan, I., Bath, K. G., Dagar, K., Perez-Castro,
R., Plummer, M. R., Lee, F. S., and Chao, M. V. (2010) The BDNF
Val66Met polymorphism impairs NMDA receptor-dependent synaptic
plasticity in the hippocampus, J. Neurosci., 26,
8866-8870.
17.Jing, D., Lee, F. S., and Ninan, I. (2016) The
BDNF Val66Met polymorphism enhances glutamatergic transmission but
diminishes activity-dependent synaptic plasticity in the dorsolateral
striatum, Neuropharmacology, pii: S0028-3908(16)30283-0.
18.Nasca, C., Zelli, D., Bigio, B., Piccinin, S.,
Scaccianoce, S., Nistico, R., and McEwen, B. S. (2015) Stress
dynamically regulates behavior and glutamatergic gene expression in
hippocampus by opening a window of epigenetic plasticity, Proc.
Natl. Acad. Sci. USA, 112, 4960-4965.
19.Lu, B., Nagappan, G., and Lu, Y. (2014) BDNF and
synaptic plasticity, cognitive function, and dysfunction, Handbook
Exp. Pharmacol., 220, 223-250.
20.Thompson, R. M., Weickert, C. S., Wyatt, E., and
Webster, M. J. (2011) Decreased BDNF, trkB-TK+ and GAD67
mRNA expression in the hippocampus of individuals with schizophrenia
and mood disorders, J. Psychiatry Neurosci., 36,
195-203.
21.Carvalho, A. L., Caldeira, M. V., Santos, S. D.,
and Duarte, C. B. (2008) Role of the brain-derived neurotrophic factor
at glutamatergic synapses, Br. J. Pharmacol., 153,
S310-324.
22.Chang, L. C., Jamain, S., Lin, C. W., Rujescu,
D., Tseng, G. C., and Sibille, E. (2014) A conserved BDNF, glutamate-
and GABA-enriched gene module related to human depression identified by
coexpression meta-analysis and DNA variant genome-wide association
studies, PLoS One, 9, e90980.
23.Martínez-Turrillas, R., Del Rio, J., and
Frechilla, D. (2005) Sequential changes in BDNF mRNA expression and
synaptic levels of AMPA receptor subunits in rat hippocampus after
chronic antidepressant treatment, Neuropharmacology, 49,
1178-1188.
24.Legutko, B., Szewczyk, B., Pomierny-Chamiolo, L.,
Nowak, G., and Pilc, A. (2006) Effect of MPEP treatment on
brain-derived neurotrophic factor gene expression, Pharmacol.
Rep., 58, 427-430.
25.Gulyaeva, N. V. (2016) Studies on stress-induced
modulation of long-term potentiation in rodent hippocampus: what can we
learn about pathogenesis of depression? Translat. Brain Rhythm.,
1, doi: 10.15761/TBR.1000107.
26.Lindholm, J. S., Autio, H., Vesa, L., Antila, H.,
Lindemann, L., Hoener, M. C., Skolnick, P., Rantamaki, T., and Castren,
E. (2012) The antidepressant-like effects of glutamatergic drugs
ketamine and AMPA receptor potentiator LY 451646 are preserved in
bdnf⁺/⁻ heterozygous null mice, Neuropharmacology,
62, 391-397.
27.Liu, W. X., Wang, J., Xie, Z. M., Xu, N., Zhang,
G. F., Jia, M., Zhou, Z. Q., Hashimoto, K., and Yang, J. J. (2016)
Regulation of glutamate transporter 1 via BDNF-TrkB signaling plays a
role in the anti-apoptotic and antidepressant effects of ketamine in
chronic unpredictable stress model of depression, Psychopharmacology
(Berl.), 233, 405-415.
28.Jia, N., Li, Q., Sun, H., Song, Q., Tang, G.,
Sun, Q., Wang, W., Chen, R., Li, H., and Zhu, Z. (2015) Alterations of
group I mGluRs and BDNF associated with behavioral abnormity in
prenatally stressed offspring rats, Neurochem. Res., 40,
1074-1082.
29.Numakawa, T., Kumamaru, E., Adachi, N., Yagasaki,
Y., Izumi, A., and Kunugi, H. (2009) Glucocorticoid receptor
interaction with TrkB promotes BDNF-triggered PLC-gamma signaling for
glutamate release via a glutamate transporter, Proc. Natl. Acad.
Sci. USA, 106, 647-652.
30.Vasquez, C. E., Riener, R., Reynolds, E., and
Britton, G. B. (2014) NMDA receptor dysregulation in chronic state: a
possible mechanism underlying depression with BDNF downregulation,
Neurochem. Int., 79, 88-97.
31.Alt, A., Nisenbaum, E. S., Bleakman, D., and
Witkin, J. M. (2006) A role for AMPA receptors in mood disorders,
Biochem. Pharmacol., 71, 1273-1288.
32.Koike, H., Fukumoto, K., Iijima, M., and Chaki,
S. (2013) Role of BDNF/TrkB signaling in antidepressant-like effects of
a group II metabotropic glutamate receptor antagonist in animal models
of depression, Behav. Brain Res., 238, 48-52.
33.Liu, C. Y., Jiang, X. X., Zhu, Y. H., and Wei, D.
N. (2012) Metabotropic glutamate receptor 5 antagonist
2-methyl-6-(phenylethynyl)pyridine produces antidepressant effects in
rats: role of brain-derived neurotrophic factor, Neuroscience,
223, 219-224.
34.Henderson, T. A. (2016) Practical application of
the neuroregenerative properties of ketamine: real world treatment
experience, Neural Regen. Res., 11, 195-200.
35.Kim, H. K., Nunes, P. V., Oliveira, K. C., Young,
L. T., and Lafer, B. (2016) Neuropathological relationship between
major depression and dementia: a hypothetical model and review,
Prog. Neuropsychopharmacol. Biol. Psychiatry, 67,
51-57.
36.Kim, Y. K., and Na, K. S. (2016) Role of
glutamate receptors and glial cells in the pathophysiology of
treatment-resistant depression, Prog. Neuropsychopharmacol. Biol.
Psychiatry, 70, 117-126.
37.Whleb, E. S., Gerhard, D., Thomas, A., and Duman,
R. S. (2016) Molecular and cellular mechanisms of rapid-acting
antidepressants ketamine and scopolamine, Curr. Neuropharmacol.,
Mar. 8.
38.Browne, C. A., and Lucki, I. (2013)
Antidepressant effects of ketamine: mechanisms underlying fast-acting
novel antidepressants, Front. Pharmacol., 4, doi:
10.3389/fphar.2013.00161.
39.Monteggia, L. M., Gideons, E., and Kavalali, E.
T. (2013) The role of eukaryotic elongation factor 2 kinase in rapid
antidepressant action of ketamine, Biol. Psychiatry, 73,
1199-1203.
40.Bjorkholm, C., and Monteggia, L. M. (2015)
BDNF – a key transducer of antidepressant effects,
Neuropharmacology, 102, 72-79.
41.Scheuing, L., Chiu, C. T., Liao, H. M., and
Chuang, D. M. (2015) Antidepressant mechanism of ketamine: perspective
from preclinical studies, Front. Neurosci., 9, doi:
10.3389/fnins.2015.00249.
42.Lepack, A. E., Fuchikami, M., Dwyer, J. M.,
Banasr, M., and Duman, R. S. (2014) BDNF release is required for the
behavioral actions of ketamine, Int. J. Neuropsychopharmacol.,
18, doi: 10.1093/ijnp/pyu033.
43.Pochwat, B., Sowa-Kucma, M., Kotarska, K.,
Misztak, P., Nowak, G., and Szewczyk, B. (2015) Antidepressant-like
activity of magnesium in the olfactory bulbectomy model is associated
with the AMPA/BDNF pathway, Psychopharmacology (Berl.),
232, 355-367.
44.Duman, R. S. (2014) Pathophysiology of depression
and innovative treatments: remodeling glutamatergic synaptic
connections, Dialogues Clin. Neurosci., 16, 11-27.