REVIEW: Investigation of Ribosomes Using Molecular Dynamics Simulation Methods
G. I. Makarov1,2, T. M. Makarova1,2, N. V. Sumbatyan1,2, and A. A. Bogdanov1,2*
1Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; E-mail: bogdanov@belozersky.msu.ru2Lomonosov Moscow State University, Chemistry Department, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received August 5, 2016
The ribosome as a complex molecular machine undergoes significant conformational changes while synthesizing a protein molecule. Molecular dynamics simulations have been used as complementary approaches to X-ray crystallography and cryoelectron microscopy, as well as biochemical methods, to answer many questions that modern structural methods leave unsolved. In this review, we demonstrate that all-atom modeling of ribosome molecular dynamics is particularly useful in describing the process of tRNA translocation, atomic details of behavior of nascent peptides, antibiotics, and other small molecules in the ribosomal tunnel, and the putative mechanism of allosteric signal transmission to functional sites of the ribosome.
KEY WORDS: ribosome, molecular dynamics simulations, allosteric signal transmissionDOI: 10.1134/S0006297916130010
Abbreviations: MD, molecular dynamics; NPET, nascent peptide exit tunnel; PTC, peptidyl transferase center of the ribosome; REMD, replica exchange molecular dynamics.
The ribosome is a molecular machine that synthesizes all cellular
proteins according to the program encoded in nucleotide sequences of
mRNAs. In recent years, ribosome structure research has yielded
spectacular progress. Due to improvement in methods of X-ray structure
analysis and cryoelectron microscopy in application to this huge
RNA–protein complex, all-atom structures of not only bacterial
ribosomes, but also eukaryotic mitochondrial and cytoplasmic ones have
become accessible (for references, see [1-3]). The resulting structures represent in detail most
of the functional states of the ribosome with its ligands (tRNA, mRNA,
elongation factors, nascent peptide, translation inhibitors) at
different stages of translation. One of the most important inferences
that has arisen from the multitude of structure data analyses is that
the ribosome acquires a particular conformation at every stage of
polypeptide chain synthesis. As generally assumed, and in certain cases
convincingly proved by biochemical and genetic methods, functional
centers of the ribosome exchange allosteric signals [4, 5] during its activity,
providing tight coordination between the stages of translation.
However, the mechanism of such signal transmission and other dynamic
aspects of ribosome function remain unclear. Moreover, it is impossible
or extremely complicated to answer these questions by means of modern
structure methods as they describe only certain discrete frozen states
of the ribosome. To overcome this, computational methods, especially
molecular dynamics (MD) simulation, are employed [6].
MD simulation can be used to observe temporally and at atomic resolution the evolution of conformation and relative arrangement of amino acids and nucleotide residues, to evaluate the power of monomer interactions, probability of hydrogen bond formation between them, and to display their interactions with water, ions and low molecular weight ligands. In this review, the physical basis and technical details of various MD simulation methods are not discussed, (they are discussed in [7, 8]), and only new data on the ribosome and its function obtained by these methods (and, apparently, only obtainable in this way) are presented.
MD simulation became broadly applied to huge and complicated objects like the ribosome rather recently. However, three directions of this research can be distinguished: MD simulation of the ribosome as a molecular machine, nascent peptide exit tunnel (NPET) investigation, and study of the molecular mechanisms of binding of ligands, especially ribosomal antibiotics.
MD SIMULATION OF RIBOSOMES AS MOLECULAR MACHINES
Research in this area is mainly devoted to modeling of aa-tRNA recognition, peptidyl transferase center (PTC) motions, and the translocation process.
Modeling of aa-tRNA recognition. As established in Ramakrishnan’s classical experiments, universally conserved residues A1492 and A1493 in 16S rRNA (according to Escherichia coli numeration, here and further) that outloop from helix h44 and, in the case of cognate (completely Watson–Crick paired) codon–anticodon complex, incorporate into minor groove of RNA stem, that additionally stabilizes the complex [9]. It is obviously a dynamical process, so it is not surprising that it attracted the attention of MD researchers. The energy profile of the process was rather minutely studied by Zeng et al. [10]. Using umbrella sampling and the thermodynamic integration method [11], they assessed the free energy barrier of the outlooping of these residues as 7 kcal/mol. Cognate tRNA binding gains 9 kcal/mol of free energy, which is exceedingly enough to ensure spontaneous outlooping: the general energetic effect of it becomes 2 kcal/mol. Noncognate tRNA binding can provide only 6 kcal/mol energetic advantage, which that does not make up energy consumption for outlooping of A1492 and A1493. Besides, exploiting classical MD simulation, they confirmed that the aminoglycoside antibiotic paromycin stabilizes the outlooped conformation of residues A1492 and A1493 and provides energetic advantage irrespective of cognation of aa-tRNA bound to the decoding center. Nevertheless, in the case of a cognate codon–anticodon pair, the energetic advantage is more than in the case of the noncognate one.
Another issue, namely how tRNA modifications participate in distinguishing AUA (the Ile-codon) from AUG (the Met-codon), was recently revealed by Satpati et al. [12]. The fact of the matter is that in bacteria and archaea one of tRNAIle carries the CAU-anticodon, which could be recognized by the methionine codon if there were no modification of C34, in the third anticodon position, by lyzidine for bacteria and agmatidine for archaea. Employing the free energy perturbation method, it was established that in the case of replacement of an AUA codon by AUG, unmodified C34 provides 3 kcal/mol free energy advantage for tRNA binding, while agmatidine-modified C34 requires 7 kcal/mol of energy loss for this. The loss is nearly equal for the amino- and imino-forms of modified cytidine. In sum, binding of tRNAIle with agmatidine-modified C34 to incorrect methionine AUG codon appears to be 10 kcal/mol less advantageous than cognate binding to tRNAMet, which maintains the necessary selectivity.
The same authors [13] examined a question, why keto–enol tautomerization of nucleobases does not entail decoding errors, and they gained a convincing answer. Free energies of interactions between tRNAPhe carrying various anticodons: correct ones, anticodons obtained by replacement of A by G in the first position (tRNALeu) or in the second position (tRNASer), and anticodons where A is substituted by the enol tautomer of G – with mRNA UUU-codon, native, or with first or second uridine in enol form (especially for anticodons with keto form of G) were calculated by applying the free energy perturbation (FEP) method in 51 discrete windows and compared with each other. Replacement of A with the keto form of G in the first position provides 5 kcal/mol Gibbs free energy loss, just as replacement with the enol form in G (the value is 6 kcal/mol with calculating error 1 kcal/mol). However, the same replacement of the enol form of G in the second position appeared to be substantially more advantageous, while it decreases energy loss (12.8 kcal/mol) by 4 kcal/mol. Enolization of U in the first, and, particularly, in the second position of the codon entails even larger binding energy loss.
Modeling of a peptidyl transferase center. Works devoted to conformation mobility and the functional role of PTC conserved residues are usually complex studies where MD simulations are carried out with selective chemical modifications or site-directed mutagenesis of these residues. Therefore, in studying the process of in vitro translation by ribosomes containing 23S RNA residues in the PTC where nucleobases or their defined functional groups were eliminated, Polacek et al. [14] determined that disruption of noncanonical base pair A2450–C2063 suppresses poly(Phe) biosynthesis. To explain the experimental results, MD simulations of the PTC area with the pair mentioned above distorted via substitution of A2450 by purine or elimination of nucleobases from A2450 or C2063 were performed. According to analysis of the simulation results, disruption of the pair A2450–C2063 by any means leads to dramatic decrease in A2062 conformational mobility, which is extremely high in the wild-type ribosome, and to perturbation of its interaction with the 3′-terminus A-tRNA amino acid residue due to transition of A2062 into a rather fixed conformation hindering such interaction. From a technical point of view, that article is an example of a simulations of a restricted fragment of the ribosome instead of exploiting the whole ribosome: only the radial 3.2 nm area around the concerned conserved residues was selected for calculation; besides, residues more than 2 nm distant from the center of the simulated system were fixed. The advantage of this approach is economy of computational resources.
Gonsales et al. [15] applied MD to simulate an elongation complex containing P-tRNA carrying a D-amino acid. According to the simulation results, the amino group of L-amino acids attached to A-tRNA appeared to be more remote from carboxyl group of the D-amino acid attached to P-tRNA than from the same one in the case of the L-amino acid; in this arrangement, nucleophilic attack of the amino group for the peptidyl transfer reaction is hampered, which explains extreme suppression of translation in the presence of a D-amino acid carrying tRNA that was detected in the same work by biochemical methods.
A successful example of how the ribosome may serve as an object for development of coordinated biopolymer residue movement investigation is an article of Pande et al. [16]. In this report, the 70S Thermus thermophilus 70S ribosome carrying or not carrying tRNA and mRNA was investigated; coherence of the movement of the residue was estimated by calculation of mutual information for the distance between the actual position of the atom and a moving average of its coordinates employing K-mean clustering. Thereby, independence of A-site and P-site 23S rRNA residue movement was revealed. Moreover, the A-site residues of a platform attaching to the 30S subunit and residues of stalks move independently from the other 50S subunit.
Reports where MD simulations revealed in detail alterations in PTC structure caused by binding of various antibiotics are described below.
Translocation modeling. During translocation, tightly coordinated movement of large ligands – mRNA and tRNA – over the ribosome surface proceeds. This is an extremely complicated process. It includes many transition states accompany by formation of multiple intermediates, which are not always possible to describe using present structure methods. Therefore, MD simulation of this stage of elongation cycle is of a particular interest, and it has yielded some unconventional results.
It is known that all three basic tRNA-binding sites on the ribosome (A, P, and E) are divided into two parts: one is situated on the small ribosomal subunit, the second is on the large subunit (for the bacterial ribosome they are the 30S and 50S subunits, respectively). When tRNA completely occupies one of these sites, this state is termed A/A, P/P, or E/E, where first letter refers to the small subunit, and the second to the large subunit. Nevertheless, transition of tRNA from one site to another proceeds through so-called “hybrid” states, which are designated as A/P or P/E. Moreover, when aminoacyl-tRNA is in complex with elongation factor Tu and the ribosome, it occupies a peculiar conformation termed the A/T state.
Sanbonmatsu et al. elaborated an ingenious method for transition state modeling with the example of “hybrid” tRNA–ribosome complexes [17]. The essence of the method is the following. The authors relied on X-ray structures of ribosome complexes with two tRNAs in P/P, A/A and E/E, P/P states containing also elongation factor G (EF-G). These structures correspond to basic energy minima of the investigated system. On the other hand, cryoelectron microscopy with separate image analysis yielded electron density maps for transition state analogs of ribosome complexes with EF-G and tRNAs in A/P*-P/E and A/P-P/E states (in the A/P* state, the CCA-terminus of tRNA does not attain its canonical position). The three-dimensional structure of the ribosome complex with tRNAs and EF-G was fitted into this map. Then energy minimization of the ribosome complex with EF-G and tRNAs in P/P, A/A states with a special form Hamiltonian was performed. During the minimization, the complex “slides down” towards the structure corresponding the transition state. In this way, ribosome structures with hybrid A/P*-P/E and A/P-P/E states of tRNAs in the presence of EF-G were obtained.
Transition of deacylated tRNA from the P to the E-site, or, more accurately, from the P/P to the hybrid P/E state was simulated in [18]. Summarizing data from 120 trajectories, it was established that hybrid state formation began from ratcheting of the small subunit with respect to the large subunit by 5° counterclockwise, which results in formation of a stretched conformation of tRNA in the P/P state. The energy aggregated in this state is consumed at next stage of the transition. Next, the tRNA shoulder moves towards configuration corresponding to the hybrid state without any steric clashes, and only then the 3′-CCA terminus of tRNA leaves the P-site of the large subunit and is incorporated into the E-site interacting with h74 residues of 23S rRNA during its movement. MD simulation methods employed here also should be mentioned. For transition simulation, so-called targeted MD, a kind of steered MD, was applied, where a collective variable represents the remoteness of the current conformation from the conformation the system should reach, so dragging potential drives the system to a predetermined conformation. In addition, the simulation was carried out using so-called structure-based models [19] that include into the potential energy contribution so-called atom contacts, existing in an experimentally determined structure, instead of usual noncovalent interactions (in this research, the contact was taken into account if its atoms were situated at a distance less than 4 Å in the experimental structure and did not belong to neighboring residues). Force constants of all covalent interactions were defined in a way that is unconventional for MD. The model implies that the experimental structure corresponds to a global energetic minimum for the biopolymer, so even a completely unfolded chain is expected to move rapidly towards this conformation upon MD simulation. Two such the models were constructed: A/A-P/P complex for one and P/P-E/E for the other, and the total model was defined as their weighted sum, thus ascribing to the system two “global” energetic minima between which the transition occurred.
Translocation kinetics was investigated earlier by Sanbonmatsu’s group [20]. To pursue this goal, a record large calculation (1.2 µs) of the 70S E. coli ribosome carrying two tRNAs in A/A-P/P states was performed. Three generalized coordinates describing the translocation process were defined: θbody, describing rotation of the small subunit relative to the large one; θhead, indicating diversion between the small and the large subunits; rtRNA, representing the distance between shoulders of the two tRNAs. For these coordinates, fluctuation diffusion coefficients and barrier crossing attempt frequencies were estimated; the latter were about 0.2 µs–1. According to the estimations, the energetic barriers separating the different translocation stages do not exceed 9-12 kcal/mol.
Bock et al. revealed a probable function of certain 50S proteins in the translocation process. The 70S E. coli ribosome was simulated starting from 17 structures obtained by adjustment of X-ray data to cryoelectron microscopy density maps. As a result of their simulation, kinetics of passage through 17 stages of translocation was evaluated, and coordinated movements of the tRNA and ribosomal subunit functional elements were observed. In particular, the L1-stalk was found to push the tRNA into the E-site [21].
MD simulations of quite recently found reverse translocation, or back-translocation, provided abundant knowledge about basic phases of the direct translocation [22]. Electron density of the complex containing 70S T. thermophilus 70S ribosome, two tRNAs in P/P-E/E states (POST state), mRNA, and EF-G was fitted into electron density of the similar complex containing 70S E. coli ribosome and tRNAs in the A/P*-P/E states. Free energy calculation along the reaction pathway was performed with the umbrella sampling method concurrently with the fitting. According to the results, the pre-translocation state is 2-3 kcal/mol higher on the free energy scale than the POST state, and the barrier overcome by the ribosome during back-translocation exceeds the POST state by 8 kcal/mol. In addition, clockwise ratcheting of the small subunit around the large one was detected (direct translocation implies counterclockwise ratcheting). At the same time, opening and closing of so-called “P/E gates” built by G1338 and A1339 on one side and A790 of 16S rRNA on another were discerned in trajectories. The “P/E gates” were widely opened in the pre-translocation state, but became shut in the following transitional and terminal POST configurations. Conformational changes and whole-object movement of EF-G were detected as well.
In work [23], interaction between the small and large subunits during translocation was investigated. For this purpose, 13 structures of translocation intermediates were obtained by fitting ribosome structures from X-ray analysis data containing tRNAs and mRNA to cryoelectron microscopy density maps, and for each of them 100-ns simulation was carried out. The total list of groups of 16S and 23S rRNA residues forming intersubunit contacts was compiled, and the enthalpy of these contacts was estimated relying on analysis of the simulation data. Seven of these intersubunit bridges were of evidently high and stable enthalpy values. During macromolecular intersubunit relative movement, the contacts are rearranged due to replacement of residues in the interaction, but not to conformational changes near the bridges. The contribution of the tRNAs and mRNA to the enthalpy of the intersubunit interaction was assessed as well; it revealed that tRNAs are preferably connected to the large subunit, while mRNA is known to be bound only with the small one, so the tRNAs–mRNA interaction makes a crucial contribution to tRNAs enthalpy share in 70S ribosome stabilization. Furthermore, the contribution of P-tRNA to the 70S ribosome stabilization is more than that of A-tRNA or E-tRNA.
INVESTIGATIONS OF PHYSICOCHEMICAL PROPERTIES OF THE RIBOSOMAL
TUNNEL
The MD simulation method provided peculiar insights into the properties of the ribosomal tunnel (NPET) that hardly could have emerged using experimentally methods. For example, Pande et al. [24] examined properties of the water inside the tunnel. The simulations were carried out in a rectangular fragment of the large ribosomal subunit containing the entire NPET filled with water. Potential of mean force, rotational entropy, diffusion coefficient, and dipole moment fluctuation tensor were calculated for water relying on the trajectory data. The diffusion coefficient appeared to be decreased, as the rotational entropy indicated sufficient ordering of the water molecules in the NPET (in comparison with extensive solvent state). The same indicates decrease in fluctuation tensor trace as much as it is more proximate to the border of the tunnel. In general, only at about 0.5 nm distance from the border water properties approximate those in the unhindered volume.
The same research group performed simulation of amino acid side chain interactions with the NPET walls [25]. Like in the article described above, only a parallelepiped fragment covering the NPET was included in the simulated system. The inner volume of the tunnel was covered by a grid with 0.1 nm step in whose nodes probe molecules imitating Ala, Ile, Lys, Asn, and Trp side chains were posited, while backbone atoms were substituted by hydrogen atoms to preclude their contribution to interaction energy. The probes were fixed in grid nodes by a harmonic potential with high force constant and were situated such that distances between them exceeded cutoff radius of noncovalent interactions. The potential mean force maps were calculated from the trajectories. The calculation revealed an energetic barrier of about 6 kcal/mol on exit from the tunnel for residues with bulky side groups (Ile, Lys, Trp), while for residues with short side chains (Ala, Asn) the barrier was only 1 kcal/mol. For apparently hydrophobic residues (Ile, Trp), mean force potential is evenly distributed along the axis of the tunnel and has comparatively higher value, thus impelling the residues to move out of the tunnel. The energy landscape for charged residues (Lys, Asn) has more complicated form dependent on the charge: the maximum for Lys is situated at the minimum for Asn. Furthermore, analysis of the trajectories revealed existence of peculiar “gates” built by the A497 23S rRNA and Arg residues of L39 protein at the exit from the tunnel, which can block the bottom region of the tunnel.
The passage of the nascent peptide through the tunnel was investigated in [26]. The poly(Ala) chain was chosen as a model. The advancement of the nascent chain was modeled by introduction of a force driving the chain towards the exit. Multiple trajectory data displayed the passage of the polypeptide chain under Arg92 of L22 protein, which coupled with L4 occupies sufficient space in the tunnel.
An attempt to simulate cotranslational “Zn-finger” domain folding inside the NPET was made by Nilson et al. [27]. It is known that α-helical regions can fold in the bottom region of the tunnel. In the mentioned work, independent cryoelectron microscopy data and MD simulation verified tertiary structure elements in the zone of the tunnel. Folding of peptides containing the SecM stop peptide fragment and the “Zn-finger” domain, separated by amino acid sequences of different lengths, was simulated by applying replica exchange MD (REMD) with the so-called “coarse-grained” model.
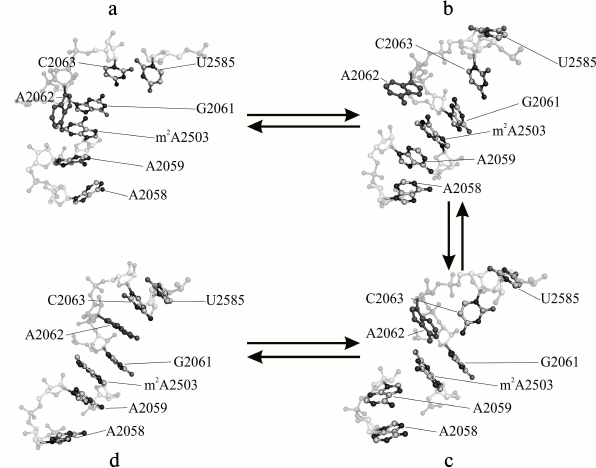
One of the most evident tasks for MD applied to study of the ribosome is development of approaches for understanding allosteric signal transmission in the NPET. The authors of this review [28] undertook MD simulation study of a core region of the large E. coli ribosomal subunit covering all the PTC and the NPET. Selection of such the fragment was governed by well-established fact of the participation of the A2062 residue (occupying the central position in this fragment) in allosteric signal transition to the PTC from a remote area of the NPET precluded by A2062 mutations [29]. For this system, trajectories from 200 to 360 ns length were obtained, and their analysis revealed conformational transitions in which some PTC residues along with a set of 2058-2063 23S rRNA (NPET region) residues were involved. These transitions are depicted in the figure. In some trajectories, they occurred in a singular manner, in others together, and in several others they built a united conformational cascade. It begins from approaching of A2058 and A2059 at 60 ns. Thus, the block of residues A2058-A2059-m2A2503-G2061 that existed before the transition and a newly formed A2062-C2063-U2585 block combine into united stacking-like structure. This transition appeared to be reversible. Coulomb and Van-der-Waals energy analysis between neighboring residues detected an energy barrier that the system overcomes between 80 and 160 ns during conformational rearrangements corresponding to the state where nucleobases G2061 and C2063 are already separated, but A2062 and U2585 had not yet formed with them stabilized stacking interaction.
Regarding the functional meaning of the observed conformational change, a hypothesis about the probability of its terminal conformation to be stabilized by any regulatory ribosome ligand causing translational arrest was suggested [28].
Major stages of a conformational transition in the A2058-C2063 region of 23S rRNA of the E. coli ribosome observed in MD simulation: a) initial (4 ns); b, c) intermediate (71 and 117 ns, respectively); d) terminal (353 ns) stages
MODELING OF INTERACTION BETWEEN RIBOSOMES AND ANTIBIOTICS
A special branch of the study of ribosomes by means of MD simulation is investigation of ribosomal antibiotic binding. A typical example of it is served by an article of Alexandrov and Simonson where tetracycline interaction with the small ribosomal subunit was investigated [30]. According to X-ray data, tetracycline has at least two binding sites on the small subunit: TET1 and TET5. MD simulation was carried out in spherical fragments of the small subunit with 2.6-nm radius with the tetracycline binding sites in its centers. The authors gained free energy difference between binding to each of these sites. With this value and experimentally obtained associating constant, occupancy of each site was calculated, which appeared to be almost 100% for TET1 and about 57% for TET5. Thus, the site having the highest affinity to tetracycline was identified.
It was formerly accepted that the antibiotic gentamycin, binding to the small subunit near the A site, fixes the A1492 and A1493 residues in outlooped conformation, which entails selection of inconsistent with codon cognate aa-tRNA and multiple mistakes in the synthesizing protein sequence. However, Vaiana and Sanbonmatsu demonstrated via molecular dynamics [31] that conformational rearrangement is induced not by the binding of the antibiotic itself. On the contrary, residues A1492 and A1493 spontaneously outloop and revert to duplex state, while gentamycin, being able to bind not so selectively in the proximity of its binding site, gradually wedges into the duplex, first replacing one of its bases and maintaining its outlooped conformation, then fixing the second one. The application of the REMD simulation method is to be particularly mentioned.
An article of Trylska et al. [32] is also dedicated to aminoglycoside antibiotics, where mechanisms of resistance to paromomycin were investigated at the molecular level. The peculiarity of the research consists in modeling antibiotic interaction not with the whole subunit, but with the 16S rRNA fragment rendering the helix h44, to which paromycin is bound in the whole ribosome. Mutations G1491A, G1491U, U1495C, and U1406C were inserted into the modeled rRNA. MD simulation in the presence and absence of paromycin provided evident observation of the way in which alterations in A1492 and A1493 flexibility destabilizes base pairing and decreases antibiotic binding.
The binding of the aminoglycoside antibiotic paromomycin to the small subunit of the bacterial ribosome and influencing the binding of K42A and R53A mutations in S12 protein mediated by conformational changes in A1492 and A1493 residues of A-site was studied in [33]. These mutations are known to result in hyper-accurate recognition of the aa-tRNA in the A site of the 30S subunit, suppressing the effect of the aminoglycoside antibiotic consisting, on the contrary, in decreasing the ribosome selectivity to aminoacyl-tRNA. Relying on conformational analysis data, the authors suggested that decreased frequency of outlooping of the residues led to reduced small subunit affinity to the incoming aminoacyl-tRNA, which increases the contribution of its firm interaction with mRNA. They suggested that the reason for the changes in A1492 and A1493 conformational dynamics is underlain in rearrangement of hydrogen bond network between 16S rRNA and S12 protein, induced by replacement of Arg or Lys with hydrogen bond donor side chain by Ala, whose side group does not possess this property.
MD simulations of complexes of antibiotics with the large subunit of the bacterial ribosome were exploited in several studies. For example, Wolf et al. examined the mode of action of thiostrepton on GTPase associated center (GAC) of the large ribosomal subunit consisting of L11 protein and helices 43-44 of 23S rRNA by means of MD simulation and circular dichroism [34]. Via treatment of data of the resulting trajectories by main component analysis, clusterization in the space of the main components and correlation analysis, it was established that thiostrepton suppresses intrinsic flexibility of protein domains, making them more rigid, and switches the conformation of the A1067 residue, stabilizing its anti-form. A1067 is in turn indispensable for tRNA and elongation factor recognition. A drawback of this investigation was the inclusion of only L11 protein and h43-44 (residues from 1051 to 1108) without any restraints into the simulated system. It is doubtful whether the system constructed in this manner can reproduce the spatial structure of the 23S rRNA during simulation, as well as to maintain the correct structure of the 23S rRNA complex with L11 protein; also it increases computational time.
The PTC and NPET of the ribosome are widely known to be the most common targets of antibiotic attack. For example, in [35] the Gibbs free energy of sparcomycin binding to the large ribosomal subunit was estimated by the application of the free energy perturbation (FEP) method. In this calculation, not the whole ribosome, but its spherical fragment with 1.5-nm radius with the large subunit P site (which is the sparsomycin-binding site) in the center, was involved. Nevertheless, the value obtained in the simulation is in line with experimental data.
At first sight, Yam and Wahab [36] determined more detailed data during MD simulations of erythromycin and roxithromycin binding to the ribosome. However, their values vary from the experimental ones. The reason for this divergence is that the authors calculated only 2.5-ns length trajectories, of which only 0.5 ns belongs to the equilibrated state of the system according to mean standard deviation diagrams. Moreover, the selected simulation method explicitly involves solvent properties, which are, in consistence with [24], demonstrated to be crucially diverged from those in unhindered volume. The conclusion about an overwhelming influence of hydrophobic interactions is doubtful for the same reason: it is unclear how water entropy is biased in the NPET upon the antibiotic binding.
The same questions emerge upon acquaintance with data obtained by Saini et al. [37], who simulated binding of linezolid and radezolid (belonging to the oxazolidinone antibiotic family) to Haloarcula marismortui and Deinococcus radiodurans ribosomes [37]. In other words, the quoted papers indicate that MD simulation study of antibiotic–ribosome interactions is still at the beginning stage of development.
Nevertheless, interesting results are expected if MD simulation would be applied to investigation of the impact of macrolide antibiotics on PTC function to unravel the mechanism of translation arrest by regulatory stop-peptides from the Erm family. In recent years, reliable data demonstrating the allosteric character of macrolide action upon the PTC were obtained [38]. Indeed, MD simulation of ribosomal complexes with erythromycin, being not directly in contact with the PTC region, displayed alteration of conformation of A2602 and U2585 residues from the PTC: these residues had propensity to form stacks with neighboring residues in a chain instead of shifting towards the NPET lumen [38]. However, no supposition about the concrete mechanism of signal transmission from the macrolide binding site (MBS) to the PTC was suggested at this stage of research.
A striking breakthrough was made in this field only recently. Mankin et al. accomplished MD simulation of a ribosome complex with the macrolide erythromycin or the ketolide telithromycin in the presence of the regulatory ErmBL stalling peptide interacting with the NPET [39]. The purpose was to endeavor to find a structural explanation for a dependence of ErmBL specificity to these antibiotics on the stalling peptide sequence, which is observed in biochemical and microbiological experiments. For example, ErmBL with Asp in the 10th position (Asp10) causes translational arrest in the presence of either erythromycin or telithromycin, while ErmBL with Glu in same position arrests translation involving erythromycin only. It should be noted that in both cases the 10th residue is on the C-terminus of the peptide bound to the 3′-terminus residue of a P site tRNA, and translational arrest occurs due to inability of the last amino acid to react with Lys11 carried on the 3′-terminus (A76) of the A site tRNA. The simulation demonstrated that in the presence of native (Asp10) peptide, the occurrence of Lys11 orientation available for nucleophilic attack of the carboxyl group of the P-site-attached peptide (and, thus, new peptide bond formation) is reduced for both erythromycin and telithromycin binding. Replacement of Asp10 by Glu10 reveals such orientation only in the case of erythromycin, but not telithromycin; then, if Asp10 is substituted by Tyr, advantageous for the peptidyl transferase reaction mutual arrangement of the substrates occurred more rarely for telithromycin in comparison with erythromycin. The authors account this effect by γ-carboxylic Glu10 group hydrogen bond formation with 4′-hydroxyl of the erythromycin cladinose, which biases orientation distribution towards disadvantageous for peptidyl transferase reaction ones in the case of the Glu10 mutant. This interaction is impossible for telithromycin due to the absence of cladinose and in the case of erythromycin with native ErmBL, when the Asp10 side chain is sufficiently distant from the cladinose. Thus, applying MD simulation provided a rational explanation for an experimentally discovered fundamental property of the ribosome – the ability to respond to binding of different low molecular weight ligands to its functional sites in a discriminative way.
An important continuation and evolution of this research is a work by Arenz et al. [40] who did per se MD simulation of one of the same systems, but relying on a more detailed structure of the regulatory ErmBL peptide in the NPET obtained by high-resolution cryoelectron microscopy. They had detailed data about positions of ErmBL amino acid side groups relative to the NPET walls near the PTC both in the presence and in the absence of erythromycin. Several interesting events were noted. While in antibiotic-free system the peptide was arranged rather uninterestedly in the NPET, in the presence of erythromycin it appeared to be clasped to the wall of the tunnel adjacent to the P site of the PTC; it is noteworthy that no immediate contact between the peptide and the antibiotic was observed. Moreover, the arrangement of the G2505-U2506/C2452 23S rRNA residues from an opposite side of the tunnel was altered, so they moved away from the axis of the tunnel. Due to this, Lys11 interacting with the same area of 23S rRNA was shifted in the same direction. Moreover, displacement of the A76 P-tRNA residue and conformational change in Asp10 attached to it was detected. All these alterations were possible due to allosteric effects, and they entirely preclude an efficient peptidyl transferase reaction. However, in our opinion their interpretation of interactions of amino acid residues of aa-tRNA in the A site with the NPET walls was too simplistically regarded as being purely electrostatic. Indeed, mutations of Lys11 into Trp, Ile, and even Gly devoid of side group charge, just like the negatively charged Gln residue, caused translational arrest as frequently as the Lys11-peptide (Fig. 7g in [41]). It is noteworthy that MD simulation performed in this work provided abundant information about positions and stability of hydrogen bonds forming by ErmBL amino acid residues with the nucleobases residues of the NPET walls.
In the end of this part of the article, one emerging direction in MD study of ribosome–antibiotic complexes should be mentioned. It consists in detailed investigation of antibiotic resistance that occurs in ribosomes. This can be exemplified by a work of Small et al. [41], where the influence of N6-methylation of the A2058 23S rRNA residue and its mutation A2058G on ketolide telithromycin affinity was estimated. The N6-methylation and mutation to guanine of A2058 are known to provide macrolide resistance. However, telithromycin partially overcomes the A2058G mutation effect, but not if it is methylated. The simulation revealed that A2058G mutation preserved the opportunity for the 2′-hydroxyl of telithromycin to participate in hydrogen bond formation with the ribosome, compelling it, however, to accept the proton from N1H of guanine instead of being a donor. The hydrogen bond of this group with A2058 N1 is retained also if the residue is dimethylated. Monomethylation of A2058 disturbs this hydrogen bond, which is apparently caused by destruction of its stacking interaction with neighboring residues. Moreover, mutation A2058G maintains stacking interaction of the imidazole-pyridine fragment of telithromycin with Watson–Crick base pair A752–U2609, while methylation of A2058 destabilizes it. This is accounted for by disturbance of A2058 stacking interactions with neighboring G2057 and A2059 by its methylation, as noted above. This disturbance is transferred from G2057 to C2611 through the Watson–Crick base pair, then through covalent bonds to U2609. Owing to this, its hydrogen bonds to A752 are weakened, and its interaction with the telithromycin is perturbed. This is the reason for the destabilization of the ribosome–telithromycin complex by methylation of A2058. An interesting approach in the simulation is noteworthy: MD calculation for a selected fragment of the hydrated ribosome (10,000 steps) alternated with water molecule placement via the Monte-Carlo method with a large canonical ensemble (10,000 steps), that owing to more advantageous solvation enabled stabilization of new conformations further optimized by MD. This technique improved representability of the system states obtained from the simulation.
Mutation G2482A in the H. marismortui ribosome (G2447A by E. coli numeration) is known to provide resistance to chloramphenicol and tiamulin binding in the so-called hydrophobic pocket formed by A2486 and C2487 (A2451 and C2452 E. coli) in the PTC. However, G2482 does not form direct contacts with these antibiotics. From MD simulations of the H. marismortui ribosome with either the presence or the absence of the G2482A mutation and its data treatment by main component and cross-correlation analysis methods, the researchers inferred that antibiotic resistance is connected with enhancement of A2486 and C2487 (A2451 and C2452 E. coli) flexibility induced by perturbation of a noncovalent interaction network maintaining spatial structure of the 23S rRNA [42].
This review of articles implementing the MD simulation method for investigation of different functional aspects of the ribosome revealed that the method is successful and productive for the study of the ribosome. MD enables dealing with system states that are hard to detect experimentally: to estimate their energetic properties, investigate physicochemical characteristics of the ribosomal tunnel and ordering of water in it, and, what is of the most importance, to model complicated coordinated movement and conformational rearrangements occurring in the ribosome during its functioning or induced by binding of antibiotics. Such structural methods as X-ray structure analysis or, to less extend, cryoelectron microscopy, render only frozen static structures of certain states of the ribosome that not in every case reproduce a detailed atomic-level mechanism of the acting processes and dynamical properties of the required ribosomal fragments. Moreover, these structure methods represent properties of the ribosome ensembles that are not in rather native solution conditions (that is particularly true for the X-ray structure method), whereas MD simulation enables reproducing (of course, relying on available structural and other experimental data) the time evolution of the ribosomal processes in water solution with required ions. The data given in this review confirm that the simulation is rather reliable and reproduces the facts observed at the macroscopic level. Thus, application of MD simulation methods for study of structure and function of the ribosome and its complexes with various ligands is reasonable and successful, though it may require large consumption of processor time due to the large-scale simulation system.
Acknowledgements
The research was supported by the Russian Science Foundation (projects Nos. 14-24-00061, sections on investigations of physicochemical properties of the ribosomal tunnel and on modeling of interaction between the ribosome and antibiotics; 15-14-00006, sections on MD simulation of the ribosome as a molecular machine).
REFERENCES
1.Melnikov, S., Ben-Shem, A., Garreau de Loubresse,
N., Jenner, L., Yusupova, G., and Yusupov, M. (2012) One core, two
shells: bacterial and eukaryotic ribosomes, Nat. Struct. Mol.
Biol., 19, 560-567.
2.Amunts, A., Brown, A., Toots, J., Scheres, S. H.
W., and Ramakrishnan, V. (2015) The structure of human mitochondrial
ribosome, Science, 348, 95-98.
3.Frank, J. (2016) Whither ribosome structure and
dynamics research? (A perspective), J. Mol. Biol., 428,
3565-3569.
4.Wang, L., Pulk, A., Wasserman, M. R., Feldman, M.
B., Altman, R. B., Doudna, C. J. H., and Blanchard, S. C. (2012)
Allosteric control of the ribosome by small-molecule antibiotics,
Nat. Struct. Mol. Biol., 19, 957-963.
5.Fei, J., Bronson, J. E., Hofman, J. M., Srinivas,
R. L., Wiggins, C. H., and Gonzalez, R. L., Jr. (2009) Allosteric
collaboration between elongation factor G and the ribosomal L1 stalk
directs tRNA movements during translation, Proc. Natl. Acad. Sci.
USA, 106, 15702-15707.
6.Sonbonmatsu, K. Y. (2012) Computational studies of
molecular machines: ribosomes, Curr. Opin. Struct. Biol.,
22, 168-174.
7.Perilla, J. R., Goh, B. C., Cassidy, C. K., Liu,
B., Bernardi, R. C., Rudack, T., Yu, H., Wu, Z., and Schulten, K.
(2015) Molecular dynamics simulations of large macromolecular
complexes, Curr. Opin. Struct. Biol., 31, 64-74.
8.Hospital, A., Coni, J. R., Orzco, M., and Gelp, J.
L. (2015) Molecular dynamics simulations: advances and applications,
Adv. Appl. Bioinform. Chem., 8, 37-47.
9.Carter, A. P., Clemons, W. M. J., Brodersen, D. E.,
Morgan-Warren, R., Wimberly, B. T., and Ramakrishnan, V. (2000)
Functional insights from the structure of the 30S ribosomal subunit and
its interaction with antibiotics, Nature, 407,
340-348.
10.Zeng, X., Chugh, J., Casiano-Negroni, A.,
Al-Hashimi, H. M., and Brooks, C. L., 3rd. (2014) Flipping of the
ribosomal A-site adenines provides a basis for tRNA selection, J.
Mol. Biol., 426, 3201-3213.
11.Knight, J. L., and Brooks, C. L. (2009)
λ-Dynamics free energy simulation methods, J. Comput.
Chem., 30, 1692-1700.
12.Satpati, P., Bauer, P., and Aqvist, J. (2014)
Energetic tuning by tRNA modifications ensures correct decoding of
isoleucine and methionine on the ribosome, Chemistry, 20,
10271-10275.
13.Satpati, P., and Aqvist, J. (2014) Why base
tautomerization does not cause errors in mRNA decoding on the ribosome,
Nucleic Acids Res., 42, 12876-12884.
14.Chirkova, A., Erlacher, M., Clementi, N.,
Zywicki, M., Aigner, M., and Polacek, N. (2010) The role of the
universally conserved A2450-C2063 base pair in the ribosomal peptidyl
transferase center, Nucleic Acids Res., 38,
4844-4855.
15.Englander, M. T., Avins, J. L., Fleisher, R. C.,
Liu, B., Effraim, P. R., Wang, J., Schulten, K., Leyh, T. S., Gonzalez,
R. L., and Cornish, V. W. (2015) The ribosome can discriminate the
chirality of amino acids within its peptidyl-transferase center,
Proc. Natl. Acad. Sci. USA, 112, 6038-6043.
16.Brandman, R., Brandman, Y., and Pande, V. S.
(2012) A-site residues move independently from P-site residues in
all-atom molecular dynamics simulations of the 70S bacterial ribosome,
PLoS One, 7, e29377.
17.Whitford, P. C., Ahmed, A., Yu, Y., Hennely, S.
P., Tama, F., Spahn, C. M., Onuchic, J. N., and Sanbonmatsu, K. Y.
(2011) Excited states of ribosome translocation revealed through
integrative molecular modeling, Proc. Natl. Acad. Sci.
USA, 108, 18943-8948.
18.Whitford, P. C., and Sanbonmatsu, K. Y. (2013)
Simulating movement of tRNA through the ribosome during hybrid-state
formation, J. Chem. Phys., 139, 121919.
19.Noel, J. K., Whitford, P. C., Sanbonmatsu, K. Y.,
and Onuchic, J. N. (2010) SMOG@ctbp: simplified deployment of
structure-based models in GROMACS, Nucleic Acids Res.,
38, W657-W661.
20.Whitford, P. C., Blanchard, S. C., Cate, J. H.
D., and Sanbonmatsu, K. Y. (2013) Connecting the kinetics and energy
landscape of tRNA translocation on the ribosome, PLoS Comput.
Biol., 9, e1003003.
21.Bock, L. V., Blau, C., Schroder, G. F., Davydov,
I. I., Fischer, N., Stark, H., Rodnina, V., Vaiana, A. C., and
Grubmuller, H. (2013) Energy barriers and driving forces in tRNA
translocation through the ribosome, Nat. Struct. Mol. Biol.,
20, 1390-1396.
22.Ishida, H., and Matsumoto, A. (2014) Free-energy
landscape of reverse tRNA: translocation through the ribosome analyzed
by electron microscopy density maps and molecular dynamics simulations,
PLoS One, 9, e101951.
23.Bock, L. V., Blau, C., Vaiana, A. C., and
Grubmuller, H. (2015) Dynamic contact network between ribosomal
subunits enables rapid large-scale rotation during spontaneous
translocation, Nucleic Acids Res., 43, 6747-6760.
24.Lucent, D., Snow, C., Aitken, C., and Pande, V.
(2010) Non-bulk-like solvent behavior in the ribosome exit tunnel,
PLoS Comput. Biol., 6, e1000963.
25.Petrone, P., Snow, C., Lucent, D., and Pande, V.
(2008) Side-chain recognition and gating in the ribosome exit tunnel,
Proc. Natl. Acad. Sci. USA, 105, 16549-16554.
26.Ishida, H., and Hayward, S. (2008) Path of
nascent polypeptide in exit tunnel revealed by molecular dynamics
simulation of ribosome, Biophys. J., 95, 5962-5973.
27.Nilsson, O. B., Hedman, R., Marino, J., Wickles,
S., Bischoff, L., Johansson, M., Muller-Lucks, A., Trovato, F.,
Puglisi, J. D., O’Brien, E. P., Beckmann, R., and Von Heijne, G.
(2015) Cotranslational protein folding inside the ribosome exit tunnel,
Cell Rep., 12, 1533-1540.
28.Makarov, G. I., Golovin, A. V., Sumbatyan, N. V.,
and Bogdanov, A. A. (2015) Molecular dynamics investigation of a
mechanism of allosteric signal transmission in ribosomes,
Biochemistry (Moscow), 80, 1047-1056.
29.Vazquez-Laslop, N., Ramu, H., Klepacki, D.,
Kannan, K., and Mankin, A. S. (2010) The key function of a conserved
and modified rRNA residue in the ribosomal response to the nascent
peptide, EMBO J., 29, 3108-3117.
30.Alexandrov, A., and Simonson, T. (2008) Molecular
dynamics simulations of the 30S ribosomal subunit reveal a preferred
tetracycline binding site, J. Amer. Chem. Soc., 130,
1114-1115.
31.Vaiana, A., and Sanbonmatsu, K. (2009) Stochastic
gating and drug–ribosome interactions, J. Mol. Biol.,
386, 648-661.
32.Romanowska, J., McCammon, J., and Trylska, J.
(2011) Understanding the origins of bacterial resistance to
aminoglycosides through molecular dynamics mutational study of the
ribosomal A-site, PLoS Comput. Biol., 7, e1002099.
33.Panecka, J., Mura, C., and Trylska, J. (2014)
Interplay of the bacterial ribosomal A-site, s12 protein mutations and
paromomycin binding: a molecular dynamics study, PLoS One,
9, e111811.
34.Wolf, A., Baumann, S., Arndt, H. D., and
Kirschner, K. N. (2012) Influence of thiostrepton binding on the
ribosomal GTPase associated region characterized by molecular dynamics
simulation, Bioorg. Med. Chem., 20, 7194-7205.
35.Ge, X., and Roux, B. (2010) Calculation of the
standard binding free energy of sparsomycin to the ribosomal
peptidyl-transferase P-site using molecular dynamics simulations with
restraining potentials, J. Mol. Recogn., 23, 128-141.
36.Yam, W. K., and Wahab, H. A. (2009) Molecular
insights into 14-membered macrolides using the MM-PBSA method, J.
Chem. Inf. Model., 49, 1558-1567.
37.Saini, J., Homeyer, N., Fulle, S., and Gohlke, H.
(2013) Determinants of the species selectivity of oxazolidinone
antibiotics targeting the large ribosomal subunit, Biol. Chem.,
394, 1529-1541.
38.Sothiselvam, S., Liu, B., Han, W., Ramu, H.,
Klepacki, D., Atkinson, G. C., Brauer, A., Remm, M., Tenson, T.,
Schulten, K., Vazquez-Laslop, N., and Mankin, A. S. (2014) Macrolide
antibiotics allosterically predispose the ribosome for translation
arrest, Proc. Natl. Acad. Sci. USA, 111,
9804-9809.
39.Gupta, P., Liu, B., Klepacki, D., Gupta, V.,
Schulten, K., Mankin, A. S., and Vazquez-Laslop, N. (2016) Nascent
peptide assists the ribosome in recognizing chemically distinct small
molecules, Nat. Chem. Biol., 12, 153-158.
40.Arenz, S., Bock, L. V., Graf, M., Innis, C. A.,
Beckmann, R., Grubmüller, H., Vaiana, A. C., and Wilson, D. N.
(2016) A combined cryo-EM and molecular dynamics approach reveals the
mechanism of ErmBL-mediated translation arrest, Nat. Commun.,
7, 12026.
41.Small, M. C., Lopes, P., Andrade, R. B., and
MacKerell, A. D., Jr. (2013) Impact of ribosomal modification on the
binding of the antibiotic telithromycin using a combined grand
canonical Monte Carlo/molecular dynamics simulation approach, PLoS
Comput. Biol., 9, e1003113.
42.Wang, Y., Shen, J. K., and Schroeder, S. J.
(2012) Nucleotide dynamics at the A-site cleft in the
peptidyltransferase center of H. marismortui 50S ribosomal
subunits, J. Phys. Chem. Lett., 8, 1007-1010.