REVIEW: Immuno-PCR: Achievements and Perspectives
D. Y. Ryazantsev*, D. V. Voronina, and S. K. Zavriev
Shemyakin–Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, 117997 Moscow, Russia; E-mail: d.yu.ryazantsev@gmail.com, szavriev@ibch.ru* To whom correspondence should be addressed.
Received April 26, 2016; Revision received July 11, 2016
The immuno-PCR (iPCR) method combines advantages of enzyme-linked immunosorbent assay and polymerase chain reaction, which is used in iPCR as a method of “visualization” of antigen–antibody interaction. The use of iPCR provides classical PCR sensitivity to objects traditionally detected by ELISA. This method could be very sensitive and allow for detection of quantities of femtograms/ml order. However, iPCR is still not widely used. The aim of this review is to highlight the special features of the iPCR method and to show the main aspects of its development and application in recent years.
KEY WORDS: immuno-PCR, quantitative PCR, antibody, antigen, DNA–antibody conjugateDOI: 10.1134/S0006297916130113
Abbreviations: BA, binding antibodies; (b)DA, (biotinylated) detecting antibodies; ELISA, enzyme-linked immunosorbent assay; iPCR, immuno-PCR; PCR, polymerase chain reaction; PLA, proximity ligation assay; qPCR, quantitative PCR (real-time PCR); RT-PCR, PCR with reverse transcription; sulfo-SMCC, sulfo-N-succinimidyl 4-(maleimidomethyl)cyclohexane-1-carboxylate.
Immuno-PCR (iPCR) was first described by Sano, Smith, and Cantor in 1992
[1]. The method combines the advantages of
polymerase chain reaction (PCR) and immunoassay, allowing the detection
of targets of interest present in a sample in extremely low
concentration.
Since the first publication in 1985 [2], PCR has proved to be an efficient technique for detection of small amounts of DNA, down to one molecule per test tube [3, 4]. Due to high sensitivity, PCR became widely used in clinical diagnostics of viral and bacterial diseases, hereditary diseases, in genetic mapping, etc. However, this method can be used only for detection of nucleic acids, while other molecules, such as hormones, antibodies, proteins, toxins, etc., apart from DNA or RNA of an organism need to be specifically detected. For these purposes, the major method of choice is ELISA: high specificity of antibodies towards their antigens allows identification of a broad range of molecules, although with much lower sensitivity compared to PCR. Thus, a method having sensitivity of PCR and the potential of ELISA is needed for a wide range of studies.
The iPCR method, which combines the two above-mentioned approaches, has become such a tool; this highly sensitive method allows the detection of a wide range of analytes. It is based on the use of a DNA–antibody conjugate with further amplification of marker DNA. In the original iPCR method proposed by Sano et al. [1], a streptavidin–protein A chimera was used as a linker between an antibody and DNA: streptavidin bound biotinylated DNA, while protein A bound an Fc-fragment of an immunoglobulin G (IgG) molecule. Such conjugate limited the possibility to use this technique only to the direct format of target molecule detection (which is not very often used), since in a “sandwich” format reaction protein A will bind primary antibodies, increasing the background signal [5]. Later, another type of conjugate was proposed, composed of a biotinylated antibody, streptavidin, and a biotinylated DNA-tag [6]. The advantage of this approach is its possible use for immunoanalysis in direct, indirect, and “sandwich” formats. Covalent conjugates, when a DNA-tag is bound directly to an antibody by means of various reagents, are also used [7]. This reduces the number of steps in a protocol, shortening the time of analysis and increasing the signal-to-background ratio.
Like ELISA, iPCR can be performed in various formats depending on the aim of the experiment. A schematic representation of the main reaction formats – direct, indirect, sandwich, indirect sandwich, and competitive – is shown in Fig. 1, as well as the standard concentration ranges detected by iPCR and ELISA. In the direct format [7], an antigen is immobilized on a support and is directly detected by a conjugated antibody. In the indirect format, immobilized antigen first forms a complex with a specific antibody, which is then detected by secondary detecting antibodies (DA) conjugated with a DNA-tag [8, 9]. In the direct sandwich format, an antigen is detected by two types of antibodies: primary antibodies (binding antibodies, BA) are immobilized on a support and bind the analyte; secondary antibodies conjugated with a DNA-tag are then used for detection [10, 11]. The first steps of the indirect sandwich format are like those of a direct sandwich except that secondary antibodies carry no DNA-tag, and the detection is carried out by tertiary antibodies [12, 13]. The competitive format is generally used for detection of low molecular mass compounds [14-17]: studied material, containing an antigen (antibody) of interest, is added simultaneously with a detecting conjugate to the antigen (antibody) immobilized on the support. If antibodies are analyzed, a labeled antigen is used for qualitative assessment (rarely), while if antigens are analyzed – labeled antibodies are used. This review discusses only the competitive format analysis with labeled antibodies. The result of the reaction is inversely proportional to the amounts of the analyte of interest.
Fig. 1. Comparison of iPCR and ELISA methods and their sensitivity. Solid line – iPCR, dotted line – ELISA. Immunoassay formats: a, c) noncompetitive: direct, indirect, direct sandwich, indirect sandwich; b, d) competitive. See details in text.
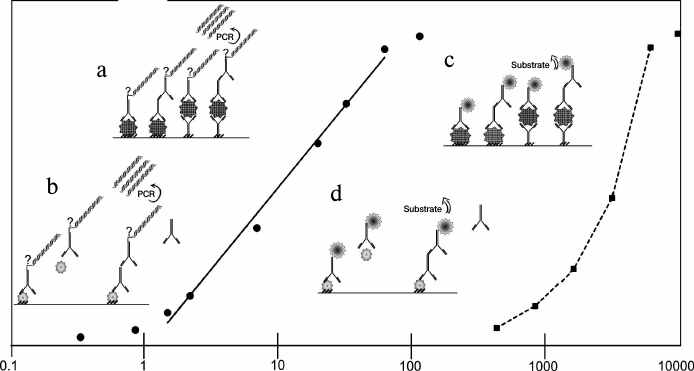
Detection of a DNA-tag is made by electrophoresis or by real time PCR (quantitative PCR, qPCR). In most published articles, the latter method is used because the exclusion of an electrophoresis step reduces substantially the time of analysis, minimizes contamination, and allows estimation of the initial amount of the template and, therefore, the analyte. Works that have used other methods of DNA amplification are not discussed in this review.
In this review, we give an overview of the use and main aspects of the development of iPCR during last 10 years.
SPECIAL ASPECTS OF iPCR METHODOLOGY
One of the variants of a universal iPCR technology is detailed elsewhere [18]; nevertheless, it is worth discussing the key steps of this process because of its complexity. High sensitivity of PCR as a method for detection of iPCR results is its main advantage and its disadvantage at the same time. Nonspecific binding of antibodies, antigens, or DNA–antibody conjugate lead to a certain level of DNA-tag amplification in all samples (background level). The main aim of optimization of the iPCR method is to increase the ratio between the level of amplification in the samples containing analyte and the level of amplification in negative control samples.
Many early studies have described a complete absence of amplification in samples not containing the analyte of interest. Thus, absence of a signal in negative control samples after 25, 35, 40, and 52 cycles of amplification has been described [7, 19-21]. It is extremely difficult and, in most cases, impossible to obtain such results, since the sensitivity of PCR allows detection of as few as a single DNA molecule in a test tube. Amplification of DNA-tag is almost always observed in negative control samples after 30-40 cycles of amplification, which is apparently a consequence of a nonspecific binding of components including DNA-tag [12, 22-24].
The problem of contamination of the working zone and the reagents must be mentioned specifically. The working zone for immunochemical manipulations, the zone for working with DNA-tag, and the zone for qPCR must be physically separated from each other.
Thus, a decrease in background level is one of the key steps in optimization and improvement of the iPCR method. At the same time, a background signal can be radically decreased by using a proximity ligation assay (PLA) [25], which will be discussed later.
Choice of support and its influence on binding. Ninety-six-well microplates are widely used for ELISA and PCR methods, which predefined the prevalence of this format in iPCR also. The characteristics of a plate used for iPCR differs from that for ELISA or PCR. The material should have high antigen binding capacity and be compatible with the thermoblock of a PCR machine to avoid an additional step of DNA-tag detachment from the antigen–antibody complex with subsequent transfer onto another plate for amplification.
Originally, polypropylene PCR microplates were used for iPCR, but they did not provide the needed level of antigen binding [26, 27]. Later, TopYield polycarbonate 8-well strips (Nunc) with improved protein-binding capacity, specially designed for iPCR, became popular [28-30]. However, Barletta [21] and Potuckova [31] claim that experiments carried out in TopYield strips showed low sensitivity. It turned out to be the result of nonuniform distribution of heat in wells during PCR, because the wells of TopYield strips did not fit the wells of the PCR machine. The problem was solved by optimization of PCR conditions – increasing denaturation temperature [21] and increasing elongation time [31]. Another drawback, associated with the form of the wells in TopYield strips, was revealed during qPCR: apparently, several zones of refraction and reflection of light appear, resulting in additional steps in a PCR curve that complicate the analysis (at least for DT-prime 96 thermal cycler). Use of TopYield strips is also difficult without application of a layer of mineral oil on the top of the reaction mixture [32-34]. Despite certain drawbacks, the TopYield 8-well strips remain the most popular option for iPCR today. Apart from TopYield strips, Corning Costar 6511 polycarbonate PCR plates and Greiner Thermoquick 651570 plates are used. Experiments made with these plates gave similar results to those obtained with the Nunc TopYield plates. With these plates, PCR curves were close to an ideally sigmoid, which means that all wells were considered in the experiment. Also, Robostrips (Roboscreen) polycarbonate plates with increased sorption were developed especially for iPCR. The Robostrips have the same form as standard 0.2-ml PCR strips that provided their tight fit to the thermocycler well, as well as uniform heat distribution [21]. The Robostrips do not have the limitations of the TopYield strips, but automatic washings in Robostrips can be difficult.
Some authors have proposed using enzymatic or thermal cleavage of DNA from an antibody with further transfer of an aliquot of the reaction mix into PCR tubes [9, 35], or doing several PCR cycles using TopYield strips with subsequent transfer of an aliquot of a reaction mix into a qPCR plate [21].
An alternative way to increase the sensitivity of antibodies is to use different microparticles (detailed in the section “Use of Nanostructures in iPCR” below).
Methods of conjugation of antibodies with DNA-tag. A fundamental difference between iPCR and ELISA is the use of DNA amplification for quantification of an analyte; for this, a DNA-tag must be conjugated with an antibody (Fig. 2).
Fig. 2. Techniques of conjugation of an antibody with a DNA-tag. a) Binding of a biotinylated antibody with a biotinylated oligonucleotide by streptavidin. b) Covalent conjugation based on a heterobifunctional linker, which reacts with different functional groups of DNA (with artificially introduced reactive group, such as SH-, NH2-, etc.) and antibody. c) Supramolecular complex of biotinylated antibody with bis-biotinylated DNA-tag. d) Biobarcode assay – bifunctional nanoparticles (gold or magnetic) covered with DA and DNA-tag. e) Phage display – an antibody fragment is exposed on the surface of a recombinant M13 phage, while phage DNA plays a role of a signal DNA. f) Biobarcode assay – magnetic particles bear BA, while gold particles bear DA and DNA-tag. g) Proximity ligation assay – after binding of PLA-probes to adjacent epitopes DNA molecules come closer to each other, so that a ligase connects two oligonucleotides, generating a new DNA strand that can be amplified.
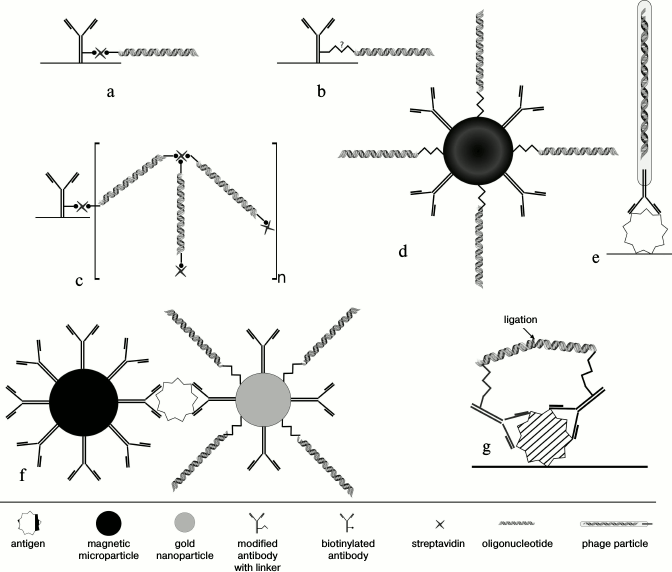
In the original iPCR format [1], streptavidin–protein A chimera provided two binding sites: streptavidin bound with biotinylated DNA, while protein A bound an Fc-fragment of an immunoglobulin G (IgG). Such linker has several drawbacks. First, it can be used only in a direct format immunoassay [5]. Second, affinity of protein A differs significantly depending on class/subclass and host species of an antibody, which reduces the versatility of this method [36]. In other early works on iPCR [37], avidin, a tetrameric protein with high affinity and specificity to biotin, was used as a linker. Avidin can form different conjugates because of its tetrameric structure, which might cause a decrease in sensitivity and reproducibility of the results. Unlike avidin, streptavidin lacks a carbohydrate fragment and has a low isoelectric point (pI = 5, whereas pI of avidin is 10), which finally leads to a decrease in nonspecific binding [38].
Conjugates based on biotin–streptavidin interactions. Zhou et al. [6] proposed a way to conjugate DNA to an antibody in situ (Fig. 2a). This approach consists in consecutive addition to a test tube of bDA, streptavidin and biotinylated DNA. Such complex can be used in any iPCR format: direct, indirect, or sandwich, so it was referred to as “universal iPCR” [39]. Despite all positive features, use of a streptavidin–biotin conjugate has some drawbacks: numerous long washes are needed to prevent nonspecific binding, which increases the time of analysis; it is difficult to choose the optimal stoichiometric ratio of reagents. To assure complete binding, the system should reach thermodynamic equilibrium at each step of incubation, which requires an optimization of the time of each step. In most iPCR protocols, time of incubation is limited to 1 h. During this time, a heterogeneous system might not reach equilibrium, which might lead to a decrease in the number of complexes and the signal strength [40].
It should be mentioned that for a streptavidin-biotinylated conjugate it is better to use an oligonucleotide biotinylated from one end. Christophe Niemeyer [28] first described supramolecular complexes formed by biotinylated at both ends double stranded DNA fragments, streptavidin and antibodies (Fig. 2c). Use of such supramolecular complexes increases specific signal, since for one DA molecule there are dozens or even hundreds of DNA-tag molecules; moreover, it is important that such system is universal. We improved the technique of making complexes with relatively short (60-70 nt) single-stranded oligonucleotides biotinylated from both 5′- and 3′-ends and a streptavidin [32, 34]. When using streptavidin–biotin complexes, the level of nonspecific signal can be decreased by reduction in the number of steps, if pre-synthesized and purified conjugates of a biotinylated antibody/DNA or streptavidin-labeled antibody/DNA are used. In addition, preparation of an antibody–streptavidin complex takes less time than biotinylation of an antibody [41].
Covalent conjugates. Another efficient method of conjugation is covalent binding of DA with DNA (Fig. 2b). Generally, a heterobifunctional fusing agent is used containing two reaction groups that interact with different substrates: DNA (with an artificially introduced reaction group, such as SH-, NH2-, etc.) and the amino group of an antibody, binding them together. The scheme of DNA covalent conjugation of an antibody with N-succinimidyl 4-(maleimidomethyl)cyclohexane-1-carboxylate (SMCC) as a binding linker is widely used (Fig. 3). In this case, a DNA-tag bearing a thiol group and NH2-groups of an antibody can be used [42], or an SH-group of an antibody and an amino group of DNA [43-46]. As one variant of this reaction, Weissleder et al. proposed use of a photocleavable bifunctional agent, which has an advantage for the detection of cellular proteins in single living cells, since very often conditions of DNA amplification are not compatible with cellular media [47].
Fig. 3. Schematic representation of a covalent conjugation with N-succinimidyl 4-(maleimidomethyl)cyclohexane-1-carboxylate. a) Using thiolated DNA-tag. b) Using DNA-tag with introduced amino group. Two main steps can be designated: 1) binding of a linker sulfo-SMCC to an NH2-group of antibodies; 2) binding of activated antibody with thiolated DNA-tag and generation of a final conjugate. When a DNA-tag with an artificially introduced amino group is used, an additional step is needed: reduction of disulfide bonds of an antibody by reducing agents, such as DTT, etc. [62], N-succinimidyl-S-acetyl-thioacetate [44, 46], tris-(2-carboxyethyl)-phosphine hydrochloride [45].
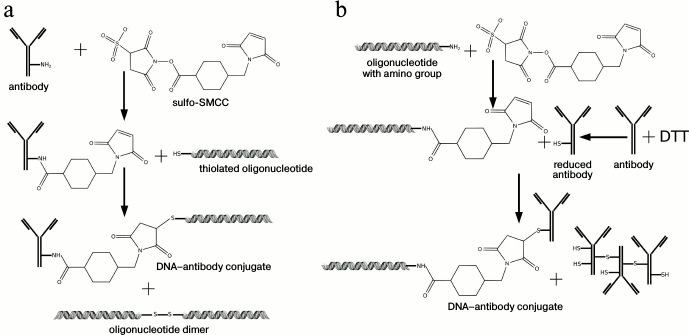
Unfortunately, in the literature there is not much data about the final conjugate yield and about the purification procedure, including purification from reaction byproducts: oligonucleotide dimers in case of thiolated oligonucleotide, or aggregates of reduced antibody molecules with disulfide bonds formation. Gel filtration under high pressure can be used for purification [7], which allows separation of the final conjugate from unused antibodies or oligonucleotide, although it is not possible to separate the conjugate from the reaction byproducts with this method. Hashimoto et al. [48] described conjugation of an antibody with DNA by means of hydrazone through benzaldehyde-modified 5’-end of DNA.
Functionalization of an antibody by introduction of genetically coded non-natural amino acid (p-acetylphenylalanine or p-azidophenylalanine) into the recombinant antibody has been described [49]. The oligonucleotide bearing a thiol group is modified with an aminooxy-maleimide linker, and conjugation is achieved through an aminooxy-group of an oligonucleotide and a modified amino acid.
Promising methods of conjugate production are known as “click chemistry”; they comprise a reaction of azide-alkyne cycloaddition, a [4+2]-cycloaddition reaction between the dienophile and a conjugated diene (inverse-electron demand Diels–Alder reaction), etc. [50]. It was shown that azide-alkyne cycloaddition catalyzed by monovalent copper can lead to precipitation of an antibody; use of a reaction triggered by strained cyclooctene or a [4+2]-cycloaddition reaction between dienophile and conjugated dienes (“Cu-free click”) is better [51]. Production of an antibody–DNA conjugate using several modifying agents has been described [51]. Tetrazine is first added to the amino group of an antibody through a succinimide ether; trans-cyclooctene is added to an oligonucleotide through the reaction of cycloaddition of azides and alkynes; and finally, conjugation is achieved in phosphate buffer through a [4+2]-cycloaddition reaction between dienophiles and conjugated dienes.
Like streptavidin–biotin conjugates, covalent conjugates are suitable for all iPCR formats. Preliminary preparation of DNA–antibody conjugates simplifies the reaction, eliminating several incubation steps and decreasing the level of nonspecific binding of reagents [38]. Commercial ready-to-use covalent conjugates (Imperacer®; Chimera Biotec) or kits for their production (Thunder-Link®, Innova Biosciences; Imperacer®, Chimera Biotec) are now available. The Chimera Biotec company provides antibody–DNA conjugates with broad specificity. For example, an anti-biotin conjugate, which can be used as a tertiary antibody specific towards biotinylated DA [52, 53], or an anti-digoxygenin conjugate are known [13]. In addition, Chimera Biotec produces conjugates with specificity needed for a customer: anti-rotavirus [54], anti-tau-protein [55], etc. Thus, researchers are freed from time- and labor-consuming procedures of preparation of antibody–DNA complex, and, in the case of ready-to-use conjugates, there is no need for optimization of its synthesis and purification. Use of covalent conjugates shortens even more the time of analysis (even if compared with pre-assembled biotin–streptavidin complexes).
The PLA method (Fig. 2g) should be described separately. This is a technology that uses “double recognition of a target molecule” for detection of proteins, protein–protein interactions, and posttranslational modifications [56]. Two antibodies specific to different epitopes of one antigen carry different DNA-tags referred to as PLA-probes. When the probes are bound to adjacent epitopes, DNA molecules are situated near each other and can be hybridized. Addition of a ligase seals the gap between two strains, and the newly generated DNA molecule can be amplified with oligonucleotides homologous to the initially separated chains. This method is highly sensitive since the level of a background signal is extremely low; the probability that two DNA molecules will accidental meet each other and be ligated under these conditions is extremely low [25, 57, 58].
In addition to antigen–antibody interaction, specific interactions of other molecules are used for preparation of conjugates for iPCR, for example, interaction between ganglioside GM1 and cholera toxin or Tus–Ter interaction. Tus protein is a terminator of replication in prokaryotes; it stably binds specific Ter DNA sequences. Schaeffer et al. [59, 60] used a Tus–Ter complex for the detection of antibodies. For this purpose, they combined the Tus–Ter construct with immunoglobulin-binding proteins such as streptococcal protein G and staphylococcal protein A [60], or with secondary antibodies [59]. The Tus–Ter-lock iPCR protocol uses the minimal number of washes, which is advantageous compared to the “universal iPCR” with streptavidin–biotin conjugates. According to the authors, covalent binding of an antibody to a DNA-tag is technically more difficult, and, therefore, cannot be easily adapted for application in diagnostic laboratories, unlike the Tus–Ter complex.
Using nanostructures in iPCR. iPCR variants are not limited to a simple combination of ELISA and PCR. During recent years, nanotechnologies have been intensively used for iPCR design, particularly magnetic or gold nanoparticles, liposomes, and viral particles.
Magnetic or gold nanoparticles are used for delivery of BA or DA with a DNA-tag. Barletta [10] and Li [61] used magnetic nanoparticles coated with polystyrene as a carrier for BA. This modification of the iPCR method is used to decrease the influence of other components of the probe (“matrix” effect): after short incubation of an antigen and DA with magnetic particles carrying BA, non-bound components are discarded, while the magnetic particles are resuspended and used further. Other authors made experiments using gold magnetic particles whose function was similar to that of nanoparticles coated with polystyrene [43, 62].
Bifunctional nanoparticles, gold or magnetic, covered with DA and DNA-tag (Fig. 2d) were described. According to Nikitina et al. [63], no negative effect on antibody affinity was detected in the system they were studying, unlike covalent binding of DNA to an antibody. Another advantage of this method is the possibility to bind several antibodies specific to different epitopes to the same particles [64]. Comparison of nanoparticle-based conjugate to other conjugates shows that production of bifunctional particles requires more time than the preparation of biotinylated antibodies, but the direct conjugation of an antibody with a DNA-tag is much more difficult [31]. Undoubtedly, many more DNA-tags are located on the surface of particles [11] than in direct conjugates with an antibody, which leads to an increase in iPCR signal when nanoparticles are used [65].
Biobarcode assay uses simultaneously magnetic microparticles and gold nanoparticles (Fig. 2f). The magnetic particles are covered with BA, while the gold particles are covered with DA and DNA-tag. The analysis is carried out in the sandwich format; immunocomplexes formed undergo magnetic separation, and DNA-tags are detached from the gold nanoparticles for further analysis. In the original format of biobarcoding assay developed by Mirkin et al. [66, 67], scanometric detection was used. Cleaved DNA was precipitated on glass chips covered with oligonucleotide complementary to half of a DNA-tag. Then, gold nanoparticles covered with silver (I) and modified DNA complementary to the other half of the DNA-tag are added. In the presence of a DNA-tag “barcode” in the initial solution, it binds to oligonucleotide and nanoparticles; silver reducing solution is then added, and the result is registered. Unlike the above-described method, in iPCR biobarcoding the DNA-tag is detected by means of qPCR [67-70]. Biobarcoding is a promising method because of the possibility of multiplex analysis, i.e. simultaneous detection of several analytes in one sample, since nanoparticles covered by different DA can be “coded” by oligonucleotides with unique sequences.
Mason et al. [71] described an unusual iPCR format where a liposome with DNA molecules and monosialoganglioside GM receptor imbedded in the lipid bilayer was used as a detecting agent. The ganglioside interacts specifically with cholera toxin and is also a binding site for some other compounds. After binding of such a construct with an antigen, the liposome was lysed, and the DNA-tag released was detected. The authors claim that detection using a liposome has advantages compared to standard iPCR. First, each liposome contains up to 60 DNA molecules, which increases the sensitivity of the method. Second, addition of DNase just before the lysis of the liposomes decreases contamination of microplate wells, since the enzyme is not able to get through the lipid bilayer.
Viral particles are also used as carriers of DA and DNA-tags: iPCR mediated by phage display was first described by Zhang et al. [72] in 2006 (Fig. 2e). This technique combines advantages of iPCR and phage display. On the surface of a recombinant bacteriophage M13, the fragment of an antibody [73, 74] or a protein specifically interacting with an analyte [75] are exposed, while phage DNA acts as a DNA-tag. The use of phage particles in iPCR obviates the difficult and long preparation of an antibody–DNA conjugate.
Blocking of vacant binding sites. One way to solve the problem of nonspecific binding of components and to decrease the background signal is to choose a suitable blocking reagent to cover the microplate surface and add in buffer solutions. The best blocking agent for each analysis is selected during iPCR optimization. Such compounds as BSA [14, 31, 67], casein [75-77], nonfat milk [9, 11, 78], salmon sperm DNA [54] or calf thymus DNA [78], and normal goat serum [79] are widely used. Blocking reagent is also added at the step of incubation with DA or sera [35, 78].
Detection and analysis of results. In iPCR there are two ways to amplify the marker DNA. The first is a one-tube method, when all reactions are carried out in the same tube. This technique requires careful choice of the microplate material, since it must bind efficiently an antibody or an antigen and be thermostable to suit a PCR machine [80]. Another approach consists of cleavage of a DNA-tag from the immune complex and its transfer in a PCR tube. The main methods of DNA-tag cleavage are either addition of a restriction site in a DNA-tag or heating to 95-100°C, resulting in denaturation of the antibody–DNA complex and the release of the DNA-tag [81, 82].
He et al. [41, 80] showed a 1000-fold decrease in iPCR sensitivity when all steps were carried out in one tube compared to iPCR with a cleavable DNA-tag. They hypothesize that this can be explained by a negative impact of ELISA components on amplification.
To date, there are two major approaches to analyze iPCR results: agarose gel electrophoresis and qPCR. Increasing the number of samples make gel electrophoresis complicated and time-consuming. Moreover, it can lead to contamination of the working zone with PCR products, which might give false positive results. Often, a simple detection of the presence or absence of the analyte in the probe is not sufficient; quantitative analysis of a bound DNA-tag, which correlates with the amount of the analyte, is needed. In qPCR, an intercalating fluorescent dye or “hydrolyzable” probe (known as TaqMan®) is added to the reaction mix. The probe bears a fluorophore and a fluorescence quencher; during PCR, it is annealed to the DNA-tag and is degraded by Taq-polymerase, resulting in separation of the fluorophore and quencher. During qPCR, fluorescence level is measured during each cycle of amplification. The number of a cycle when the level of fluorescence exceeds the basic level is inversely proportional to the quantity of a DNA-tag in a well. Therefore, it is possible to use qPCR for precise quantitative analysis of the original template. This approach reduces the number of manipulations one sample must undergo, reduces the time of analysis, and minimizes the risk of contamination, especially in case of using one tube for all reactions, which makes qPCR the main method for analysis of iPCR results.
For analysis and qPCR results, representation of a threshold cycle (Cq) value is calculated – the first PCR cycle when the level of fluorescence of the probe exceeds the basal fluorescence level (these calculations are made by the software provided with the thermocycler). For representation of the results, the following scheme can be used [18]. For all samples the threshold cycle value is subtracted from the total number of amplification cycles; these values are used to calculate mean values and standard deviations. The threshold value (TV) is calculated using the equation: TV = [Mean (40 – Cq)without analyte + 3·Standard deviation (40 – Cq)without analyte], and normalized values (Cq) are calculated using the equation: Cqsample = [(Mean (40 – Cq)sample – TV]. Samples with normalized values lower than 0 are considered negative, and with values above 0 – positive. In various experiments, only normalized values can be compared [18]. To analyze the amount of the molecule of interest in the sample, a standard curve is created, using regression analysis of Ct values from the value log of known concentration of the analyte.
DETECTION OF VARIOUS MOLECULES USING iPCR
It is obvious that iPCR, being as sensitive as PCR and flexible as ELISA, can be used to detect of a wide range of molecules from viral and bacterial antigens to drugs and non-protein toxins. The table summarizes some data about analyzed objects, sensitivity of iPCR method (compared to ELISA), immunoanalysis format, and the type of the conjugate used. In this section, we discuss some difficulties related to structure specificity and/or specificity of purification of certain objects, as well as ways of overcoming them; we will compare detection of one analyte with different iPCR methods. We will provide several of the most specific and important examples for each group of objects detected by iPCR.
iPCR detection sensitivity for different objects*
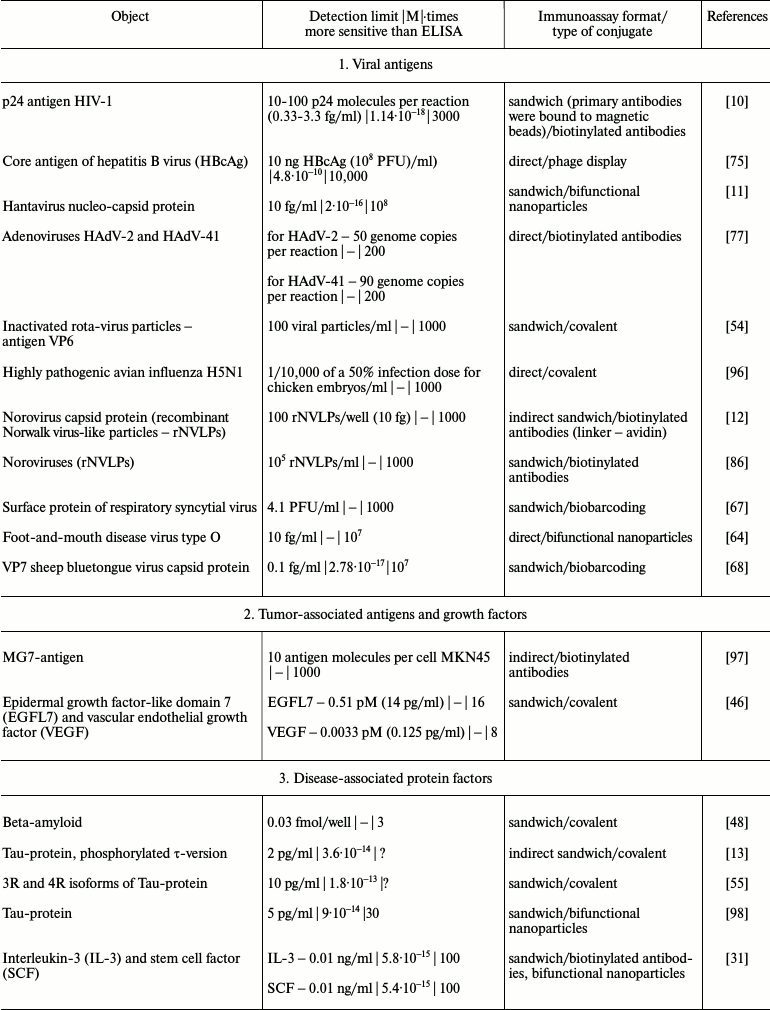
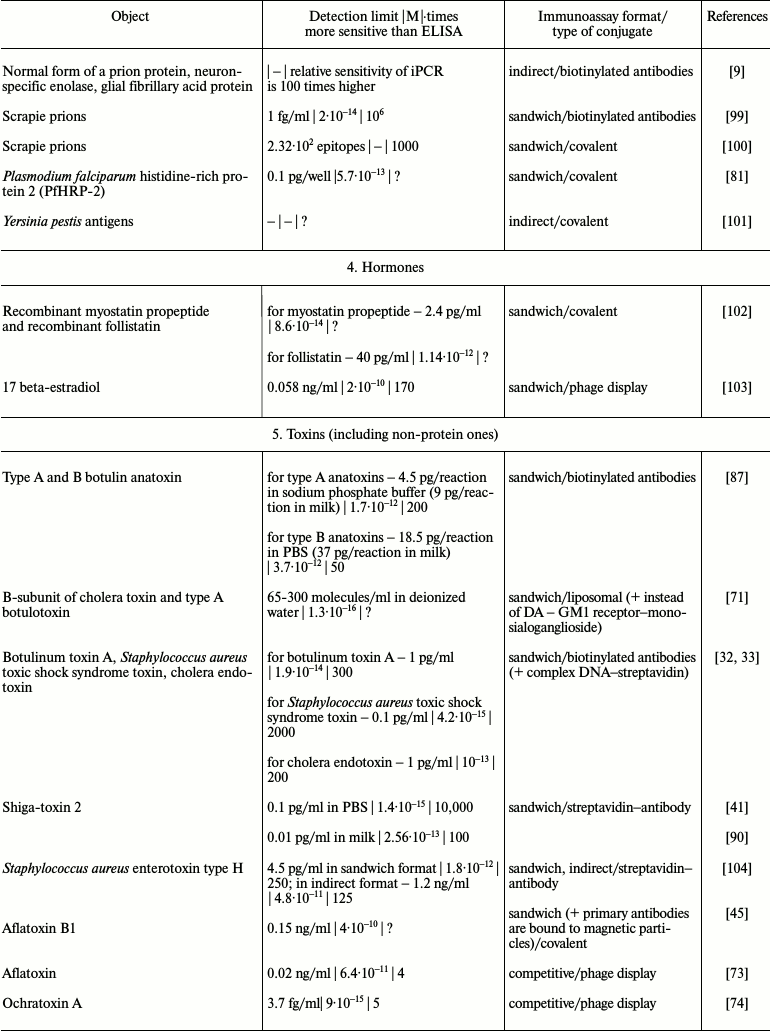
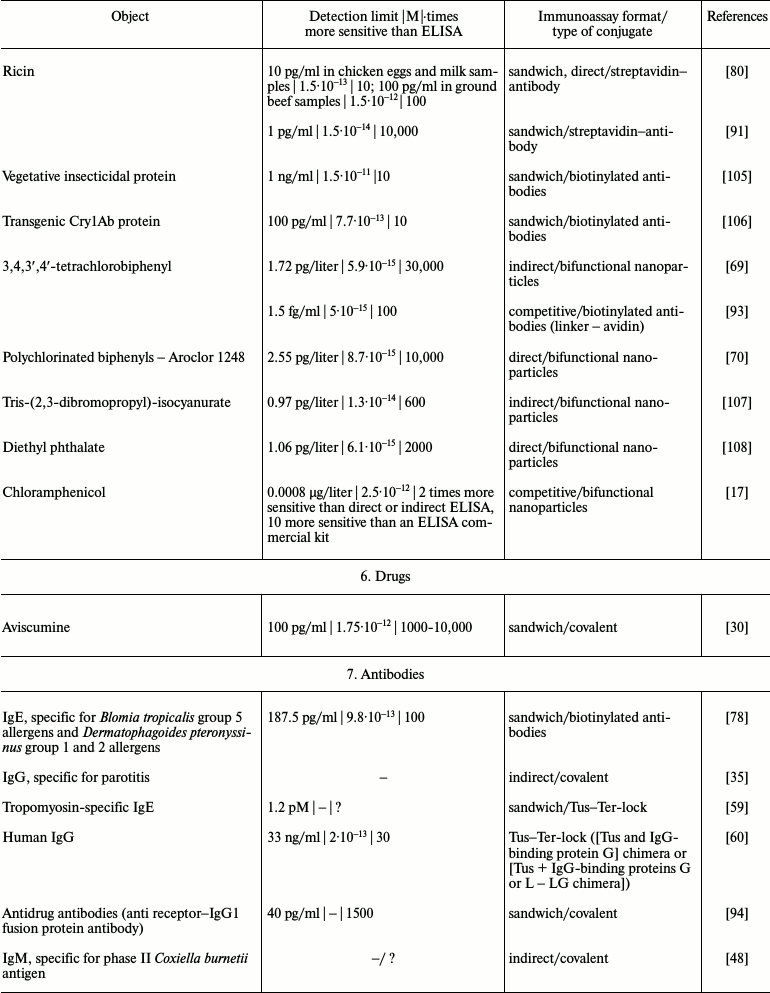
Notes: –, sensitivity was not determined by the authors or is not
possible to be assessed; ?, absence of direct comparison of ELISA and
iPCR.
* To facilitate comparison, sensitivity is presented in two units: first
as in the original article, and then recalculated to moles/liter format
(for those cases when single molecules were detected and not cells or
virions).
Viral proteins are one of the most widespread “targets” for iPCR, while an extremely low number of virions is replicated at early stages of a disease according to data about the pathogenesis of most viral infections. Thus, highly sensitive methods play an exclusive role in early diagnostics of viral diseases, as well as in control of the efficiency of antiviral therapy.
Early diagnosis is very important for detection of the human immunodeficiency virus (HIV). During the acute phase of HIV infection, which lasts up to one month, no specific antibodies are detected in the blood of the patient, but viral RNA is found, which suggests that the main approach in HIV diagnosis in the prodromal stage is PCR with the reverse transcription. The most remarkable results using iPCR were obtained by Barletta et al. [10]: in this study the limit of detection of antigen p24, a HIV-1 capsid component, was as few as 10-100 molecules per reaction, which corresponds to less than one virion (one HIV-1 virion contains about 1200-3000 molecules of p24 protein). For comparison, a limit of detection of HIV using PCR is about a half of a virion (HIV-1 genome consists of two RNA molecules), although in practice a detection limit using PCR with reverse transcription is 50 virions/ml [83].
Detection of adenoviruses in stool samples was described by Bonot et al. [77]. Adenoviruses are usually detected by qPCR. Extraction and purification of nucleic acids is a necessary and critical step in PCR, which can result in analyte loss. Moreover, stool samples contain Taq-polymerase inhibitors, which complicates diagnostics and gives false-negative results. iPCR does not require DNA purification, and inhibitors are removed during numerous washes. The frequency of adenovirus identification in stool samples increased from 9.5 to 59% when compared qPCR with iPCR.
Rotaviruses cause serious diarrhea in children; 10 virus particles are enough for infection [84]. Adler et al. [54] proposed the use of iPCR for rotavirus infection diagnosis in stool samples. Despite the advantages of iPCR compared to qPCR described above, the sensitivity of iPCR described by Adler [54] was similar to the limit of detection of rotavirus particles by RT-PCR described earlier [85] – 100 viral particles/ml (probably, the absence of increase in sensitivity is due to the use of different samples for the analysis: stool samples for iPCR and water samples for RT-PCR).
Noroviruses are the cause of sporadic and epidemic gastrointestinal diseases. Noroviruses include many strains, which complicates the design of PCR primers, making diagnostics more difficult. At the same time, ELISA cannot assure sensitivity sufficient of their detection. In 2006, Tian and Mandrell [12] reported the use of iPCR for detection of noroviruses in fecal and food samples using as an example norovirus-like particles. About 100 purified virus-like particles can be detected by quantitative iPCR. Detection of noroviruses in fecal samples was 10 times more sensitive using iPCR than using PCR. Matsushita et al. [86] in 2013 also used iPCR and norovirus-like particles for detection of noroviruses in water samples.
Detection of proteins and peptides playing a major role in pathogenesis of some diseases, such as Alzheimer’s disease, prion diseases, etc., also requires a highly sensitive method for precise diagnosis at early stages. Although the pathogenesis of Alzheimer’s disease is not fully understood, the aggregation of beta-amyloid and hyper-phosphorylation of tau-protein are supposed to trigger the process [13, 47]. Low concentration of tau-protein in cerebrospinal liquid (100-2000 pg/ml) limits the possibility to use ELISA for Alzheimer’s disease diagnostics. An iPCR variant proposed by Singer et al. [13] allows detection of 2 pg/ml of tau-protein in liquor using the indirect sandwich format.
Unlike viral and bacterial diseases that can be diagnosed by means of PCR due to the presence of viruses and bacteria in biological samples and the possibility of extraction of its DNA/RNA, the presence of toxins and their level does not correlate directly with the presence of an etiological agent, which makes iPCR unique for highly sensitive detection of toxins in a studied sample. The iPCR approaches for detection of bacterial, plant, and mycotoxins in food samples, important for monitoring of food quality to prevent poisoning, have been described.
Botulotoxin is the strongest organic toxin; it is produced by the anaerobic bacterium Clostridium botulinum. Type A botulotoxin was detected by Mason and coauthors [71] by means of their method of liposomal iPCR: DNA marker was imbedded inside a liposome lipid bilayer, where the receptor of a monosialoganglioside GM1 was inserted. Potentially, ganglioside receptor can be used for the detection of not only toxins of botulism and cholera, but also toxins of pertussis, tetanus, ricin, Shiga toxin, and thermolabile enterotoxin. The authors claimed that the level of detection of botulotoxin in deionized water was 12 ± 4 molecules or 0.02 fg/ml. The lower limit of the detectable concentration depends on affinity of BA and ganglioside receptor to biotoxin. BA should not compete for binding of ganglioside with an antigen determinant. For this reason, polyclonal antibodies are more efficient as BA, unlike monoclonal antibodies that compete with ganglioside receptor (unpublished data mentioned for ricin [71]).
The possibility to use iPCR in detection of a botulin neurotoxin was studied by Andreja et al. [87]. As a model, they used type A and B botulin anatoxins. The sensitivity determined for anatoxin A in sodium phosphate buffer (PBS) was 90 pg/ml. The rather low sensitivity in this case is explained using anatoxin. However, these values are in the range of a standard method of detection and identification of botulin neurotoxins – the intraperitoneal injections in a mouse with a detection limit at 10 pg for type A botulotoxin [88, 89]. The limit of detection of type A botulin toxin in semi-skimmed milk was 3.75 pg/ml, which is 20 times lower than the limit of detection for anatoxin. Type B botulin anatoxin was detected at 370 pg/ml in PBS and 750 pg/ml in semi-skimmed milk.
The group of Shiga toxins produced by Escherichia coli is the most common cause of human food intoxication. Type 2 Shiga toxin is considered the main cause of hemolytic uremic syndrome and other changes in the organism associated with Shiga toxins. The sensitivity of the iPCR method for toxin detection described by He et al. [90] is 0.1 pg/ml in PBS and 0.01 pg/ml in milk samples. This difference in sensitivity can be probably explained by the use of commercial antibodies in the first case, and more specific, produced and carefully selected by authors monoclonal antibodies in the second.
He et al. [80] detected ricin – an extremely toxic plant protein found in castor oil plant – in samples of eggs, milk, and ground beef. Their data showed that the sensitivity of a sandwich format is higher than that of a direct iPCR. The best result was achieved using polyclonal antibodies as both BA and DA, which might have reduced the cross-reactivity and nonspecific interactions between couples of different antibodies. In another work, the presence of ricin was analyzed in samples of serum and excrements of mice [91] using, as described above, polyclonal antibodies for binding and detection of ricin. Similar techniques showed a comparable limit of detection: 10-100 pg/ml in samples from eggs, milk, and ground beef against 1 pg/ml in serum and excrements of mice.
The iPCR detection of both protein and non-protein toxins, as well as organic compounds that are dangerous pollutants of the environment, is discussed below.
Aflatoxin is a carcinogenic mycotoxin produced by some fungi of the Aspergillus genus (Aspergillus flavus, A. parasiticus, etc.), which enters the food chain from fungal infection of bread grains and forages. Immunomagnetic iPCR was developed by Babu and Muriana [45] as a tool for detection of aflatoxin B1. Several reactions were made with various order of binding of antibodies and antigen with poly-/monoclonal antibodies. The best result (a threshold cycle – 33.9) was obtained with monoclonal antibodies in indirect binding (antibodies were absorbed on magnetic beads after binding with antigen); however, the worst result (40.1) was also obtained with monoclonal antibodies, but in direct binding (first, antibodies are attached to magnetic particles and after the interaction with antigen takes place). They assumed that binding of monoclonal antibodies with a protein G results in the loss of their antigen-binding capacity due to steric problems.
Liu et al. described the detection of aflatoxin based on phage display technology [73]. The M13 phage carried an anti-idiotypic nano-antibody to aflatoxin and a DNA-tag. In this study, the authors met some difficulties associated with isolation of toxins from samples of grain and forages. Aflatoxins are purified from samples by treatment with high concentration of methanol, as they are not very soluble in water, which leads to a decrease in the activity of antibodies and results in decrease in sensitivity. They optimized the concentration of methanol for effective extraction of aflatoxin and maintenance of high sensitivity. In addition, during the analysis of corn, rice, or complete feed samples the matrix effect was eliminated by addition of 3% BSA, and during analysis of peanut samples – by addition of 0.05% Tween 20.
Polychlorinated biphenyls compose a group of organic compounds included in the list of stable organic pollutants determined by the Stockholm Convention [92]. They are widespread in the environment and can enter food chains. Yang et al. [69] developed a method for detection of such substances using gold nanoparticles as a carrier for an antibody and a DNA-tag, where monoclonal antibodies played the role of DA, and another approach was competitive iPCR with biotinylated polyclonal antibodies with avidin as a linker between DNA-tag and antibody [93]. The latter experiment was three orders of magnitude less sensitive.
Another area in which the iPCR method can be used is the detection of antibodies. Although it can be done by means of classical immunological methods, there are some exceptions. For example, determination of allergen-specific IgE is usually done by skin tests, which are not always possible, or by means of a classical immunoanalysis. Sometimes skin tests are positive, while immunoanalysis (test strips are generally used) is negative, which demonstrates some limitations of the latter technique. As shown by Lee et al. [78], who studied IgE specific to group 5 allergens of Blomia tropicalis and group 1 and 2 allergens of Dermatophagoides pteronyssinus, iPCR is a reliable method for detection of specific IgE in serum with detection limit of 200 pg/ml.
Johnston et al. [59] developed a Tus–Ter-lock iPCR approach for detection of human IgG and tropomyosin-specific IgE with sensitivity equal to 1.2 pM (180 pg/ml). This value is similar to the detection limit of the ImmunoCAP method that is a widespread commercial test providing specific and quantitative detection of IgE. However, they claim that their approach has the possibility of simultaneous detection of several allergens and can become a less expensive and simpler alternative platform for allergen profiling compared to methods based on the use of microchips.
Use of therapeutic proteins for treatment of various diseases can cause immune response and development of antibodies reducing or abolishing the effect of a drug. Thus, for safe and efficient therapy, monitoring of antibodies to therapeutic proteins in clinical samples is needed. Jani et al. [94] compared three different methods of assessment of immunogenic reaction: ELISA, electrochemiluminescence (ECL), and iPCR. All experiments were performed in sandwich format. The role of DA in ELISA was played by monoclonal antibodies conjugated with horseradish peroxidase; an enzyme marker was detected by measuring optical density after addition of 3,3′,5,5′-tetramethylbenzidine. In the ECL method, the role of DA was played by IgG conjugated with ruthenium. Correlation between the results obtained by ELISA and iPCR was observed. At the same time, iPCR showed as positive those samples that were determined as negative by ELISA. iPCR in this study was 1500 times more sensitive than ELISA and 500 times more sensitive than ECL.
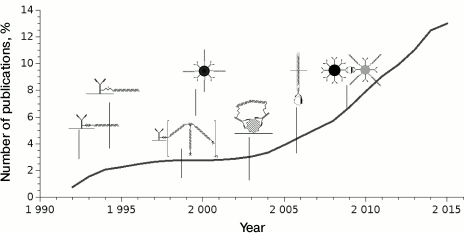
The iPCR technique has been used for nearly 25 years, although it became popular only since 2009 (Fig. 4); the number of publications using this method worldwide is not large – about 400.
Fig. 4. Dynamics of publications starting from the first article describing iPCR. Dates of publication of major types of conjugates are presented.
The sensitivity of a method depends on many factors, and we did not manage to reveal a considerable increase in sensitivity in experiments with covalent conjugates compared to biotinylated antibodies; sensitivity is apparently defined by the properties of an antibody. When using covalent antibody–DNA conjugates, sensitivity was never higher than 10–14 mol/liter, probably because of a decrease in affinity of antibodies [63]. We had a case when from six pairs of biotinylated antibodies to staphylococcal toxins having similarly low constants of affinity and showing similar background level and sensitivity in ELISA, only three pairs showed a high level of sensitivity in iPCR (unpublished data).
Many studies (35%) used biotin–streptavidin conjugates, although the most spectacular results in terms of sensitivity were obtained using bifunctional nanoparticles and a biobarcoding system (down to 0.1 fg/ml or 1.14·10–18 mol/liter). However, the detection limit in the range of 10–16-10–15 mol/liter was reached also with other iPCR variants with the use of biotinylated antibodies on nanoparticles [31], liposomes [71] and phage display [15]. Some researchers report a slightly higher sensitivity of this method compared with ELISA (less than 10-fold) [46, 48, 74].
The iPCR method has some drawbacks. Generally, it is the duration and the complexity of optimization of the method and the analysis itself. Good experimental practice is needed to achieve high sensitivity. A protocol comprises 20 washes (in the protocol of Malou [52] there were 31 washes in an automatic washer), about half of them done after incubation with a reagent containing a DNA-tag [18, 21]; the whole experiment can take from 26 h [35] to 2 days [18]. It is possible to reduce the time of analysis by excluding a step of binding of antigen/primary antibodies (depending on type of immunoanalysis) on the plates. This can be achieved using magnetic particles with covalently bound BA or using biobarcoding technology. The duration of analysis can be shortened by preassembly of biotinylated DNA–streptavidin-biotinylated antibody complexes or using covalent conjugates. Automation of the analysis can further shorten its duration. It was shown that use of automatic washers alone reduces deviation of results and improves reproducibility [48]. One direction for further development is to create a robotic station for carrying out of a full iPCR cycle.
Despite the mentioned drawbacks, the advantages of iPCR are obvious: they are high sensitivity and broad range of concentrations that can be determined (several orders of magnitude) [10, 64, 75]. An important advantage of iPCR compared to ELISA is the possibility to detect an antigen in a small quantity of a sample (also in case of significant dilution to decrease the influence of “matrix effect”), which is essential for analysis of food samples [80, 95], in pediatrics (where sometimes it is not possible to get enough analyzed material), and other spheres. Many aliquots for repetitive and more precise analysis can be prepared, since a small amount of analyzed material is sufficient for the reaction. Highly sensitive multiplex detection of substances (analyte) in extremely small concentration [7], including in a biobarcoding system [67-80], is also important. Immuno-PCR, combining the advantages of ELISA and PCR, overcomes the limitations of both methods. Considering the great potential of iPCR, we predict wider use of this method with further optimization and simplification of protocols.
Acknowledgements
This work was done with a financial support by the Russian Science Foundation (project No. 16-14-00136) and a scholarship of the President of RF SP-4413.2015.4.
REFERENCES
1.Sano, T., Smith, C. L., and Cantor, C. R. (1992)
Immuno-PCR: very sensitive antigen detection by means of specific
antibody–DNA conjugates, Science, 258, 120-122.
2.Saiki, R. K., Scharf, S., Faloona, F., Mullis, K.
B., Horn, G. T., Erlich, H. A., and Arnheim, N. (1985) Enzymatic
amplification of beta-globin genomic sequences and restriction site
analysis for diagnosis of sickle cell anemia, Science,
230, 1350-1354.
3.Nakamura, S., Katamine, S., Yamamoto, T., Foung, S.
K., Kurata, T., Hirabayashi, Y., Shimada, K., Hino, S., and Miyamoto,
T. (1993) Amplification and detection of a single molecule of human
immunodeficiency virus RNA, Virus Genes, 7, 325-338.
4.Li, H., Cui, X., and Arnheim, N. (1990) Direct
electrophoretic detection of the allelic state of single DNA molecules
in human sperm by using the polymerase chain reaction, Proc. Natl.
Acad. Sci. USA, 87, 4580-4584.
5.Sano, T., Smith, C. L., and Cantor, C. R. (1993)
Response, Science, 260, 699.
6.Zhou, H., Fisher, R. J., and Papas, T. S. (1993)
Universal immuno-PCR for ultra-sensitive target protein detection,
Nucleic Acids Res., 21, 6038-6039.
7.Hendrickson, E. R., Truby, T. M., Joerger, R. D.,
Majarian, W. R., and Ebersole, R. C. (1995) High sensitivity
multianalyte immunoassay using covalent DNA-labeled antibodies and
polymerase chain reaction, Nucleic Acids Res., 11,
522-529.
8.Wang, T., Lu, H., Lou, P., and Lin, F. H. (2008)
Application of highly sensitive, modified glass substrate-based
immuno-PCR on the early detection of nasopharyngeal carcinoma,
Biomaterials, 29, 4447-4454.
9.Kuczius, T., Becker, K., Fischer, A., and Zhang, W.
(2012) Simultaneous detection of three CNS indicator proteins in
complex suspensions using a single immuno-PCR protocol, Anal.
Biochem., 431, 4-10.
10.Barletta, J., Bartolome, A., and Constantine, N.
(2009) Immunomagnetic quantitative immuno-PCR for detection of less
than one HIV-1 virion, J. Virol. Methods, 157,
122-132.
11.Chen, L., Wei, H., Guo, Y., Cui, Z., Zhang, Z.,
and Zhang, X. E. (2009) Gold nanoparticle enhanced immuno-PCR for
ultrasensitive detection of Hantaan virus nucleocapsid protein, J.
Immunol. Methods, 346, 64-70.
12.Tian, P., and Mandrell, R. (2006) Detection of
norovirus capsid proteins in fecal and food samples by a real time
immuno-PCR method, J. Appl. Microbiol., 100, 564-574.
13.Singer, D., Soininen, H., Alafuzoff, I., and
Hoffmann, R. (2009) Immuno-PCR-based quantification of multiple
phosphorylated tau-epitopes linked to Alzheimer’s disease,
Anal. Bioanal. Chem., 395, 2263-2267.
14.Dan, B., Huisheng, Zh., Guangxin, Y., and
Xianyin, P. (2015) A real-time immuno-PCR assay for the detection of
tetrabromobisphenol A, Anal. Methods., 7, 99-106.
15.Zhuang, H. S., and Zhou, C. (2009) Determination
of anthracene by real-time immuno-polymerase chain reaction assay,
Anal. Chim. Acta, 633, 278-282.
16.Meng, X. Y., Li, Y. S., Zhou, Y., Zhang, Y. Y.,
Qiao, B., Sun, Y., Yang, L., Hu, P., Lu, S. Y., Ren, H. L., Zhang, J.
H., Wang, X. R., and Liu, Z. S. (2015) Real-time immuno-PCR for
ultrasensitive detection of pyrene and other homologous PAHs, Bio
Sens. Bioelectron., 70, 42-47.
17.Tao, X., He, Z., Cao, X., Shen, J., and Li, H.
(2014) Development of a highly sensitive real-time immuno-PCR for the
measurement of chloramphenicol in milk based on magnetic bead
capturing, Anal. Methods, 6, 9340-9347.
18.Niemeyer, C. M., Adler, M., and Wacker, R. (2007)
Detecting antigens by quantitative immuno-PCR, Nature Protocols,
2, 1918-1930.
19.Liang, H., Cordova, S. E., Kieft, T. L., and
Rogelj, S. (2003) A highly sensitive immuno-PCR assay for detecting
group A Streptococcus, J. Immunol. Methods, 279,
101-110.
20.Chao, H. Y., Wang, Y. C., Tang, S. S., and Liu,
H. W. (2004) A highly sensitive immuno-polymerase chain reaction assay
for Clostridium botulinum neurotoxin type A, Toxicon,
43, 27-34.
21.Barletta, J. (2006) Applications of real-time
immuno-polymerase chain (RT-iPCR) for the rapid diagnoses of viral
antigens and pathologic proteins, Mol. Aspects Med., 27,
224-253.
22.Fischer, A., Von Eiff, C., Kuczius, T., Omoe, K.,
Peters, G., and Becker, K. (2007) A quantitative real-time immuno-PCR
approach for detection of staphylococcal enterotoxins, J. Mol.
Med. (Berl.), 85, 461-469.
23.Niemeyer, C. M., Wacker, R., and Adler M. (2003)
Combination of DNA-directed immobilization and immuno-PCR: very
sensitive antigen detection by means of self-assembled
DNA–protein conjugates, Nucleic Acids Res., 31,
e90.
24.Pinzani, P., Lind, K., Malentacchi, F., Nesi, G.,
Salvianti, F., Villari, D., Kubista, M., Pazzagli, M., and Orlando, C.
(2008) Prostate-specific antigen mRNA and protein levels in laser
microdissected cells of human prostate measured by real-time reverse
transcriptase-quantitative polymerase chain reaction and
immuno-quantitative polymerase chain reaction, Hum. Pathol.,
39, 1474-1482.
25.Gullberg, M., Gustafsdottir, S., Schallmeiner,
E., Jarvius, J., Bjarnegård, M., Betsholtz, C., Landegren, U.,
and Fredriksson, S. (2004) Cytokine detection by antibody-based
proximity ligation, Proc. Natl. Acad. Sci. USA, 101,
8420-8424.
26.Case, M., Burt, A., Hughes, J., Palmer, J.
M., Collier, J. D., Bassendine, M. F., Yeaman, S. J.,
Hughes, M. A., and Major, G. N. (1999) Enhanced ultrasensitive
detection of structurally diverse antigens using a single immuno-PCR
assay protocol, J. Immunol. Methods, 223, 93-106.
27.Numata, Y., and Matsumoto, Y. (1997) Rapid
detection of alpha-human atrial natriuretic peptide in plasma by a
sensitive immuno-PCR sandwich assay, Clin. Chim. Acta,
259, 169-176.
28.Niemeyer, C. M., Adler, M., Pignataro, B.,
Lenhert, S., Gao, S., Lifeng, C., Fuchs, H., and Dietmar, B. (1999)
Self-assembly of DNA–streptavidin nanostructures and their use as
reagents in immuno-PCR, Nucleic Acids Res., 27,
4553-4561.
29.Saito, K., Kobayashi, D., Sasaki, M., Araake, H.,
Kida, T., Yagihashi, A., Yajima, T., Kameshima, H., and Watanabe, N.
(1999) Detection of human serum tumor necrosis factor-alpha in healthy
donors, using a highly sensitive immuno-PCR assay, Clin. Chem.,
45, 665-669.
30.Adler, M., Langer, M., Witthohn, K., Eck, J.,
Blohm, D., and Niemeyer, C. M. (2003) Detection of rViscumin in plasma
samples by immuno-PCR, Biochem. Biophys. Res. Commun.,
300, 757-763.
31.Potuskova, L., Franko, F., Bambouskova, M., and
Draber, P. (2011) Rapid and sensitive detection of cytokines using
functionalized gold nanoparticle-based immuno-PCR, comparison with
immuno-PCR and ELISA, J. Immunol. Methods, 371,
38-47.
32.Ryazantsev, D. Yu., Petrova, E. E., Kalinina, N.
A., Valyakina, T. I., Grishin, E. V., and Zavriev, S. K. (2012)
Application of supramolecular DNA–streptavidin complexes for
ultrasensitive detection of several toxins by immuno-PCR, Global J.
Anal. Chem., 3, e17.
33.Ryazantsev, D. Yu., and Zavriev, S. K. (2011)
Immuno-PCR: application of DNA–streptavidin supramolecular
complexes for hypersensitive detection of antigens, FEBS J.,
238, 161-162.
34.Maerle, A. V., Ryazantsev, D. Yu., Dmitrenko, O.
A., Petrova, E. E., Komaleva, R. L., Sergeeva, I. V., Trofimov, D. Yu.,
and Zavriev, S. K. (2014) Detection of Staphylococcus aureus
toxins using immuno-PCR, Russ. J. Bioorg. Chem., 40,
526-531.
35.McKie, A. A., Samuel, D., Cohen, B., and
Saunders, N. A. (2002) A quantitative immuno-PCR assay for the
detection of mumps-specific IgG, J. Immunol. Methods,
270, 135-141.
36.Harlow, E., and Lane, D. (1988) Antibodies: A
Laboratory Manual, Cold Spring Harbor Laboratory Press, N. Y., p.
559.
37.Ruzicka, V., Marz, W., Russ, A., and Gross, W.
(1993) Immuno-PCR with a commercially available avidin system,
Science, 260, 698-699.
38.Malou, N., and Raoult, D. (2011) Immuno-PCR: a
promising ultrasensitive diagnostic method to detect antigens and
antibodies, Trends Microbiol., 19, 295-302.
39.Niemeyer, C. M., Adler, M., and Wacker, R. (2005)
Immuno-PCR: high sensitivity detection of proteins by nucleic acid
amplification, Trends Biotechnol., 23, 208-216.
40.Crowther, J. R. (1995) ELISA: Theory and
Practice, Humana Press Inc.
41.He, X., Qi, W., Quinones, B., McMahon, S.,
Cooley, M., and Mandrell, R. E. (2011) Sensitive detection of Shiga
toxin 2 and some of its variants in environmental samples by a novel
immuno-PCR assay, Appl. Environ. Microbiol., 77,
3558-3564.
42.Sims, P. W., Vasser, M., Wong, W. L., Williams,
P. M., and Meng, Y.-J. G. (2000) Immuno-polymerase chain reaction using
real-time polymerase chain reaction for detection, Anal.
Biochem., 281, 230-232.
43.Deng, M., Xiao, X., Ji, X., Sun, T., Wu, Z. X.,
Zheng, X. L., Wang, Q., and Zhu, L. H. (2014) Immuno-PCR for detection
of Giardia lamblia Cy in water, J. AOAC Int., 97,
561-566.
44.Allen, R., Rogelj, S., Cordova, S., and Kieft, T.
L. (2006) An immuno-PCR method for detecting Bacillus
thuringiensis Cry1Ac toxin, J. Immunol. Methods, 205,
364-367.
45.Babu, D., and Muriana, P. (2011) Immunomagnetic
bead-based recovery and real time quantitative PCR (RT iq-PCR) for
sensitive quantification of aflatoxin B(1), J. Microbiol.
Methods, 86, 188-194.
46.Zhang, J., Vernes, J., Ni, J., Nelson, C., Wong,
A., Chen, S. T., Asundi, A., Vandlen, R., and Meng, Y. G. (2014)
Real-time immuno-polymerase chain reaction in a 384-well format:
detection of vascular endothelial growth factor and epidermal growth
factor-like domain 7, Anal. Biochem., 463, 61-66.
47.Agasti, S. S., Liong, M., Peterson, V. M., Lee,
H., and Weissleder, R. (2012) Photocleavable DNA barcode–antibody
conjugates allow sensitive and multiplexed protein analysis in single
cells, J. Am. Chem. Soc., 134, 18499-184502.
48.Hashimoto, M., Aoki, M., Winblad, B., and
Tjernberg, L. O. (2012) A novel approach for Αβ1-40
quantification using immuno-PCR, J. Neurosci. Methods,
205, 364-367.
49.Kazane, S., Sok, D., Cho, E. H., Uson, M. L.,
Kuhn, P., Schultz, P. G., and Smider, V. V. (2012) Site-specific
DNA–antibody conjugates for specific and sensitive immuno-PCR,
Proc. Natl. Acad. Sci. USA, 109, 3731-3736.
50.Spicer, C. D., and Davis, B. G. (2014) Selective
chemical protein modification, Nat. Commun., 5, 4740.
51.Van Buggenum, J. A., Gerlach, J. P., Eising, S.,
Schoonen, L., van Eijl, R. A., Tanis, S. E., Hogeweg, M., Hubner, N.
C., Van Hest, J. C., Bonger, K. M., and Mulder, K. W. (2016) A covalent
and cleavable antibody–DNA conjugation strategy for sensitive
protein detection via immuno-PCR, Sci. Rep., 6,
22675.
52.Malou, N., Renvoise, A., Nappez, C., and Raoult,
D. (2012) Immuno-PCR for the early serological diagnosis of acute
infectious diseases: the Q fever paradigm, Eur. J. Clin. Microbiol.
Infect. Dis., 31, 1951-1960.
53.Spengler, M., Adler, M., Jonas, A., and Niemeyer,
C. M. (2009) Immuno-PCR assays for immunogenicity testing, Biochem.
Biophys. Res. Commun., 387, 278-282.
54.Adler, M., Schulz, S., Fisher, R., and Niemeyer,
C. M. (2005) Detection of rotavirus from stool samples using a
standardized immuno-PCR (“Imperacer”) method with end-point
and real-time detection, Biochem. Biophys. Res. Commun.,
333, 1289-1294.
55.Luk, C., Compta, Y., Magdalinou, N., Marti, M.
J., Hondhamuni, G., Zetterberg, H., Blennow, K., Constantinescu, R.,
Pijnenburg, Y., Mollenhauer, B., Trenkwalder, C., Van Swieten,
J., Chiu, W. Z., Borroni, B., Camara, A., Cheshire,
P., Williams, D. R., Lees, A. J., and De Silva, R. (2012)
Development and assessment of sensitive immuno-PCR assays for the
quantification of cerebrospinal fluid three- and four-repeat tau
isoforms in tauopathies, J. Neurochem., 123, 396-405.
56.Soderberg, O., Gullberg, M., Jarvius, M.,
Ridderstrale, K., Leuchowius, K.-J., Jarvius, J., Wester, K., Hygbring,
P., Bahram, F., Larsson, L.-G., and Landegren, U. (2006) Direct
observation of individual endogenous protein complexes in situ
by proximity ligation, Nat. Methods, 3, 995-1000.
57.Gehwolf, R., Band, E., Trost, A., Iglseder, B.,
Trinka, E., Haschke-Becher, E., Kraus, J., and Harrer, A. (2014)
TaqManR proximity ligation technology for the detection of
heterodimeric adhesion receptors on lymphocytes, J. Immunol.
Methods, 404, 81-86.
58.Jiang, X., Cheng, S., Chen, W., Wang, L., Shi,
F., and Zhu, C. (2012) Comparison of oligonucleotide-labeled antibody
probe assays for prostate-specific antigen detection, Anal.
Biochem., 424, 1-7.
59.Johnston, E., Kamath, S., Lopata, A., and
Schaeffer, P. M. (2014) Tus–Ter-lock immuno-PCR assays for the
sensitive detection of tropomyosin-specific IgE antibodies,
Bioanalysis, 6, 465-476.
60.Morin, I., Askin, S. P., and Schaeffer, P. M.
(2011) IgG-detection devices for the Tus–Ter-lock immuno-PCR
diagnostic platform, Analyst, 136, 4815-4821.
61.Li, Z., Wang, X., Chang, J., Xie, W. B., Liu, T.
F., Zhang, Q. L., Deng, Y. J., and Ding, Y. Q. (2012) The establishment
of supramolecular immunobead real-time PCR and the identification of
Cox-2 as a metastasis-related marker in colorectal carcinoma, Oncol.
Rep., 28, 977-984.
62.Deng, M., Long, L., Xiao, X., Wu, Z., Zhang, F.,
Zhang, Y., Zheng, X., Xin, X., Wang, Q., and Wu, D. (2011) Immuno-PCR
for one step detection of H5N1 avian influenza virus and Newcastle
disease virus using magnetic gold particles as carriers, Vet.
Immunol. Immunopathol., 141, 183-189.
63.Nikitina, I., Sabirova, E., Solopova, O.,
Surzhikov, S. A., Grineva, E. N., Karpov, V. L., Lisitsyn, N. A., and
Beresten’, S. F. (2014) A new immunoPCR format for serological
diagnosis of colon cancer, Mol. Biol. (Moscow), 48,
117-123.
64.Ding, Y., Liu, Y., Zhou, J., Chen, H. T., Zhang,
J., Ma, L. N., and Wei, G. (2011) A highly sensitive detection for
foot-and-mouth disease virus by gold nanoparticle improved immuno-PCR,
Virol. J., 8, 148.
65.Nam, J. M., Thaxton, C. S., and Mirkin, C. A.
(2003) Nanoparticle-based bio-barcodes for the ultrasensitive detection
of proteins, Science, 301, 1884-1886.
66.Nam, J. M., Park, S. J., and Mirkin, C. A. (2002)
Bio-barcodes based on oligonucleotide-modified nanoparticles, J. Am.
Chem. Soc., 124, 3820-3821.
67.Perez, J., Vargis, E., Russ, P., Haselton, F. R.,
and Wright, D. W. (2011) Detection of respiratory syncytial virus using
nanoparticle amplified immuno-polymerase chain reaction, Anal.
Biochem., 410, 141-148.
68.Yin, H., Jia, M., Shi, L., Yang, S., Zhang, L.
Y., Zhang, Q. M., Wang, S. Q., Li, G., and Zhang, J. G. (2011)
Nanoparticle-based bio-barcode assay for the detection of bluetongue
virus, J. Virol. Methods, 178, 225-228.
69.Yang, G., Zhuang, H., Chen, H. Y., Ping, X. Y.,
and Bu, D. (2014) A sensitive immunosorbent bio-barcode assay based on
real-time immuno-PCR for detecting
3,4,3′,4′-tetrachlorobiphenyl, Anal. Bioanal. Chem.,
406, 1693-1700.
70.Yang, G., Zhuang, H., Chen, H., Ping, X., and Bu,
D. (2015) A gold nanoparticle based immunosorbent bio-barcode assay
combined with real-time immuno-PCR for the detection of polychlorinated
biphenyls, Sens. Actuator B Chem., 214, 152-158.
71.Mason, J., Xu, L., Sheng, Z., He, J., and
O’Leary, T. J. (2006) Liposome polymerase chain reaction assay
for the sub-attomolar detection of cholera toxin and botulinum
neurotoxin type A, Nat. Protoc., 1, 2003-2011.
72.Guo, Y., Zhou, Y., Zhang, X., Zhang, Z., Qiao,
Y., Bi, L., Wen, J., Liang, M., and Zhang, J. (2006) Phage display
mediated immuno-PCR, Nucleic Acids Res., 34, e62.
73.Lei, J., Li, P., Zhang, Q., Wang, Y., Zhang, Z.,
Ding, X., and Zang, W. (2014) An anti-idiotypic nanobody-phage based
real-time immuno-PCR for detection of hepatocarcinogen aflatoxin in
grains and feedstuffs, Anal. Chem., 86, 10841-10846.
74.Liu, X., Xu, Y., Xiong, Y., Tu, Z., Li, Y. P.,
He, Z. Y., Qiu, Y. L., Fu, J. H., Gee, S. J., and Hammock, B. D. (2014)
VHH phage-based competitive real-time immuno-polymerase chain reaction
for ultrasensitive detection of ochratoxin A in cereal, Anal.
Chem., 86, 7471-7477.
75.Monjezi, R., Tan, S., Tey, B., Sieo, C. C., and
Tan, W. S. (2013) Detection of hepatitis B virus core antigen by phage
display mediated TaqMan real-time immuno-PCR, J. Virol. Methods,
187, 121-126.
76.Kasai, N., Kobayashi, K., Shioya, S., Yoshikawa,
Y., Yotsumoto, F., Miyamoto, S., Mekada, E., and Enokizono, J. (2012)
Soluble heparin-binding EGF-like growth factor (HB-EGF) detected by
newly developed immuno-PCR method is a clear-cut serological biomarker
for ovarian cancer, Am. J. Transl. Res., 4, 415-421.
77.Bonot, S., Ogorzaly, L., El Moualij, B., Zorzi,
W., and Cauchie, H. M. (2014) Detection of small amounts of human
adenoviruses in stools: comparison of a new immuno real-time PCR assay
with classical tools, Clin. Microbiol. Infect., 20,
O1010-6.
78.Lee, K., Hur, B., Chua, K., Kuo, I., Song, S.,
and Cha, S. (2008) Detection of serum IgE specific to mite allergens by
immuno-PCR, Immune Network, 8, 82-89.
79.Kakizaki, E., Yoshida, T., Kawakami, H., Oseto,
M., Sakai, T., and Sakai, M. (1996) Detection of bacterial antigens
using immuno-PCR, Lett. Appl. Microbiol., 23,
101-103.
80.He, X., McMahon, S., McKeon, T., and Brandon, D.
L. (2010) Development of a novel immuno-PCR assay for detection of
ricin in ground beef, liquid chicken egg, and milk, J. Food.
Prot., 73, 695-700.
81.Mikita, K., Thakur, K., Anstey, N., Piera, K. A.,
Pardo, C. A., Weinberg, J. B., Mukemba, J., Florence, S., Mwaikambo, E.
D., Granger, D. L., and Sullivan, D. J. (2014) Quantification of
Plasmodium falciparum histidine-rich protein-2 in cerebrospinal
spinal fluid from cerebral malaria patients, Am. J. Trop. Med.
Hyg., 91, 486-492.
82.Peroni, L., Reis, J., Coletta-Filho, H., De
Souza, A. A., Machado, M. A., and Stach-Machado, D. R. (2008)
Assessment of the diagnostic potential of immmunocapture-PCR and
immuno-PCR for citrus variegated chlorosis, J. Microbiol.
Methods, 75, 302-307.
83.Barletta, J. M., Edelman, D. C., and Constantine,
N. T. (2004) Lowering the detection limits of HIV-1 viral load using
real-time immuno-PCR for HIV-1 p24 antigen, Am. J. Clin.
Pathol., 122, 20-27.
84.http://edoc.rki.de/documents/rki_fv/rewfsQVGV4llc/PDF/20H9Hx8sK,
(2002) Epidemiologisches Bulletin, No. 10, 77-80.
85.Kittigul, L., Ekchaloemkiet, S., Utrarachkij, F.,
Siripanichgon, K., Sujirarat, D., Pungchitton, S., and Boonthum, A.
(2004) An efficient virus concentration method and RT-nested PCR for
detection of rotaviruses in environmental water samples, J. Virol.
Methods, 124, 117-122.
86.Matsushita, T., Shirasaki, N., Tatsuki, Y., and
Matsui, Y. (2013) Investigating norovirus removal by microfiltration,
ultrafiltration, and precoagulation-microfiltration processes using
recombinant norovirus virus-like particles and real-time immuno-PCR,
Water Res., 47, 5819-5827.
87.Andreja, R., Benaissa, E., Youssef, F., Dierick,
K., Zorzi, W., Heinen, E., Uner, A., and Uyttendaele, M. (2012)
Detection of Clostridium botulinum neurotoxins A and B in milk
by ELISA and immuno-PCR at higher sensitivity than mouse bio-assay,
Food Anal. Methods, 5, 319-326.
88.Scarlatos, A., Welt, B. A., Cooper, B. Y.,
Archer, D., DeMarse, T., and Chau, K. V. (2005) Methods for detecting
botulinum toxin with applicability to screening foods against
biological terrorist attacks, J. Food Sci., 70,
r121-r130.
89.Sharma, S. K., Ferreira, J. L., Eblen, B. S., and
Whiting, R. C. (2006) Detection of type A, B, E, and F clostridium
botulinum neurotoxins in foods by using an amplified enzyme-linked
immunosorbent assay with digoxigenin-labeled antibodies, Appl.
Environ. Microbiol., 72, 1231-1238.
90.He, X., McMahon, S., Skinner, C., Merrill, P.,
Scotcher, M. C., and Stanker, L. H. (2013) Development and
characterization of monoclonal antibodies against Shiga toxin 2 and
their application for toxin detection in milk, J. Immunol.
Methods, 389, 18-28.
91.He, X., McMahon, S., Henderson, T., Griffey, S.
M., and Cheng, L. W. (2010) Ricin toxicokinetics and its sensitive
detection in mouse sera or feces using immuno-PCR, PLoS One,
5, e12858.
92.Stockholm Convention on Persistent Organic
Pollutants, http://chm.pops.int.
93.Chen, H., and Zhuang, H. (2011) A real-time
immuno-PCR method for detecting
3,3’,4,4’-tetrachlorobiphenyl, Microchim. Acta,
172, 233-239.
94.Jani, D., Savino, E., and Goyal, J. (2015)
Feasibility of immuno-PCR technology platforms as an ultrasensitive
tool for the detection of anti-drug antibodies, Bioanalysis,
7, 285-294.
95.Tao, X., Jiang, H., Zhu, J., Wang, X., Wang, Z.,
Niu, L., Wu, X., Shi, W., and Shen, J. (2014) An ultrasensitive
chemiluminescent ELISA for determination of chloramphenicol in milk,
milk powder, honey, eggs and chicken muscle, Food Agric.
Immunol., 25, 137-148.
96.Deng, M., Xiao, X., Zhang, Y., Wu, X. H., Zhu, L.
H., Xin, X. Q., and Wu, D. L. (2011) A highly sensitive immuno-PCR
assay for detection of H5N1 avian influenza virus, Mol. Biol.
Rep., 38, 1941-1948.
97.Chen, Z., Hong, L., Liu, L., Peng, D., Li, Q.,
Jin, B., Qiao, T., Wu, K., and Fan, D. (2010) Monoclonal antibody MG7
as a screening tool for gastric cancer, Hybridoma, 29,
27-30.
98.Stegurova, L., Draberova, E., Bartos, A., Draber,
P., Ripova, D., and Draber, P. (2014) Gold nanoparticle-based
immuno-PCR for detection of tau protein in cerebrospinal fluid, J.
Immunol. Methods, 406, 137-142.
99.Barletta, J., Edelman, D., Highsmith, W., and
Constantine, N. T. (2005) Detection of ultra-low levels of pathologic
prion protein in scrapie infected hamster brain homogenates using
real-time immuno-PCR, J. Virol. Methods, 127,
154-164.
100.Reuter, T., Gilroyed, B., Alexander, T.,
Mitchell, G., Balachandran, A., Czub, S., and McAllister, T. A. (2009)
Prion protein detection via direct immuno-quantitative real-time PCR,
J. Microbiol. Methods, 78, 307-311.
101.Malou, N., Tran, T. N., Nappez, C., Signoli,
M., Le Forestier, C., Castex, D., Drancourt, M., and Raoult, D. (2012)
Immuno-PCR – a new tool for paleomicrobiology: the plague
paradigm, PLoS One, 7, e31744.
102.Diel, P., Schiffer, T., Geisler, S., Hertrampf,
T., Mosler, S., Schulz, S., Wintgens, K. F., and Adler, M. (2010)
Analysis of the effects of androgens and training on myostatin
propeptide and follistatin concentrations in blood and skeletal muscle
using highly sensitive immuno PCR, Mol. Cell. Endocrinol.,
330, 1-9.
103.Dong, J., Hasan, S., Fujioka, Y., and Ueda, H.
(2012) Detection of small molecule diagnostic markers with phage-based
open-sandwich immuno-PCR, J. Immunol. Methods, 377,
1-7.
104.Kwon, K., Hwang, S., Park, Y., Yoon, J., Kim,
S., and Hong, J. (2014) A quantitative real-time immuno-PCR assay for
detection of Staphylococcus aureus enterotoxin H, J.
Food Saf., 34, 249-256.
105.Kumar, R. (2011) A quantitative
immuno-polymerase chain reaction method for detection of vegetative
insecticidal protein in genetically modified crops, J. Agric. Food
Chem., 59, 10448-10453.
106.Kumar, R. (2012) A real-time immuno-PCR assay
for the detection of transgenic Cry1Ab protein, Eur. Food Res.
Technol., 234, 101-108.
107.Bu, D., and Zhuang, H. (2014) A real-time
immuno-PCR assay for the flame retardant tris(2,3-dibromopropyl)
isocyanurate using a probe DNA conjugated to gold nanoparticles,
Microchim. Acta, 182, 1863-1868.
108.Sun, R., and Zhuang, H. (2015) An
ultrasensitive gold nanoparticles improved real-time immuno-PCR assay
for detecting diethyl phthalate in foodstuff samples, Anal.
Biochem., 480, 49-57.