Neonatal Proinflammatory Stress Induces Accumulation of Corticosterone and Interleukin-6 in the Hippocampus of Juvenile Rats: Potential Mechanism of Synaptic Plasticity Impairments
M. V. Onufriev*, S. V. Freiman, D. I. Peregud, I. V. Kudryashova, A. O. Tishkina, M. Yu. Stepanichev, and N. V. Gulyaeva
Institute of Higher Nervous Activity and Neurophysiology, Russian Academy of Sciences, 117485 Moscow, Russia; E-mail: mikeonuf1@rambler.ru* To whom correspondence should be addressed.
Received October 25, 2016; Revision received November 17, 2016
Infectious diseases in early postnatal ontogenesis can induce neuroinflammation, disrupt normal central nervous system development, and contribute to pathogenesis of cerebral pathologies in adults. To study long-term consequences of such early stress, we induced neonatal proinflammatory stress (NPS) by injecting bacterial lipopolysaccharide into rat pups on postnatal days 3 and 5 and then assessed the levels of corticosterone, proinflammatory cytokines and their mRNAs, and neurotrophins and their mRNAs in the hippocampus and neocortex of the one-month-old animals. Long-term potentiation (LTP) was studied in hippocampal slices as an index of synaptic plasticity. NPS-induced impairments of LTP were accompanied by the accumulation of corticosterone and IL-6 in the hippocampus. In the neocortex, a decrease in exon IV BDNF mRNA was detected. We suggest that excessive corticosterone delivery to hippocampal receptors and proinflammatory changes persisting during brain maturation are among the principal molecular mechanisms responsible for NPS-induced neuroplasticity impairments.
KEY WORDS: neonatal proinflammatory stress, lipopolysaccharide, corticosterone, proinflammatory cytokines, neurotrophins, long-term potentiationDOI: 10.1134/S0006297917030051
Abbreviations: BDNF, brain-derived neurotrophic factor; CNS, central nervous system; CS, corticosterone; GR, glucocorticoid receptor; HPA, hypothalamic–pituitary–adrenal axis; IL, interleukin; LPS, lipopolysaccharide; LTP, long-term potentiation; MR, mineralocorticoid receptor; NGF, nerve growth factor; NPS, neonatal proinflammatory stress; TNF, tumor necrosis factor.
Earlier, we have shown that neonatal proinflammatory stress (NPS)
induced by injection of bacterial lipopolysaccharide (LPS) on postnatal
days 3 and 5 (corresponding to months 5 and 8 in humans) dysregulates
the hypothalamic–pituitary–adrenal (HPA) axis, which
results in an increase in corticosterone (CS) blood levels, attenuation
of corticosteroid response to moderate stress, and promotion of
depression-like behavior in juvenile (1-month-old) and adult
(3-month-old) rats [1]. Our data confirmed the
hypothesis that NPS causes malfunctioning of “perinatal
programming” and induces behavioral and physiological deviations
in adult organisms because of impaired neuroplasticity [2]. However, molecular mechanisms underlying the
adverse effects of NPS remain unknown.
Many types of cells, such as macrophages, microglia, epithelial cells of blood vessels, fibroblasts, and neurons secrete proinflammatory cytokines IL-1β, IL-6, and TNFα in response to systemic administration of LPS [3]. The peripheral pool of proinflammatory cytokines affects the central nervous system (CNS) and activates the HPA axis with the following release of glucocorticoids by the adrenal grands [4, 5], thereby stimulating expression of proinflammatory cytokines in immunocompetent brain cells and promoting neuroinflammation and neurodegeneration [6-8]. The HPA axis is activated via direct stimulation by proinflammatory cytokines of the production and release of corticotropin-releasing hormone in the hypothalamus and of the adrenocorticotropic hormone in the hypophysis [9, 10].
Glucocorticoids interact with two types of receptors: glucocorticoid receptors (GR) and mineralocorticoid receptors (MRs). Both GRs and MRs are expressed in the brain, but the affinity of MRs for the ligands is about 10 times higher than the affinity of GRs [11]. After binding CS, GRs and MRs function as gene expression regulators [12, 13]. Brain regions that are involved in stress response and play an important role in emotion formation and cognitive functions (hippocampal CA1 region, dentate gyrus, medial prefrontal cortex, and amygdala) usually coexpress both types of receptors [13]. Corticosteroids can rapidly modulate neuronal activity by a nongenomic mechanism via interacting with the membrane GRs and MRs [14, 15]. In the hippocampus, MR activation supports neuronal activity, whereas post-stress activation of GRs suppresses excitability and synaptic plasticity in neurons [16, 17]. Neurotrophins, including BDNF (brain-derived neurotrophic factor), NGF (nerve growth factor), NT-3, and NT-4, regulate various cell functions, including synaptic plasticity in the peripheral and central nervous systems [18, 19]. Dysregulation of the HPA axis, as well as increase in the level of proinflammatory cytokines, impair neuronal plasticity via direct or mediated impact on neurotrophin expression and functions in the CNS [20, 21].
The goal of this study was to evaluate the role of glucocorticoids, proinflammatory cytokines, and neurotrophic factors in the mechanism of brain plasticity impairments caused by neonatal proinflammatory stress.
MATERIALS AND METHODS
NPS model. Newborn male Wistar rats (n = 19) were divided into two groups. On postnatal days 3 and 5, animals of the experimental group (n = 9) were injected subcutaneously with LPS at a dose of 50 µg/kg body weight; animals of the control group (n = 10) were injected with an equivalent volume of physiological saline. When the rats achieved 1 month of age, they were sacrificed by decapitation, and their cortices and hippocampi were isolated. Peripheral blood was collected from the tail vein and used for plasma isolation.
Brain tissue homogenization. Samples of brain tissues from the isolated brain regions were homogenized in a Potter tissue grinder in PBS buffer containing 1% Nonidet P-40, 1 mM EDTA, 10% glycerol, and protease inhibitor cocktail (Thermo Scientific, USA). The homogenates were centrifuged at 13,000g for 30 min at 4°C. The resulting supernatants were aliquoted and stored at –80°C.
Corticosterone assay. The levels of CS (both free and bound to transport proteins) in plasma and brain tissue supernatants were determined with an enzyme immunoassay kit (DRG, Germany) using the competitive ELISA technique.
Growth factor assay. BDNF and NGF were assayed by the ELISA method using commercially available kits (Millipore, USA).
Proinflammatory cytokine assay. The contents of IL-1β, IL-6, and TNF-α in the supernatants of brain homogenates were determined using commercial ELISA kits (PeproTech, Germany) as recommended by the manufacturer.
Evaluation of IL-1β, IL-6, TNF-α, and BDNF mRNA expression. Expression of mRNAs for proinflammatory cytokines and BDNF in the neocortex and hippocampus was evaluated by real-time RT-qPCR as described in [22].
Electrophysiological studies. Long-term potentiation (LTP) was registered in hippocampal slices from 17-33-day-old rats. The slices were perfused with medium containing 124 mM NaCl, 5 mM KCl, 1.3 mM MgSO4·7 H2O, 2.5 mM CaCl2, 1 mM NaH2PO4, 26 mM NaHCO3, and 10 mM D-glucose (pH 7.3-7.4). The medium was saturated with oxygen–CO2 mixture (95% O2/5% CO2); all procedures were performed at 34°C. Focal potentials were registered in the CA1 pyramidal cell layer with a glass microelectrode filled with 0.33 M NaCl. The stimulating electrode was placed in the Schaffer collaterals of the radial layer. Before LTP registration, the stimulation threshold value and the dependence of the response amplitude on the stimulation intensity were determined. The stimulation intensity that induced a response equaling 40-45% of the maximal amplitude was used for LTP registration. LTP was induced by high-frequency stimulation (1 s, 100 Hz) of the Schaffer collaterals. The responses of the CA1 neurons were recorded before the stimulation and within 1 h after stimulation with a 30-s interval. A total of 30 slices from 18 rats (n =10 for control group; n = 8 for experimental group) were analyzed.
Statistical analysis. Statistical analysis was performed with the Statistica 8 program. The results on long-term potentiation were analyzed by dispersion analysis (ANOVA). Factors “groups” and “time” were used to evaluate the effect of NPS and the development of LTP after induction, respectively. The significance of differences between the groups was estimated by nonparametric methods using the Mann–Whitney U-test. The results are presented as M ± SEM. Differences were considered significant at p < 0.05.
RESULTS AND DISCUSSION
In our experiments, the CS levels in blood plasma and brain of juvenile rats showed no difference between the control and experimental groups: 291.8 ± 29.9 vs. 236.1 ± 16.2 ng/ml in blood plasma, 10.67 ± 0.33 vs. 8.20 ± 0.92 ng/g tissue in neocortex, and 10.83 ± 0.77 vs. 9.21 ± 0.69 ng/g tissue in hippocampus, respectively. Using a similar NPS model, Shanks et al. [23] showed that adult rats displayed stronger response (increase in adrenocorticotropic hormone and CS blood levels) after immobilization-induced stress, but the basal CS levels in the blood of these animals did not differ from the CS levels in the control group. In another study, increased CS blood levels were observed in young animals 4 h after a LPS injection on postnatal day 5, while in adult rats, this parameter did not exceed the control values [24].
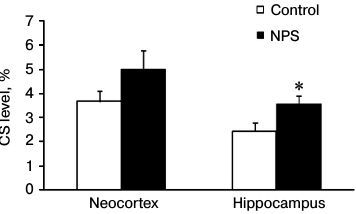
To estimate the efficiency of CS delivery to the brain, for each experimental animal we calculated the ratio between the CS contents in the brain and blood plasma (Fig. 1). We found that NPS increased the CS delivery to the brain 1.5-fold. The observed changes were statistically significant in the hippocampus, but not in the neocortex (due to a great variation in the results). Apparently, the efficiency of CS delivery to the brain determines the intensity of GR- and MR-mediated signal transduction. An increased expression of GRs in the prefrontal cortex and hippocampus caused by early postnatal stress [25] may play an important role in this process. This serves as evidence that the NPS-induced deteriorations in HPA axis regulation [1] manifest themselves as more efficient ligand binding to the receptor and potentially stronger signal transduction via GRs and/or MRs.
Fig. 1. Effect of NPS on the ratio between CS levels in brain and blood plasma (taken as 100%) in 1-month-old rats; * p < 0.05.
To evaluate the extent of neuroinflammation in the neocortex and hippocampus after NPS, we determined gene expression and immunoreactivity for three major proinflammatory cytokines – IL-1β, IL-6, and TNFα (Fig. 2). We found that NPS increased only the content of IL-6 in the hippocampus (1.3-fold); excessive IL-6 secretion is typical for chronic neuroinflammation. The levels of IL-1β and TNFα, usually elevated at the acute stages of stress [26], did not differ in the control and experimental groups.
Fig. 2. Effect of NPS on levels of proinflammatory cytokine proteins (left) and mRNAs (right) in rat brain regions; * p < 0.05.
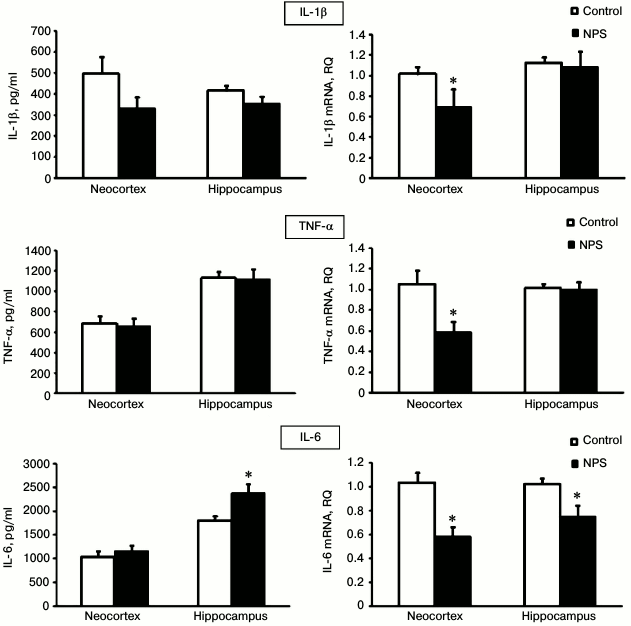
It should be noted that the directionality of changes in the immunoreactivity of the studied cytokines in no case coincided with the changes in the expression of the corresponding mRNAs. Thus, the levels of IL-6 mRNA were significantly decreased after NPS in the neocortex and hippocampus, while the immunoreactivity of this cytokine did not change in the neocortex and, as mentioned above, it even increased in the hippocampus. Statistically significant suppression of the IL-1β and TNFα mRNA expression after NPS was observed in the neocortex, but not in the hippocampus, while the immunoreactivity of these proteins in the neocortex did not change.
At first glance, the data on the protein and mRNA expression of cytokines contradict each other. However, the observed increased content of the IL-6 protein with simultaneously decreased expression of its gene indicates that, despite adaptive antiinflammatory response that is regulated at the gene expression level, the proinflammatory system can be supported, for example, by decrease in the rate of IL-6 protein metabolism. Note that it is immunoreactivity of proinflammatory cytokines that is of functional significance, and we observed the effect of NPS on this immunoreactivity namely in the hippocampus, where this effect is mediated by corticosteroid regulation [27].
The “contradictory” effects of NPS on proinflammatory cytokine immunoreactivity and mRNA expression have also been observed by other researchers. Thus, the levels of IL-1β, IL-6, and TNFα proteins in the hippocampus of adult rats were not affected by NPS and increased only after subsequent immobilization-induced stress [24]. However, NPS activated IL-1β mRNA expression in the fascia dentata of the hippocampus in adult animals [28]. Expression of IL-1β, IL-6, and TNFα mRNAs in the prefrontal cortex of juvenile rats did not change after NPS, while in the hippocampus NPS increased TNFα mRNA expression but did not affect expression of IL-1β and IL-6 mRNAs [29].
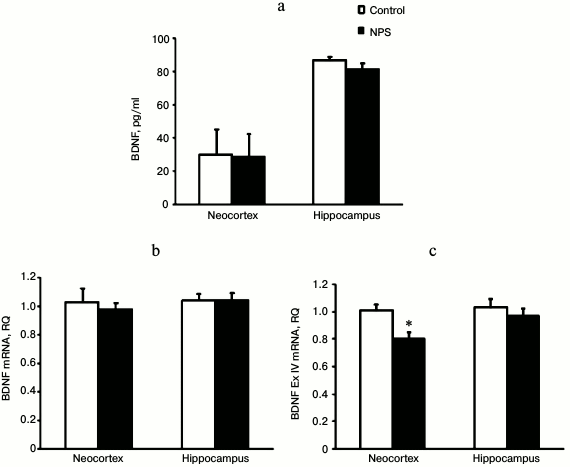
NPS did not affect the levels of NGF protein and mRNA in both the neocortex and hippocampus (data not shown). Similarly, BDNF immunoreactivity and mRNA levels remained unchanged in the neocortex and hippocampus after NPS (Fig. 3). However, when we analyzed expression of mRNAs for individual BDNF exons, we found that NPS significantly decreased (1.3-fold) expression of BDNF exon IV in the neocortex. Exon IV is known to be the most stress-responsive exon of BDNF [30]. Both neocortex and hippocampus are stress-sensitive brain areas. The decrease in the expression of BDNF and its exons in these structures might be caused by an increase in the levels of CS [31] and/or proinflammatory cytokines [32]. However, in our study expression of BDNF exon IV mRNA was decreased only in the neocortex of juvenile rats subjected to NPS and, it was rather prolonged even in the absence of noticeable changes in the levels of expression-regulating factors.
Fig. 3. Effect of NPS on levels of BDNF protein (a), BDNF mRNA (b), and BDNF exon IV mRNA (c) in rat brain regions; * p < 0.05.
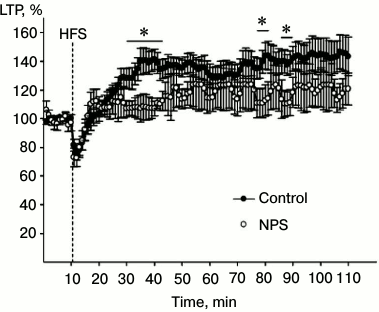
To evaluate the synaptic plasticity of the hippocampus, we used the LTP paradigm. LTP is an increase in the synaptic efficiency induced by high-frequency stimulation of the afferent inputs. It is one of the well-known phenomena of synaptic plasticity that represents the ability of chemical synapsis to maintain a high level of signal transduction as a result of a particular stimulation pattern. LTP is often studied in hippocampal slices, since hippocampus is involved in learning and memory. Changes in the magnitude of the population spike are often studied in pyramidal neurons that receive afferent signals from Schaffer collaterals after high-frequency stimulation of the latter. We found that high-frequency stimulation of Schaffer collaterals resulted in LTP induction in synapses of CA3–CA1 neurons (factor “time” p < 0.01) in hippocampal slices from both control animals and rats subjected to NPS (Fig. 4; factor “group” p > 0.1). ANOVA analysis of the increase in the response after high-frequency stimulation revealed statistically significant correlation between factors “group” × “time” (p < 0.001), which indicates distinctions in the LTP development in animals subjected to NPS. Indeed, NPS considerably changed the LTP properties in the hippocampus of juvenile rats, the most noticeable effect being in the early phase of potentiation. In particular, comparison of the dynamics of synaptic modifications within the first 15 min after tetanization revealed statistically significant correlation between factors “group” × “time” (p < 0.001), which was related to the properties of the stimulation-induced changes, especially in rats subjected to NPS. Both in control and experimental rats, high-frequency stimulation was followed by a period of post-activation synaptic depression. In all slices of the control group and in seven slices of the experimental group, the magnitude of the response increased after the period of depression. However, in nine slices of the experimental group, the early stage of potentiation was absent. However, when potentiation was induced, the stage of its maintenance in slices from rats subjected to NPS did not differ from that in the control animals (correlation between factors “group” × “time” p > 0.7). As demonstrated earlier, both CS and IL-6 inhibit LTP in the hippocampus [32, 33]. It is possible that the results of our study on the stimulating effect of NPS on the IL-6 levels and CS delivery to the hippocampus might reflect mechanisms of neuroplasticity impairments in this brain region.
Fig. 4. Effect of NPS on LTP development in hippocampal slices: filled circles, control (average value for all slices of control group); open circles, NPS (average value for all slices of experimental group). HFS, high-frequency stimulation; * p < 0.05.
In conclusion, we found that NPS induces long-term region-specific changes in neurochemical systems of the brain. NPS activates CS delivery to the hippocampus, where neuroinflammation takes place. This neuroinflammation might be the cause of the observed impairments in brain synaptic plasticity. In this work, we did not study the neurophysiology of the neocortical plasticity, but the changes in BDNF exon IV expression in this brain area indicate possible plasticity impairments in the neocortex too. Our data confirm the concept that regional disturbances in the corticosteroid and neurotrophin signaling, as well as neuroinflammation, underlie the mechanisms of psycho-emotional and cognitive defects in people that experienced early proinflammatory stress.
Acknowledgements
This work was supported by the Russian Science Foundation (project No. 14-25-00136).
REFERENCES
1.Tishkina, A., Stepanichev, M., Kudryashova, I.,
Freiman, S., Onufriev, M., Lazareva, N., and Gulyaeva, N. (2016)
Neonatal proinflammatory challenge in male Wistar rats: effects on
behavior, synaptic plasticity, and adrenocortical stress response,
Behav. Brain Res., 304, 1-10.
2.Walker, A., Nakamura, T., Byrne, R. J., Naicker,
S., Tynan, R. J., Hunte, M., and Hodgson, D. M. (2009) Neonatal
lipopolysaccharide and adult stress exposure predisposes rats to
anxiety-like behaviour and blunted corticosterone responses:
implications for the double-hit hypothesis,
Psychoneuroendocrinology, 34, 1515-1525.
3.Silverman, M. N., Pearce, B. D., Biron, C. A., and
Miller, A. H. (2005) Immune modulation of the
hypothalamic-pituitary-adrenal (HPA) axis during viral infection,
Viral Immunol., 18, 41-78.
4.Gwosdow, A. R., Kumar, M. S., and Bode, H. H.
(1990) Interleukin 1 stimulation of the hypothalamic-pituitary-adrenal
axis, Am. J. Physiol., 258, E65-E70.
5.Turnbull, A. V., and Rivier, C. L. (1999)
Regulation of the hypothalamic-pituitary-adrenal axis by cytokines:
actions and mechanisms of action, Physiol. Rev., 79,
1-71.
6.Gatti, S., and Bartfai, T. (1993) Induction of
tumor necrosis factor-alpha mRNA in the brain after peripheral
endotoxin treatment: comparison with interleukin-1 family and
interleukin-6, Brain Res., 624, 291-294.
7.Dantzer, R. (2004) Cytokine-induced sickness
behavior: a neuroimmune response to activation of innate immunity,
Eur. J. Pharmacol., 500, 399-411.
8.Chung, D. W., Yoo, K. Y., Hwang, I. K., Kim, D. W.,
Chung, J. Y., Lee, C. H., Choi, J. H., Choi, S. Y., Youn, H. Y., Lee,
I. S., and Won, M. H. (2010) Systemic administration of
lipopolysaccharide induces cyclooxygenase-2 immunoreactivity in
endothelium and increases microglia in the mouse hippocampus, Cell
Mol. Neurobiol., 30, 531-541.
9.Chida, D., Imaki, T., Suda, T., and Iwakura, Y.
(2005) Involvement of corticotropin-releasing hormone- and interleukin
(IL)-6-dependent proopiomelanocortin induction in the anterior
pituitary during hypothalamic-pituitary-adrenal axis activation by
IL-1α, Endocrinology, 146, 5496-5502.
10.Kageyama, K., Tamasawa, N., and Suda, T. (2011)
Signal transduction in the hypothalamic corticotropin-releasing factor
system and its clinical implications, Stress, 14,
357-367.
11.De Kloet, E. R., Fitzsimons, C. P., Datson, N.
A., Meijer, O. C., and Vreugdenhil, E. (2009) Glucocorticoid signaling
and stress-related limbic susceptibility pathway: about receptors,
transcription machinery and microRNA, Brain Res., 1293,
129-141.
12.Herman, J. P., Figueiredo, H., Mueller, N. K.,
Ulrich-Lai, Y., Ostrander, M. M., and Choi, D. C. (2003) Central
mechanisms of stress integration: hierarchical circuitry controlling
hypothalamo-pituitary-adrenocortical responsiveness, Front.
Neuroendocrinol., 24, 151-180.
13.Reul, J. M., and De Kloet, E. R. (1985) Two
receptor systems for corticosterone in rat brain: microdistribution and
differential occupation, Endocrinology, 117,
2505-2511.
14.Dallman, M. F. (2005) Fast glucocorticoid actions
on brain: back to the future, Front. Neuroendocrinol.,
26, 103-108.
15.Tasker, J. G., Di, S., and Malcher-Lopes, R.
(2006) Minireview: rapid glucocorticoid signaling via
membrane-associated receptors, Endocrinology, 147,
5549-5556.
16.Joëls, M., Karst, H., Krugers, H., and
Lucassen, P. (2007) Chronic stress: implications for neuronal
morphology, function and neurogenesis, Front. Neuroendocrinol.,
28, 72-96.
17.Kim, J. J., and Diamond, D. M. (2002) The
stressed hippocampus, synaptic plasticity and lost memories, Nature
Rev. Neurosci., 3, 453-462.
18.Chao, M. V., Rajagopal, R., and Lee, F. S. (2006)
Neurotrophin signaling in health and disease, Clin. Sci.
(Lond.), 110, 167-173.
19.Ohira, K., and Hayashi, M. (2009) A new aspect of
the TrkB signaling pathway in neural plasticity, Curr.
Neuropharmacol., 7, 276-285.
20.Wosiski-Kuhn, M., Erion, J. R., Gomez-Sanchez, E.
P., Gomez-Sanchez, C. E., and Stranahan, A. M. (2014) Glucocorticoid
receptor activation impairs hippocampal plasticity by suppressing BDNF
expression in obese mice, Psychoneuroendocrinology, 42,
165-177.
21.Tong, L., Prieto, G. A., Kramar, E. A., Smith, E.
D., Cribbs, D. H., Lynch, G., and Cotman, C. W. (2012) Brain-derived
neurotrophic factor-dependent synaptic plasticity is suppressed by
interleukin-1β via p38 mitogen-activated protein kinase, J.
Neurosci., 32, 17714-17724.
22.Peregud, D. I., Yakovlev, A. A., Stepanichev, M.
Y., Onufriev, M. V., Panchenko, L. F., and Gulyaeva, N. V. (2016)
Expression of BDNF and TrkB phosphorylation in the rat frontal cortex
during morphine withdrawal are NO dependent, Cell Mol.
Neurobiol., 36, 839-849.
23.Shanks, N., Larocque, S., and Meaney, M. J.
(1995) Neonatal endotoxin exposure alters the development of the
hypothalamic-pituitary-adrenal axis: early illness and later
responsivity to stress, J. Neurosci., 15, 376-384.
24.Walker, A. K., Nakamura, T., and Hodgson, D. M.
(2010) Neonatal lipopolysaccharide exposure alters central cytokine
responses to stress in adulthood in Wistar rats, Stress,
13, 506-515.
25.Meaney, M. J., Aitken, D. H., van Berkel, C.,
Bhatnagar, S., and Sapolsky, R. M. (1988) Effect of neonatal handling
on age-related impairments associated with the hippocampus,
Science, 239, 766-768.
26.Himmerich, H., Fischer, J., Bauer, K., Kirkby, K.
C., Sack, U., and Krugel, U. (2013) Stress-induced cytokine changes in
rats, Eur. Cytokine Netw., 24, 97-103.
27.Piskunov, A. K., Yakovlev, A. A., Stepanichev, M.
Yu., Onufriev, M. V., and Gulyaeva, N. V. (2011) Selective sensitivity
of hippocampus to interoceptive stress: effects of interleukin-1β
and erythropoietin, Neirokhimiya, 28, 216-219.
28.Bilbo, S. D., Barrientos, R. M., Eads, A. S.,
Northcutt, A., Watkins, L. R., Rudy, J. W., and Maier, S. F. (2008)
Early-life infection leads to altered BDNF and IL-1β mRNA
expression in rat hippocampus following learning in adulthood, Brain
Behav. Immun., 22, 451-455.
29.Bland, S. T., Beckley, J. T., Watkins, L. R.,
Maier, S. F., and Bilbo, S. D. (2010) Neonatal Escherichia coli
infection alters glial, cytokine, and neuronal gene expression in
response to acute amphetamine in adolescent rats, Neurosci.
Lett., 474, 52-57.
30.Nair, A., Vadodaria, K. C., Banerjee, S. B.,
Benekareddy, M., Dias, B. G., Duman, R. S., and Vaidya, V. A. (2007)
Stressor-specific regulation of distinct brain-derived neurotrophic
factor transcripts and cyclic AMP response element-binding protein
expression in the postnatal and adult rat hippocampus,
Neuropsychopharmacology, 32, 1504-1519.
31.Dwivedi, Y., Rizavi, H. S., and Pandey, G. N.
(2006) Antidepressants reverse corticosterone-mediated decrease in BDNF
expression: dissociation in regulation of specific exons by
antidepressants and corticosterone, Neuroscience, 139,
1017-1029.
32.Sharvit, A., Segal, M., Kehat, O., Stork, O., and
Richter-Levin, G. (2015) Differential modulation of synaptic plasticity
and local circuit activity in the dentate gyrus and CA1 regions of the
rat hippocampus by corticosterone, Stress, 18,
319-327.
33.Li, A. J., Katafuchi, T., Oda, S., Hori, T., and
Oomura, Y. (1997) Interleukin-6 inhibits long-term potentiation in rat
hippocampal slices, Brain Res., 748, 30-38.