REVIEW: Mechanisms of Non-canonical Activation of Ataxia Telangiectasia Mutated
S. V. Khoronenkova1,2
1University of Cambridge, Department of Biochemistry, Cambridge CB2 1GA, UK; E-mail: sk870@cam.ac.uk2Lomonosov Moscow State University, Department of Chemistry, 119991 Moscow, Russia
Received March 12, 2016
ATM is a master regulator of the cellular response to DNA damage. The classical mechanism of ATM activation involves its monomerization in response to DNA double-strand breaks, resulting in ATM-dependent phosphorylation of more than a thousand substrates required for cell cycle progression, DNA repair, and apoptosis. Here, new experimental evidence for non-canonical mechanisms of ATM activation in response to stimuli distinct from DNA double-strand breaks is discussed. It includes cytoskeletal changes, chromatin modifications, RNA–DNA hybrids, and DNA single-strand breaks. Noncanonical ATM activation may be important for the pathology of the multisystemic disease Ataxia Telangiectasia.
KEY WORDS: ATM, Ataxia Telangiectasia mutated, DNA damage, DNA single-strand breaks, DNA double-strand breaks, R-loopsDOI: 10.1134/S0006297916130058
Abbreviations: A-T, Ataxia Telangiectasia; ATM, Ataxia Telangiectasia mutated; ATR, ATM- and Rad3-related kinase; DNA-PKcs, DNA-dependent protein kinase catalytic subunit; DSB, DNA double-strand break; MRN, Mre11-Rad50-Nbs1 complex; R-loop, RNA–DNA hybrid; SSB, DNA single-strand break; Top1, DNA topoisomerase I; Top1cc, Top1–DNA intermediate.
ATM (Ataxia Telangiectasia mutated, EC 2.7.11.1) is a serine/threonine
protein kinase that belongs to a family of phosphatidylinositol
3-kinase related kinases (PIKK). The ATM gene encodes a 350-kDa
protein consisting of 3056 amino acids. The domain structure of ATM
includes HEAT repeats, FAT (FRAM/ATM/TRRAP), C-terminal FATC and kinase
domains as described elsewhere [1]. ATR (ATM- and
Rad3-related kinase) and DNA-PKcs (catalytic subunit of DNA-dependent
protein kinase) display significant homology with ATM. ATM, ATR, and
DNA-PKcs are all important for the cellular response to DNA damage,
although specific functions of these kinases differ significantly [2].
The primary function of ATM is coordination of the cellular response to DNA damage caused by ionizing radiation. The mechanism of ATM kinase activation in response to ionizing radiation, initially proposed by Kastan’s laboratory, involves intramolecular autophosphorylation of ATM at serine 1981 followed by its monomerization [3]. However, subsequent studies showed that many other amino acid residues undergo posttranslational modification. These include acetylation of lysine 3016 by the Tip60 histone acetyltransferase required for induction of kinase activity followed by autophosphorylation [4, 5], as well as autophosphorylation of serines 367, 1893, and 2996 and tyrosine 1885 [6-8]. These modifications are functionally important in human cell lines, but their relevance in mouse models and Xenopus extracts remains controversial [9, 10]. The canonical model for activation invokes a specific role for DNA double-strand breaks (DSBs), which are highly mutagenic DNA lesions induced by ionizing radiation. ATM activation requires the presence of the MRN (Mre11–Rad50–Nbs1) complex that ensures the initial localization of ATM in a complex with Tip60 at sites of DSB formation [4, 5, 11-14]. In response to ATM activation, a cascade of kinase activities leads to the phosphorylation of over a thousand substrates that are required for the coordination of various cellular processes such as chromatin remodeling, transcription and splicing, cell cycle progression, DNA repair, and apoptosis (for reviews see [15-17]).
It has also been proposed that ATM is activated following oxidative stress by a fundamentally different mechanism [18]. In this case, an active ATM dimer contains a disulfide bond formed upon oxidation of two cysteine 2991 residues. In contrast to DSB-dependent activation of ATM in the nucleus, this oxidative-dependent ATM activation mechanism also occurs in the cytoplasm and is important for the coordination of insulin signaling and mitochondrial and peroxisome functions [19-21]. Consistent with these proposals, several cytoplasmic substrates of ATM have been identified by quantitative proteomics [22]. The mechanisms of ATM activation in response to DSBs and oxidative stress are discussed in detail in several comprehensive reviews [17, 23-26].
Given the functional importance of ATM in the cellular landscape, it is not surprising that inactivation of its function underpins the disease Ataxia Telangiectasia (A-T), also referred to as Louis–Bar syndrome [27-29]. A-T is a rare autosomal recessive multisystemic disorder (one case per 40,000-100,000) that develops in early childhood [30, 31]. A-T is characterized by immunodeficiency, progressive neurodegeneration, and increased predisposition to cancer. There are currently no treatments for A-T [32].
In recent years, the exclusivity of DNA double-strand breaks and oxidative stress as inducers of ATM activation has been questioned. In this review, new experimental data are discussed that indicate a wider cellular role for ATM as a sensor and regulator of the cellular response to DNA single-strand breaks, RNA–DNA hybrids, as well as changes in the structure of chromatin and the cytoskeleton.
NON-CANONICAL ACTIVATORS OF ATM
DNA lesions (except for DNA double-strand breaks). DNA single-strand breaks. More than 20 years ago, Tomas Lindahl (2015 Nobel Laureate in Chemistry) proposed that the number of DNA double-strand breaks arising due to the inherent instability of DNA is significantly lower (10-20 lesions per cell per day) than that of endogenous DNA single-strand breaks (15,000-20,000 lesions/cell/day) [33]. In addition, DNA single-strand breaks (SSBs) form as intermediates during the repair of damaged DNA bases (the so-called base excision repair pathway) [34]. The repair of SSBs is crucial for a cell as the replication of SSB-containing DNA leads to the formation of highly mutagenic DSBs [35]. Moreover, the transcription of SSB-containing DNA is inefficient and can be blocked [36, 37]. Defects in SSB repair have been linked to various diseases, including neurodegeneration and cancer [38-42].
Unrepaired SSBs activate ATM in the absence of DSBs [43]. This activation is important for promoting a G1 cell cycle delay, thus providing time for the controlled repair of SSBs prior to the replication of DNA and therefore preventing the formation of replication-associated DSBs. In addition, ATM-dependent signaling is important for regulating the capacity of DNA base damage and SSB repair [44, 45]. Consequently, inadequate signaling of unrepaired SSBs in the absence of ATM leads to replication of the damaged DNA followed by accumulation of mutagenic DSBs, thus contributing to the genetic instability phenotype characteristic of A-T (Fig. 1).
Fig. 1. ATM-dependent coordination of SSB repair. Activation of ATM in response to unrepaired SSBs coordinates their repair by increasing the efficiency of DNA repair and promoting a G1 cell cycle delay that provides additional time for multiple rounds of repair. This results in coordinated and timely repair of SSBs prior to DNA replication, thus supporting the stability of the genome (left panel). In the absence of ATM (A-T), the detection of SSBs is abrogated, leading to replication of damaged DNA followed by the formation of DSBs and accumulation of mutations (right panel).
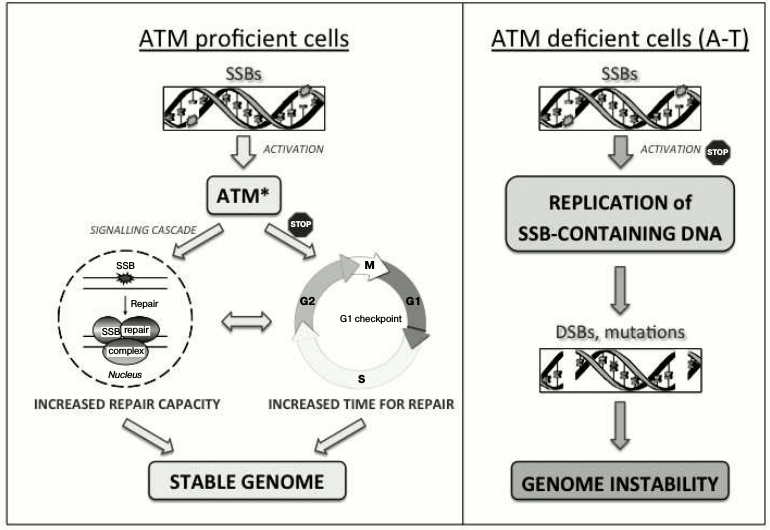
A role for ATM in SSB signaling is consistent with the sensitivity of A-T cells to DNA-damaging agents that cause DNA base damage and SSB formation [46, 47]. However, the mechanism of ATM activation in response to SSBs – its dependence on the presence of MRN complex components or the oxidation of disulfide bonds, or the existence of an independent mechanism – is presently unknown. It also remains unclear whether SSB-dependent ATM activation contributes to ATM activation in response to ionizing radiation, a treatment that induces a significant number of SSBs and DNA base damage in addition to DSBs [48].
Covalent DNA topoisomerase I–DNA adducts. DNA topoisomerase I (Top1) catalyzes the relaxation of DNA supercoiling that is produced during DNA replication and transcription. The mechanism of relaxation involves formation of a Top1–DNA intermediate (Top1cc), cleavage of one DNA strand of DNA, followed by ligation [49]. Top1 inhibitors, such as camptothecin, stabilize Top1cc adducts and thus prevent DNA ligation, inducing transcription defects [50]. ATM is activated in response to treatment of quiescent (nonreplicating) human cells and post-mitotic mouse cortical neurons with camptothecin [51]. SSBs with a covalent link between the 3′-phosphoryl end and a tyrosine residue of the Top1 active site peptide have been suggested as possible inducers of ATM activity. Such unconventional SSBs form during partial proteasomal degradation of Top1 within camptothecin-stabilized Top1cc adducts. Consequently, the accumulation of Top1cc adducts and ATM autophosphorylation can be rescued upon inhibition of transcription and proteasomal degradation using DRB (5,6-dichloro-1-β-D-ribofuranosylbenzimidazole) and MG-132 proteasome, respectively. The formation of DSBs that could contribute to ATM activation was excluded using DNA Comet assays.
The mechanism of activation and signaling for activation will require further investigation, but it is possible that activation results from local changes in chromatin structure at sites of arrested transcription complexes [51]. It is also possible that ATM activation involves DNA single-strand breaks with a 3′-modification. Interestingly, endogenous Top1cc adducts aberrantly accumulate in the brain cells of Atm–/– mice and in human A-T cells [52]. This effect is independent of ATM kinase activity, and it is related to abrogated proteasomal degradation of Top1 adducts in the absence of ATM.
R-loops. More recently, R-loops, or RNA–DNA heteroduplexes [53], that form upon inhibition of Top1 activity during transcription [54], have been implicated in ATM activation. However, it remains controversial whether R-loops can activate ATM directly.
ATM is activated in non-replicating cells (primary lymphocytes, rat cortical neurons, and synchronized human primary fibroblasts) treated with camptothecin [55, 56]. However, in contrast to some observations [51], DSBs were detected by the accumulation of γH2AX and 53BP1 foci, which serve as DSB markers, and by neutral Comet assays. Both ATM activation and DSB formation were rescued upon inhibition of transcription or expression of RNase H1 that cleaves RNA within R-loops, indicating an indirect role for R-loops in this activation. It has been proposed that transcription-blocking Top1cc lesions lead to R-loop formation, which are then processed into DSBs that activate ATM [55, 57]. The mechanism by which R-loops can be converted into DSBs is unknown. However, it is thought that the transcription-coupled nucleotide excision repair endonucleases XPG and XPF-ERCC1 can cleave both DNA strands during resolution of R-loops, resulting in replication-independent DSBs [58]. Alternatively, SSBs with partially cleaved Top1 peptide at the 3′-end might act as precursors of replication-independent DSBs. These DSBs could result from a proximal SSB formed by repair of another Top1cc adduct, an endogenous DNA lesion, or an R-loop (cleavage of a single DNA strand during classical transcription-coupled nucleotide excision repair) [56]. It therefore appears that R-loops can mediate ATM activation indirectly, through the formation of replication-independent DSBs.
More recently, R-loops were proposed to be the primary inducers of ATM activity, which regulates alternative pre-mRNA splicing [59]. R-loop formation and ATM activation have been observed in UV-treated nonreplicating human skin fibroblasts, and ATM activation can be rescued by the treatment of cells with inhibitors of transcription elongation and overexpression of RNase H1. Importantly and in contrast to [55, 56], ATM activation occurs in the absence of DSBs, as determined by the absence of γH2AX and 53BP1 focus formation. The role for R-loops in ATM activation and its mechanism require further investigation [59].
Structural changes. Changes in chromatin structure. DSBs are known to initiate significant changes in the structure of chromatin (see [60] for review), and it is possible that chromatin remodeling is a direct inducer of ATM activation in response to ionizing radiation [3]. Indeed, the importance of chromatin remodeling has been demonstrated by the activation of ATM in the absence of DSBs. This was shown by treatment of human cells in hypotonic solution with chloroquine, using inhibitors of histone deacetylases that promote chromatin decompaction and a siRNA-mediated knockdown of the heterochromatin protein 1α [3, 61]. MRN-independent activation of ATM under hypotonic stress conditions was found to be dependent on the interaction of ATM with the ATMIN protein, whereas ATM activation in response to irradiation is ATMIN-independent [62]. Further details of a role for chromatin remodeling in ATM activation is discussed elsewhere [63].
Mechanical stress. The activity of ATR kinase, a close homolog of ATM, is canonically induced in the presence of nucleofilaments of single-stranded DNA and the replicative protein A induced in response to mechanical stress [64]. The localization of ATR to the nuclear envelope, together with the induction of its kinase activity, was observed in human cells under hypertonic conditions that induce osmotic shock and mechanical (membrane) stress in the absence of DSBs. Similar results were obtained upon mechanical stretching of cells and on cell compression, within the physiological range of mechanical forces, using a compressive-load system. The mechanism of ATR activation in response to mechanical stress requires further investigation, although it appears to be distinct from the canonical mode of activation. The activation of ATR is important for regulation of the plasticity of the nuclear envelope and the association of chromatin with the nuclear envelope [64]. It remains to be established whether mechanical stress plays a role in ATM activation.
Activation of ATM in the absence of DSB sensors. Two independent protein complexes are important for the detection of DSBs in eukaryotes [65]; DSB recognition by the MRN-complex results in the activation of ATM, whereas the Ku complex (Ku70–Ku80) promotes the induction of DNA-PKcs activity, leading to the repair of DSBs via nonhomologous end-joining (NHEJ) [66]. In recent elegant work [67], activated DNA-PKcs was shown to functionally substitute for ATM in the absence of MRN in mouse embryonic fibroblasts, whereas MRN-dependent ATM activation was observed in the absence of Ku. Unexpectedly, ATM-dependent phosphorylation of histone H2AX, as well as a G2M cell cycle delay in response to ionizing radiation, were observed in cells deficient in both DSB sensors. The mechanism of such MRN-independent ATM activation remains unclear [67].
ETIOLOGY OF A-T
The clinical symptoms of A-T that relate to the functions of ATM in the coordination of the cellular response to DSBs include immunodeficiency, sensitivity to ionizing radiation (radiosensitivity) and other DNA damaging agents, and an increased risk of tumorigenesis [68, 69]. In addition, A-T patients present with progressive neurodegeneration, including atrophy of the spinal cord, cerebellum, and brain stem, coupled with the loss of Purkinje cells, as well as granular neurons and cells of the molecular layer [70-72], and ataxia.
The neurodegenerative phenotype of A-T is likely to be multifactorial in accordance with the variety of cellular functions of ATM, and the reasons for neurodegeneration are yet to be established. Our progress in understanding the molecular basis underlying A-T has been limited due to the absence of a good animal model for the disease – the progressive neurodegeneration phenotype that is observed in Atm–/– mice is rather mild compared to that in humans [73-75]. Interestingly, this mild phenotype is partly rescued using antioxidants, indicating a role for oxidative stress [76, 77]. Increased levels of oxidative stress are also detected in A-T patient cell lines [78, 79]. It is worth noting that no increase in the levels of R-loops has been observed in brain tissues of Atm–/– mice, rejecting a role for RNA–DNA heteroduplexes in the etiology of the disease [80]. The neurodegenerative pathology of A-T can also be linked to defects in the elimination of cells containing unrepaired DSBs during development of the neural system [81]. Moreover, the novel role for ATM in coordination of the repair of SSBs indicates a possible contribution of transcription-inhibiting SSBs to the etiology of ataxia telangiectasia.
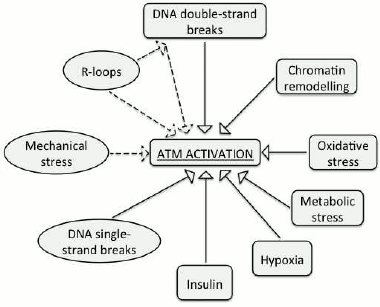
The experimental data demonstrate that ATM can be activated by a few non-canonical inducers (i.e. distinct from DNA double-strand breaks). These include DNA single-strand breaks, RNA–DNA hybrids, and changes in chromatin structure (Fig. 2). However, despite our progress in understanding functions of the ATM kinase, it is yet to be established, whether there exists a universal inducer of ATM activation, such as, for example, DNA lesions or chromatin remodeling. In addition, whether ATM gets activated in response to various discrete types of stress and the mechanisms of its activation are yet to be studied.
Fig. 2. Inducers of ATM activity. Non-canonical activators that are discussed in this review are designated with ovals.
REFERENCES
1.Lavin, M. F., Scott, S., Gueven, N., Kozlov, S.,
Peng, C., and Chen, P. (2004) Functional consequences of sequence
alterations in the ATM gene, DNA Rep. (Amst.), 3,
1197-1205.
2.Falck, J., Coates, J., and Jackson, S. P. (2005)
Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA
damage, Nature, 434, 605-611.
3.Bakkenist, C. J., and Kastan, M. B. (2003) DNA
damage activates ATM through intermolecular autophosphorylation and
dimer dissociation, Nature, 421, 499-506.
4.Sun, Y., Jiang, X., Chen, S., Fernandes, N., and
Price, B. D. (2005) A role for the Tip60 histone acetyltransferase in
the acetylation and activation of ATM, Proc. Natl. Acad. Sci.
USA, 102, 13182-13187.
5.Sun, Y., Xu, Y., Roy, K., and Price, B. D. (2007)
DNA damage-induced acetylation of lysine 3016 of ATM activates ATM
kinase activity, Mol. Cell. Biol., 27, 8502-8509.
6.Kozlov, S., Gueven, N., Keating, K., Ramsay, J.,
and Lavin, M. F. (2003) ATP activates ataxia-telangiectasia mutated
(ATM) in vitro. Importance of autophosphorylation, J. Biol.
Chem., 278, 9309-9317.
7.Kozlov, S. V., Graham, M. E., Peng, C., Chen, P.,
Robinson, P. J., and Lavin, M. F. (2006) Involvement of novel
autophosphorylation sites in ATM activation, EMBO J., 25,
3504-3514.
8.Kozlov, S. V., Graham, M. E., Jakob, B., Tobias,
F., Kijas, A. W., Tanuji, M., Chen, P., Robinson, P. J.,
Taucher-Scholz, G., Suzuki, K., So, S., Chen, D., and Lavin, M. F.
(2011) Autophosphorylation and ATM activation: additional sites add to
the complexity, J. Biol. Chem., 286, 9107-9119.
9.Pellegrini, M., Celeste, A., Difilippantonio, S.,
Guo, R., Wang, W., Feigenbaum, L., and Nussenzweig, A. (2006)
Autophosphorylation at serine 1987 is dispensable for murine ATM
activation in vivo, Nature, 443, 222-225.
10.Daniel, J. A., Pellegrini, M., Lee, J. H., Paull,
T. T., Feigenbaum, L., and Nussenzweig, A. (2008) Multiple
autophosphorylation sites are dispensable for murine ATM activation
in vivo, J. Cell Biol., 183, 777-783.
11.Uziel, T., Lerenthal, Y., Moyal, L., Andegeko,
Y., Mittelman, L., and Shiloh, Y. (2003) Requirement of the MRN complex
for ATM activation by DNA damage, EMBO J., 22,
5612-5621.
12.Carson, C. T., Schwartz, R. A., Stracker, T. H.,
Lilley, C. E., Lee, D. V., and Weitzman, M. D. (2003) The Mre11 complex
is required for ATM activation and the G2/M checkpoint, EMBO J.,
22, 6610-6620.
13.Sun, Y., Jiang, X., Xu, Y., Ayrapetov, M. K.,
Moreau, L. A., Whetstine, J. R., and Price, B. D. (2009) Histone H3
methylation links DNA damage detection to activation of the tumour
suppressor Tip60, Nat. Cell Biol., 11, 1376-1382.
14.Deshpande, R. A., Williams, G. J., Limbo, O.,
Williams, R. S., Kuhnlein, J., Lee, J. H., Classen, S., Guenther, G.,
Russell, P., Tainer, J. A., and Paull, T. T. (2014) ATP-driven Rad50
conformations regulate DNA tethering, end resection, and ATM checkpoint
signaling, EMBO J., 33, 482-500.
15.Bhatti, S., Kozlov, S., Farooqi, A. A., Naqi, A.,
Lavin, M., and Khanna, K. K. (2011) ATM protein kinase: the linchpin of
cellular defenses to stress, Cell. Mol. Life Sci., 68,
2977-3006.
16.Shiloh, Y., and Ziv, Y. (2013) The ATM protein
kinase: regulating the cellular response to genotoxic stress, and more,
Nat. Rev. Mol. Cell Biol., 14, 197-210.
17.Paull, T. T. (2015) Mechanisms of ATM activation,
Annu. Rev. Biochem., 84, 711-738.
18.Guo, Z., Kozlov, S., Lavin, M. F., Person, M. D.,
and Paull, T. T. (2010) ATM activation by oxidative stress,
Science, 330, 517-521.
19.Yang, D. Q., and Kastan, M. B. (2000)
Participation of ATM in insulin signalling through phosphorylation of
eIF-4E-binding protein 1, Nat. Cell Biol., 2,
893-898.
20.Valentin-Vega, Y. A., Maclean, K. H.,
Tait-Mulder, J., Milasta, S., Steeves, M., Dorsey, F. C., Cleveland, J.
L., Green, D. R., and Kastan, M. B. (2012) Mitochondrial dysfunction in
ataxia-telangiectasia, Blood, 119, 1490-1500.
21.Zhang, J., Tripathi, D. N., Jing, J., Alexander,
A., Kim, J., Powell, R. T., Dere, R., Tait-Mulder, J., Lee, J. H.,
Paull, T. T., Pandita, R. K., Charaka, V. K., Pandita, T. K., Kastan,
M. B., and Walker, C. L. (2015) ATM functions at the peroxisome to
induce pexophagy in response to ROS, Nat. Cell Biol., 17,
1259-1269.
22.Kozlov, S. V., Waardenberg, A. J.,
Engholm-Keller, K., Arthur, J. W., Graham, M. E., and Lavin, M. F.
(2015) ROS-activated ATM-dependent phosphorylation of cytoplasmic
substrates identified by large scale phosphoproteomics screen, Mol.
Cell. Proteomics, 15, 1032-1047.
23.McKinnon, P. J. (2012) ATM and the molecular
pathogenesis of ataxia telangiectasia, Annu. Rev. Pathol.,
7, 303-321.
24.Di Domenico, E. G., Romano, E., Del Porto, P.,
and Ascenzioni, F. (2014) Multifunctional role of ATM/Tel1 kinase in
genome stability: from the DNA damage response to telomere maintenance,
Biomed. Res. Int., 2014, 787404.
25.Shiloh, Y. (2014) ATM: expanding roles as a chief
guardian of genome stability, Exp. Cell Res., 329,
154-161.
26.Lavin, M. F., Kozlov, S., Gatei, M., and Kijas,
A. W. (2015) ATM-dependent phosphorylation of all three members of the
MRN complex: from sensor to adaptor, Biomolecules, 5,
2877-2902.
27.Syllaba, L., and Henner, K. (1926) Contribution a
le tude de l’inde pendance de l’athe tose double
idiopathique et conge nitale. Atteinte familiale, syndrome
dystrophique, signe du re sau vasculaire conjonctival, inte grite
psychique, Rev. Neurol. (Paris), 1, 541-560.
28.Louis-Bar, D. (1941) Sur un syndrome progressif
cormprenant des telangiectasies capillaires cutanees et conjonctivales
symetriques, a disposition naevoide et des troubles cerebelleux,
Confin. Neurol., 4, 32-42.
29.Boder, E., and Sedgwick, R. P. (1958)
Ataxia-telangiectasia; a familial syndrome of progressive cerebellar
ataxia, oculocutaneous telangiectasia and frequent pulmonary infection,
Pediatrics, 21, 526-554.
30.Su, Y., and Swift, M. (2000) Mortality rates
among carriers of ataxia-telangiectasia mutant alleles, Ann. Intern.
Med., 133, 770-778.
31.Swift, M., Morrell, D., Cromartie, E.,
Chamberlin, A. R., Skolnick, M. H., and Bishop, D. T. (1986) The
incidence and gene frequency of ataxia-telangiectasia in the United
States, Am. J. Hum. Genet., 39, 573-583.
32.Teive, H. A., Moro, A., Moscovich, M., Arruda, W.
O., Munhoz, R. P., Raskin, S., and Ashizawa, T. (2015)
Ataxia-telangiectasia – a historical review and a proposal for a
new designation: ATM syndrome, J. Neurol. Sci., 355,
3-6.
33.Lindahl, T. (1993) Instability and decay of the
primary structure of DNA, Nature, 362, 709-715.
34.Dianov, G., Price, A., and Lindahl, T. (1992)
Generation of single-nucleotide repair patches following excision of
uracil residues from DNA, Mol. Cell. Biol., 12,
1605-1612.
35.Kuzminov, A. (2001) Single-strand interruptions
in replicating chromosomes cause double-strand breaks, Proc. Natl.
Acad. Sci. USA, 98, 8241-8246.
36.Zhou, W., and Doetsch, P. W. (1993) Effects of
abasic sites and DNA single-strand breaks on prokaryotic RNA
polymerases, Proc. Natl. Acad. Sci. USA, 90,
6601-6605.
37.Kathe, S. D., Shen, G. P., and Wallace, S. S.
(2004) Single-stranded breaks in DNA but not oxidative DNA base damages
block transcriptional elongation by RNA polymerase II in HeLa cell
nuclear extracts, J. Biol. Chem., 279, 18511-18520.
38.Date, H., Onodera, O., Tanaka, H., Iwabuchi, K.,
Uekawa, K., Igarashi, S., Koike, R., Hiroi, T., Yuasa, T., Awaya, Y.,
Sakai, T., Takahashi, T., Nagatomo, H., Sekijima, Y., Kawachi, I.,
Takiyama, Y., Nishizawa, M., Fukuhara, N., Saito, K., Sugano, S., and
Tsuji, S. (2001) Early-onset ataxia with ocular motor apraxia and
hypoalbuminemia is caused by mutations in a new HIT superfamily gene,
Nat. Genet., 29, 184-188.
39.Moreira, M. C., Barbot, C., Tachi, N., Kozuka,
N., Uchida, E., Gibson, T., Mendonca, P., Costa, M., Barros, J.,
Yanagisawa, T., Watanabe, M., Ikeda, Y., Aoki, M., Nagata, T.,
Coutinho, P., Sequeiros, J., and Koenig, M. (2001) The gene mutated in
ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein
aprataxin, Nat. Genet., 29, 189-193.
40.Takashima, H., Boerkoel, C. F., John, J., Saifi,
G. M., Salih, M. A., Armstrong, D., Mao, Y., Quiocho, F. A., Roa, B.
B., Nakagawa, M., Stockton, D. W., and Lupski, J. R. (2002) Mutation of
TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in
spinocerebellar ataxia with axonal neuropathy, Nat. Genet.,
32, 267-272.
41.Shen, J., Gilmore, E. C., Marshall, C. A.,
Haddadin, M., Reynolds, J. J., Eyaid, W., Bodell, A., Barry, B.,
Gleason, D., Allen, K., Ganesh, V. S., Chang, B. S., Grix, A., Hill, R.
S., Topcu, M., Caldecott, K. W., Barkovich, A. J., and Walsh, C. A.
(2010) Mutations in PNKP cause microcephaly, seizures and defects in
DNA repair, Nat. Genet., 42, 245-249.
42.Markkanen, E., Fischer, R., Ledentcova, M.,
Kessler, B. M., and Dianov, G. L. (2015) Cells deficient in
base-excision repair reveal cancer hallmarks originating from
adjustments to genetic instability, Nucleic Acids Res.,
43, 3667-3679.
43.Khoronenkova, S. V., and Dianov, G. L. (2015) ATM
prevents DSB formation by coordinating SSB repair and cell cycle
progression, Proc. Natl. Acad. Sci. USA, 112,
3997-4002.
44.Khoronenkova, S. V., Dianova, I. I., Ternette,
N., Kessler, B. M., Parsons, J. L., and Dianov, G. L. (2012)
ATM-dependent downregulation of USP7/HAUSP by PPM1G activates p53
response to DNA damage, Mol. Cell, 45, 801-813.
45.Khoronenkova, S. V., and Dianov, G. L. (2013)
USP7S-dependent inactivation of Mule regulates DNA damage signalling
and repair, Nucleic Acids Res., 41, 1750-1756.
46.Hoar, D. I., and Sargent, P. (1976) Chemical
mutagen hypersensitivity in ataxia telangiectasia, Nature,
261, 590-592.
47.Yi, M., Rosin, M. P., and Anderson, C. K. (1990)
Response of fibroblast cultures from ataxia-telangiectasia patients to
oxidative stress, Cancer Lett., 54, 43-50.
48.Roots, R., Kraft, G., and Gosschalk, E. (1985)
The formation of radiation-induced DNA breaks: the ratio of
double-strand breaks to single-strand breaks, Int. J. Radiat. Oncol.
Biol. Phys., 11, 259-265.
49.Champoux, J. J., and Dulbecco, R. (1972) An
activity from mammalian cells that untwists superhelical DNA – a
possible swivel for DNA replication (polyoma-ethidium
bromide-mouse-embryo cells-dye binding assay), Proc. Natl. Acad.
Sci. USA, 69, 143-146.
50.Hsiang, Y. H., Hertzberg, R., Hecht, S., and Liu,
L. F. (1985) Camptothecin induces protein-linked DNA breaks via
mammalian DNA topoisomerase I, J. Biol. Chem., 260,
14873-14878.
51.Lin, C. P., Ban, Y., Lyu, Y. L., Desai, S. D.,
and Liu, L. F. (2008) A ubiquitin-proteasome pathway for the repair of
topoisomerase I-DNA covalent complexes, J. Biol. Chem.,
283, 21074-21083.
52.Katyal, S., Lee, Y., Nitiss, K. C., Downing, S.
M., Li, Y., Shimada, M., Zhao, J., Russell, H. R., Petrini, J. H.,
Nitiss, J. L., and McKinnon, P. J. (2014) Aberrant topoisomerase-1 DNA
lesions are pathogenic in neurodegenerative genome instability
syndromes, Nat. Neurosci., 17, 813-821.
53.Huertas, P., and Aguilera, A. (2003)
Cotranscriptionally formed DNA:RNA hybrids mediate transcription
elongation impairment and transcription-associated recombination,
Mol. Cell, 12, 711-721.
54.Tuduri, S., Crabbe, L., Conti, C., Tourriere, H.,
Holtgreve-Grez, H., Jauch, A., Pantesco, V., De Vos, J., Thomas, A.,
Theillet, C., Pommier, Y., Tazi, J., Coquelle, A., and Pasero, P.
(2009) Topoisomerase I suppresses genomic instability by preventing
interference between replication and transcription, Nat. Cell
Biol., 11, 1315-1324.
55.Sordet, O., Redon, C. E., Guirouilh-Barbat, J.,
Smith, S., Solier, S., Douarre, C., Conti, C., Nakamura, A. J., Das, B.
B., Nicolas, E., Kohn, K. W., Bonner, W. M., and Pommier, Y. (2009)
Ataxia telangiectasia mutated activation by transcription- and
topoisomerase I-induced DNA double-strand breaks, EMBO Rep.,
10, 887-893.
56.Cristini, A., Park, J. H., Capranico, G., Legube,
G., Favre, G., and Sordet, O. (2015) DNA-PK triggers histone
ubiquitination and signaling in response to DNA double-strand breaks
produced during the repair of transcription-blocking topoisomerase I
lesions, Nucleic Acids Res., 44, 1161-1178.
57.Sordet, O., Nakamura, A. J., Redon, C. E., and
Pommier, Y. (2010) DNA double-strand breaks and ATM activation by
transcription-blocking DNA lesions, Cell Cycle, 9,
274-278.
58.Sollier, J., Stork, C. T., Garcia-Rubio, M. L.,
Paulsen, R. D., Aguilera, A., and Cimprich, K. A. (2014)
Transcription-coupled nucleotide excision repair factors promote
R-loop-induced genome instability, Mol. Cell, 56,
777-785.
59.Tresini, M., Warmerdam, D. O., Kolovos, P.,
Snijder, L., Vrouwe, M. G., Demmers, J. A., van IJcken, W. F.,
Grosveld, F. G., Medema, R. H., Hoeijmakers, J. H., Mullenders, L. H.,
Vermeulen, W., and Marteijn, J. A. (2015) The core spliceosome as
target and effector of non-canonical ATM signalling, Nature,
523, 53-58.
60.Price, B. D., and D’Andrea, A. D. (2013)
Chromatin remodeling at DNA double-strand breaks, Cell,
152, 1344-1354.
61.Kaidi, A., and Jackson, S. P. (2013) KAT5
tyrosine phosphorylation couples chromatin sensing to ATM signalling,
Nature, 498, 70-74.
62.Kanu, N., and Behrens, A. (2007) ATMIN defines an
NBS1-independent pathway of ATM signalling, EMBO J., 26,
2933-2941.
63.Bakkenist, C. J., and Kastan, M. B. (2015)
Chromatin perturbations during the DNA damage response in higher
eukaryotes, DNA Rep. (Amst.), 36, 8-12.
64.Kumar, A., Mazzanti, M., Mistrik, M., Kosar, M.,
Beznoussenko, G. V., Mironov, A. A., Garre, M., Parazzoli, D.,
Shivashankar, G. V., Scita, G., Bartek, J., and Foiani, M. (2014) ATR
mediates a checkpoint at the nuclear envelope in response to mechanical
stress, Cell, 158, 633-646.
65.Goodarzi, A. A., and Jeggo, P. A. (2013) The
repair and signaling responses to DNA double-strand breaks, Adv.
Genet., 82, 1-45.
66.Taccioli, G. E., Gottlieb, T. M., Blunt, T.,
Priestley, A., Demengeot, J., Mizuta, R., Lehmann, A. R., Alt, F. W.,
Jackson, S. P., and Jeggo, P. A. (1994) Ku80: product of the XRCC5 gene
and its role in DNA repair and V(D)J recombination, Science,
265, 1442-1445.
67.Hartlerode, A. J., Morgan, M. J., Wu, Y., Buis,
J., and Ferguson, D. O. (2015) Recruitment and activation of the ATM
kinase in the absence of DNA-damage sensors, Nat. Struct. Mol.
Biol., 22, 736-743.
68.Epstein, W. L., Fudenberg, H. H., Reed, W. B.,
Boder, E., and Sedgwick, R. P. (1966) Immunologic studies in
ataxia-telangiectasia. I. Delayed hypersensitivity and serum immune
globulin levels in probands and first-degree relatives, Int. Arch.
Allergy Appl. Immunol., 30, 15-29.
69.Boder, E., and Sedgwick, R. P. (1970)
Ataxia-telangiectasia (clinical and immunological aspects),
Psychiatr. Neurol. Med. Psychol. Beih., 13-14, 8-16.
70.Aguilar, M. J., Kamoshita, S., Landing, B. H.,
Boder, E., and Sedgwick, R. P. (1968) Pathological observations in
ataxia-telangiectasia. A report of five cases, J. Neuropathol. Exp.
Neurol., 27, 659-676.
71.Paula-Barbosa, M. M., Ruela, C., Tavares, M. A.,
Pontes, C., Saraiva, A., and Cruz, C. (1983) Cerebellar cortex
ultrastructure in ataxia-telangiectasia, Ann. Neurol.,
13, 297-302.
72.Vinters, H. V., Gatti, R. A., and Rakic, P.
(1985) Sequence of cellular events in cerebellar ontogeny relevant to
expression of neuronal abnormalities in ataxia-telangiectasia, Kroc
Found. Ser., 19, 233-255.
73.Barlow, C., Hirotsune, S., Paylor, R., Liyanage,
M., Eckhaus, M., Collins, F., Shiloh, Y., Crawley, J. N., Ried, T.,
Tagle, D., and Wynshaw-Boris, A. (1996) ATM-deficient mice: a paradigm
of ataxia telangiectasia, Cell, 86, 159-171.
74.Barlow, C., Ribaut-Barassin, C., Zwingman, T. A.,
Pope, A. J., Brown, K. D., Owens, J. W., Larson, D., Harrington, E. A.,
Haeberle, A. M., Mariani, J., Eckhaus, M., Herrup, K., Bailly, Y., and
Wynshaw-Boris, A. (2000) ATM is a cytoplasmic protein in mouse brain
required to prevent lysosomal accumulation, Proc. Natl. Acad. Sci.
USA, 97, 871-876.
75.Borghesani, P. R., Alt, F. W., Bottaro, A.,
Davidson, L., Aksoy, S., Rathbun, G. A., Roberts, T. M., Swat, W.,
Segal, R. A., and Gu, Y. (2000) Abnormal development of Purkinje cells
and lymphocytes in ATM mutant mice, Proc. Natl. Acad. Sci. USA,
97, 3336-3341.
76.Chen, P., Peng, C., Luff, J., Spring, K.,
Watters, D., Bottle, S., Furuya, S., and Lavin, M. F. (2003) Oxidative
stress is responsible for deficient survival and dendritogenesis in
purkinje neurons from ataxia-telangiectasia mutated mutant mice, J.
Neurosci., 23, 11453-11460.
77.Reliene, R., and Schiestl, R. H. (2007)
Antioxidants suppress lymphoma and increase longevity in ATM-deficient
mice, J. Nutr., 137, 229S-232S.
78.Rybczynska, M., Pawlak, A. L., Sikorska, E., and
Ignatowicz, R. (1996) Ataxia telangiectasia heterozygotes and patients
display increased fluidity and decrease in contents of sulfhydryl
groups in red blood cell membranes, Biochim. Biophys. Acta,
1302, 231-235.
79.Reichenbach, J., Schubert, R., Schindler, D.,
Muller, K., Bohles, H., and Zielen, S. (2002) Elevated oxidative stress
in patients with ataxia telangiectasia, Antioxid. Redox Signal.,
4, 465-469.
80.Yeo, A. J., Becherel, O. J., Luff, J. E., Cullen,
J. K., Wongsurawat, T., Jenjaroenpun, P., Kuznetsov, V. A., McKinnon,
P. J., and Lavin, M. F. (2014) R-loops in proliferating cells but not
in the brain: implications for AOA2 and other autosomal recessive
ataxias, PLoS One, 9, e90219.
81.Orii, K. E., Lee, Y., Kondo, N., and McKinnon, P.
J. (2006) Selective utilization of nonhomologous end-joining and
homologous recombination DNA repair pathways during nervous system
development, Proc. Natl. Acad. Sci. USA, 103,
10017-10022.