REVIEW: Tumor Necrosis Factor and Lymphotoxin in Regulation of Intestinal Inflammation
E. O. Gubernatorova1 and A. V. Tumanov1,2*
1Institute of Molecular Biology, Russian Academy of Sciences, 119991 Moscow, Russia; E-mail: tumanov@uthscsa.edu2University of Texas Health Science Center, Department of Microbiology, Immunology, and Molecular Genetics, San Antonio, TX 78229, USA
* To whom correspondence should be addressed.
Received July 3, 2016; Revision received August 16, 2016
Ulcerative colitis and Crohn’s disease are the major forms of inflammatory bowel disease. Cytokines of the tumor necrosis factor (TNF) family play an important role in the regulation of intestinal inflammation. In this review, we discuss the function of key cytokines of this family – TNF and lymphotoxin (LT) – in mucosal healing, IgA production, and in control of innate lymphoid cells (ILCs), novel regulators of mucosal homeostasis in the gut. TNF plays a central role in the pathogenesis of inflammatory bowel diseases (IBD). LT regulates group 3 of ILCs and IL-22 production and protects the epithelium against damage by chemicals and mucosal bacterial pathogens. In addition, we discuss major mouse models employed to study the mechanism of intestinal inflammation, their advantages and limitations, as well as application of TNF blockers in the therapy for IBD.
KEY WORDS: tumor necrosis factor, lymphotoxin, intestinal inflammation, inflammatory bowel disease, mouse modelsDOI: 10.1134/S0006297916110092
Abbreviations: CD, Crohn’s disease; DCs, dendritic cells; DSS, dextran sulfate sodium; FDA, Food and Drug Administration of USA; FDC, follicular dendritic cells; GALT, gut-associated lymphoid tissue; IBD, inflammatory bowel disease; IFN, interferon; IL, interleukin; ILCs, innate lymphoid cells; LIGHT, homologous to lymphotoxin, exhibits inducible expression and competes with HSV glycoprotein D for binding to herpesvirus entry mediator, a receptor expressed on T-lymphocytes (another name: tumor necrosis factor superfamily member 14, TNFSF14); LT, lymphotoxin; LTβR, membrane lymphotoxin receptor; MHC, major histocompatibility complex; MNP, mononuclear phagocytes; TACE, TNF-alpha converting enzyme; TNBS, 2,4,6-trinitrobenzenesulfonic acid; TNF, tumor necrosis factor; TNFR, tumor necrosis factor receptor; UC, ulcerative colitis.
CYTOKINES OF TNF SUPERFAMILY
Tumor Necrosis Factor (TNF), lymphotoxin (LT), and LIGHT (homologous to lymphotoxin, exhibits inducible expression and competes with HSV glycoprotein D for binding to herpesvirus entry mediator, a receptor expressed on T-lymphocytes, TNFSF14) are key cytokines of the TNF superfamily that perform various functions in mammalian biology. Most ligands and receptors of the TNF superfamily (over 40 ligand–receptor pairs) are expressed by immune system cells [1]. Some receptors belonging to this cytokine family, for instance, TNFR1, are able to activate mechanisms of cell death, whereas others (LTβR) predominantly promote cell survival and inflammation [2]. Most ligands of the TNF superfamily are membrane-associated trimeric molecules. However, upon activation of certain metalloproteinases, trimers may be converted into soluble form. For instance, metalloproteinase TACE (TNF-alpha converting enzyme) releases the extracellular TNF moiety [3]. Intracellular parts of receptors do not contain kinase domains and are unable to bind tyrosine kinase. Thus, signaling is realized by means of adapter molecules. Several receptors of the TNF family, such as TNFR1 and Fas, contain death domains within their cytoplasmic regions, which are able to activate and recruit caspase precursors and to induce apoptosis. Another feature of these molecules is their composition of three subunits that determines signaling stereochemistry. Thus, the listed unique structural attributes determine the connection between cytokines of the TNF superfamily and signal transduction pathways in cell proliferation, differentiation, and survival [1, 2]. This is why possible use of cytokines of the TNF superfamily and their inhibitors in therapy for various autoimmune diseases and cancer is actively studied in recent years [4].
The main ligand–receptor interactions in the TNF/LT system as well as producer and responding cells are presented in the Fig. 1. TNF and TNFR are widely produced. Membrane lymphotoxin is an LTβ2LTα1 heterotrimer that is predominantly located on lymphocytes. LTβR receptor for the membrane lymphotoxin, in turn, is expressed in epithelial and stromal cells, dendritic cells (DCs), and macrophages, but it is absent on lymphocytes [3, 5]. This expression pattern for LT and LTβR provides interaction of LT-producing lymphocytes – cells with stromal microenvironment – that is important for lymph node development and maintenance of lymphoid organ structure [3, 6]. Recent studies have demonstrated that LTα1β2 has two LTβR binding sites with different affinities, and LTβR dimerizes upon LTα1β2 binding. This dimerization is required for efficient signal transduction [7]. Soluble LTα3 homotrimer does not bind LTβR, but it interacts with TNFR1 and TNFR2, and, similarly to TNF, it is able to stimulate development of various inflammatory reactions. In contrast to TNFR1, which is constantly expressed in all cells, TNFR2 expression is more selective and inducible [8]. It is known that membrane TNF has higher affinity for TNFR2 compared to the soluble TNF [8]. Another TNF family ligand, LIGHT (TNFSF14), mainly expressed on T cells, macrophages, NK cells, and DCs, can also interact with LTβR [3]. In addition to LTβR, LIGHT can interact with herpes virus entry mediator (HVEM) receptor, which is also a ligand for receptors belonging to another cytokine family, CD160 and BTLA (B and T lymphocyte attenuator) [9]. This complex system of ligands and receptors in the TNF cytokine family provides selectivity of interaction and signal transduction between various immune system cells and implies involvement of compensatory mechanisms.
Fig. 1. Ligands and receptors of the TNF/LT family. The main ligand–receptor interactions and producer cells are indicated with arrows. The dashed line indicates action sites for metalloproteinases (MMP), whose activity converts membrane-bound ligand into a soluble form. DcR3 is a receptor-trap for LIGHT (found in some human tumors).
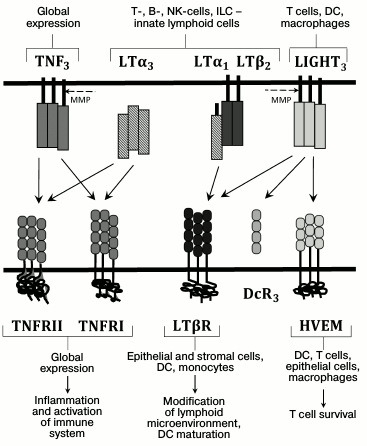
The best-studied function of LT and LTβR is their participation in lymphoid organ development, such as lymph nodes and Peyer’s patches, and in maintenance of proper spleen white pulp architecture [6, 10]. However, it was later shown that LT function is broader and not restricted to lymphoid organs and protection against infections. Thus, it was found that LT plays an important role in maintenance of lipid homeostasis and liver regeneration. It also regulates inflammation and commensal gut microbiota, proliferation, and tumor transformation [11-17]. In addition to maintenance of lymphoid organ structure, TNF also participates in defense against infections, regulation of inflammation, and autoimmune diseases [18-20]. Therefore, LT, TNF, and LIGHT form an integral signaling network that is required for efficient innate and adaptive immune responses. We will further review mechanisms of TNF and LT participation in regulation of intestinal homeostasis and describe mouse models widely used in studying the mechanisms of intestinal inflammation.
EXPERIMENTAL ANIMAL MODELS FOR INTESTINAL INFLAMMATION
The intestine is a sophisticated organ containing different cell types that contact the “outer” environment and are constantly interacting with commensal microbiota. Revealing mechanisms involved in intestinal inflammation and damage is an important step in developing therapeutic strategies and prevention of disease. It is beyond doubt that the most reliable data can only be obtained during analysis of samples from patients who suffer from various diseases. However, sampling size, ethical issues related to utilization of human samples, and genetic heterogeneity in the group significantly restrict human research trials. Therefore, studying pathogenesis of the gastrointestinal tract requires developing complex experimental models on animals that are devoid of such drawbacks as sampling limitation and genetic heterogeneity. At the same time, human diseases should be reproduced in these animals with maximum likeness [21-23]. Advantages in the genetic engineering field and well-studied genome make mice the most-used object in modeling immune processes, including those involved in intestinal inflammation [24]. Modeling disorders in mice gives a number of advantages, such as simplicity of genome and microbiome manipulations, and fast generation change. On the other hand, small size, different susceptibility to disease due to different genetic background, complexity of surgery, and high cost of keeping impose certain restrictions. Despite the fact that no animal model can perfectly reproduce a human disease, using mice to study mechanisms that regulate intestinal inflammation now seems optimal [21, 22, 25, 26].
Ulcerative colitis (UC) and Crohn’s disease (CD) are major inflammatory bowel diseases (IBD) in humans [27-31]. The number of patients with IBD grows in developed countries. Therefore, about three million people in Europe and two million in the USA suffer from these disorders [32, 33]. Despite different etiologies, these disorders are characterized by chronic intestinal inflammation, epithelial barrier damage, and misbalance in commensal microbiota, cytokines, and immune cells, which results in development of immunopathology [27, 34-36]. Like the majority of autoimmune diseases, IBD pathogenesis depends on genetic susceptibility and ambient conditions, which may include intestinal infections, diet, stress, and disruption of the balance of microbiota [37, 38].
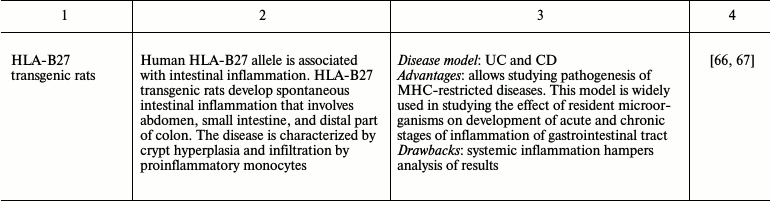
Many experimental mouse IBD models allow observing spontaneous development of chronic disease similar to that in humans. However, use of chemical and biological inducers of inflammation that provide fast development of reproducible reaction is also advantageous [21, 24, 39]. The diversity of chemical compounds and pathogenic microorganisms applied for induction of intestinal inflammation allows dissection of both acute and chronic inflammation [21, 39, 40]. One model is often insufficient for precise disease modeling. In this case, one has to utilize several mouse models of disease to see the whole picture. Thus, finding the most appropriate model for studying different aspects of intestinal inflammation is a key step in designing a research program. The most frequently used intestinal inflammation models along with their advantages and drawbacks are presented in Table 1.
Table 1. Modeling inflammatory bowel disease
in animals
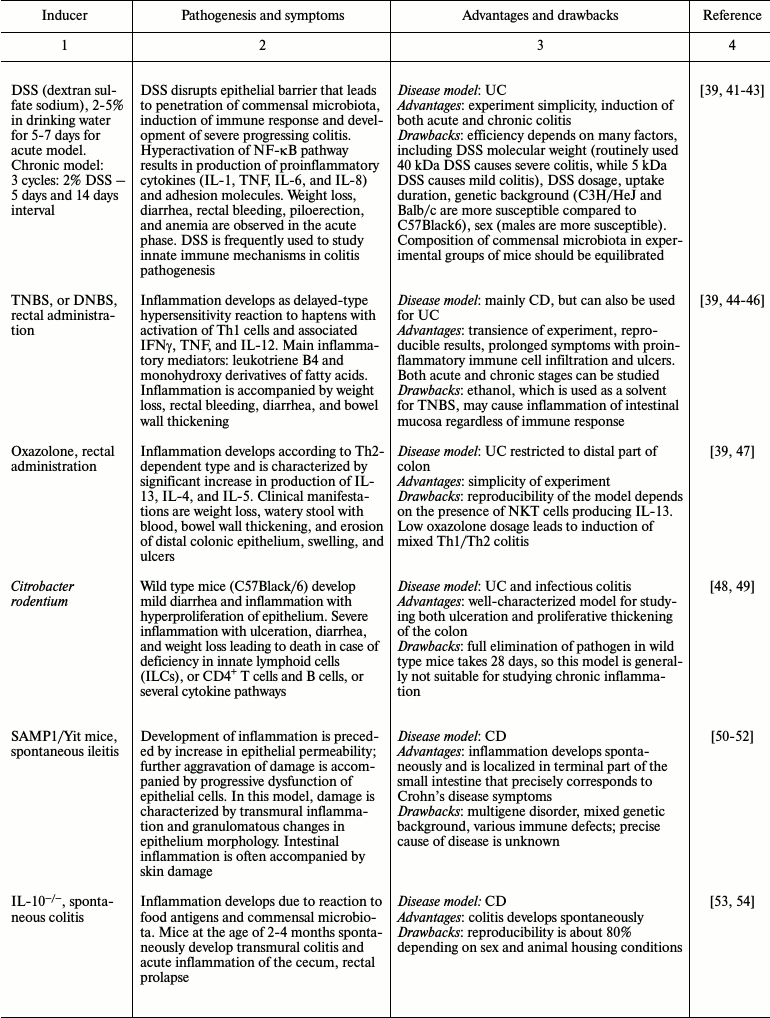

In experiments on animals, chemical inducers are used as fast, inexpensive, and efficient strategies for intestinal inflammation modeling. DSS (dextran sulfate sodium) and TNBS (2,4,6-trinitrobenzenesulfonic acid) are frequently used for inducing colitis in mice. Acetic acid and oxazolone are applied much less often. Efficiency of intestinal tissue damage induction depends on DSS molecular mass, concentration, manufacturer, and reagent batch [39, 42]. Moreover, damage severity varies depending on genetic background of mouse line, sex, age, and microbiota composition [43, 68, 69]. One must consider microbiota composition when comparing experimental animals with a control group since differences in this parameter may cause variations in mouse susceptibility to DSS-induced colitis. Hence, using littermate controls in the same cage is recommended. If this is impossible, microbiota composition is balanced before the experiment by means of bedding exchange during a week, as mice are prone to coprophagy. Despite the difficulties arising during work with chemically induced colitis models, such agents as DSS and TNBS are valuable tools for studying pathogenesis of the intestinal inflammation [21, 23].
As an alternative to chemical agents, biological inducers of inflammation such as bacteria (Escherichia coli, Citrobacter rodentium, etc.), viruses, and helminths are applied. Change in microbiota composition may result in inflammatory response. This is why use of biological inducers of inflammation is considered to be closer to natural physiological conditions.
Most genetically engineered mouse models are designed exclusively for research applications: finding functions of molecules via complete or conditional gene knockout, stimulation, or repression of intracellular signaling pathways, withdrawal of transcription factors, and immunocompetent molecules, cells, and whole organs [24]. Immunodeficient mice were also used in studying mechanisms of response to intestinal injury. Such animal models include immunodeficient SCID and Rag1–/– mice. These mice are subject to adoptive transfer of CD4+CD45RBhigh naïve T cells, which are activated by interaction with food antigens and commensal microorganisms and cause chronic transmural inflammation of the large and small intestine [57, 70]. Mouse models with spontaneous development of intestinal inflammation, such as IL-10–/– and SAMP1/Yit mice, most precisely reproduce Crohn’s disease, a chronic intestinal disease [52, 53, 71]. However, these models also have a number of drawbacks (Table 1). “Humanized” animals with introduced human genes maximally approach a model system to human disorder [72]. For instance, rats in which human HLA-B27 allele (associated with intestinal inflammation) is expressed are successfully used as a system for testing therapeutic agents [66]. Thus, genetic engineering in animals provides a universal platform for studying intestinal inflammation mechanisms.
INNATE LYMPHOID CELLS – NEW REGULATORS OF INTESTINAL
INFLAMMATION
Equilibrium between efficient immune response and pathological intestinal inflammation requires fine regulation of interactions between the intestinal epithelium and immune cells. Upon pathogen entry to intestinal mucosa, innate immune system cells act as the first line of defense. Innate lymphoid cells (ILCs) are a recently discovered family of the innate immune cells that play an important role in immune response and mucosal inflammation [73, 74]. ILCs are found in barrier tissues, where they participate in protection against pathogenic microorganisms and in damage repair. In contrast to classical lymphocytes, ILCs lack antigen-specific receptors [75-77]. Innate lymphoid cells can be divided into three groups based on variations in cytokines produced and transcription regulators involved [78-80]. The first group includes cells with the main transcription factors T-bet (ILC1) and eomesodermin (NK – natural killers); transcription factor GATA3 is important for ILC2s; innate lymphoid cells of the third group (ILC3) depend on transcription regulator RORγt [78]. Numerous studies published during the last few years have shown high polyfunctionality of ILCs. RORγt+ ILC3s are involved in regulation of intestinal inflammation, antifungal and antibacterial protection, participate in control of commensal microbiota, development and repair of mucosa-associated lymphoid tissues, and carcinogenesis [73, 81-87]. Secretion of proinflammatory cytokines by activated antigen-presenting cells in the intestine activates ILC3s for IL-17a, IL-22, and GM-CSF secretion. IL-22 secretion increases production of antimicrobial peptides by epithelial cells and production of mucins, which is important for retention of commensal microbiota and for protection from bacterial and viral pathogens [84, 88, 89]. At the same time, IL-12 produced by dendritic cells inhibits the activity of ILC3s and can result in their conversion into ILC1s, which can trigger intestinal inflammation by secretion of IFNγ [90-92]. Thus, there is a differentiation plasticity between ILC populations depending on instructive cytokines and balance between specific transcription factors [93-97]. ILC3s can also control the adaptive immune response. On one hand, ILC3s are able to boost T- and B-cell immune responses via expression of costimulatory molecules CD40L and OX40L [98, 99]. On the other hand, ILC3s induce elimination of autoreactive CD4+ T cells specific for commensal microbiota antigens by expressing MHCII and presenting antigen. This suppressor phenomenon has been called intestinal T cell selection, similarly to negative selection in the thymus [85].
Recent studies have shown that ILC3s are important regulators of the intestinal inflammation. For example, epithelial damage in the DSS-induced colitis model in mice results in activation of ILC3s and production of IL-22 [17, 82]. IL-22, in turn, interacts with IL-22R on the intestinal epithelial cell and stimulates cell proliferation and production of mucins and antimicrobial peptides [88]. Moreover, it was shown that production of IL-22 protects intestinal stem cells from damage caused by donor immune cells in GVHD (graft-versus-host disease) reaction [100, 101]. However, under certain circumstances, ILC3s are also able to promote intestinal inflammation. For instance, high IL-23 levels can trigger production of IL-17 and IFNγ by ILC3s [102-104]. Thus, both secretion of certain cytokines by ILCs and specific ILC subpopulations can either inhibit or promote intestinal inflammation depending on the tissue microenvironment.
Identification of ILCs as key regulators of intestinal inflammation provides new opportunities in designing therapeutic approaches for human diseases. So, during recent studies of RORγt inhibitors, a selective blocker of RORγt was found for Th17 cells that does not affect the function of RORγt+ ILC3s [105]. Moreover, it was suggested that low efficiency of a number of experimental therapeutic drugs for IBD is linked with their inability to target ILCs, important regulators of intestinal inflammation [106]. Thus, the design of specific stimulators and antagonists of ILC function for use in therapy for IBD seems to be a timely problem in this field.
LYMPHOTOXIN – A NEW REGULATOR OF ILC3s AND IL-22
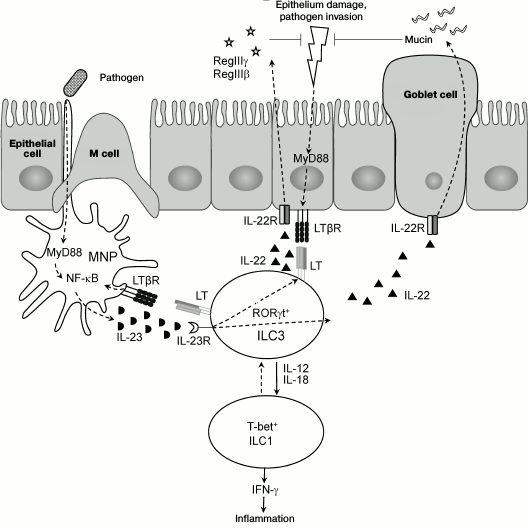
Recent studies revealed LT as a key regulator of ILC3s in the intestine [16, 81, 107], while LTβR not only mediates interaction between epithelial cells and immune cells, but also stimulates migration of neutrophils into sites of infection and promotes regeneration of epithelial cells via activation of IL-22 producing ILC3s [17, 81, 107]. These mechanisms are required for repair of damaged epithelium and for protection from bacterial intestinal pathogens. Therefore, LTβ- and LTβR-deficient mice develop severe intestinal inflammation upon C. rodentium infection and die during the first two weeks after infection [81, 108]. Use of mice bearing a specific deletion of the Ltb gene in RORγt expressing cells (RORγt-Ltb–/–) revealed a key role of membrane LT on ILC3s for providing communication between ILC3s and mucosal cells, and production of IL-22 and antimicrobial peptides for protection against C. rodentium [107]. Interestingly, LT regulates IL-22 secretion by ILC3s indirectly via activation of accessory cells, mononuclear phagocytes (MNP) [109, 110]. The intestinal mononuclear phagocyte system includes functionally connected cell populations of bone marrow precursor cells (dendritic cells, DC), blood monocytes, and tissue macrophages [109, 110]. DCs and macrophages are a major source of IL-23, which is required for induction of IL-22 expression by ILC3s. Upon interaction between ILC3s expressing membrane LT complex and MNP expressing LTβR, activation of IL-23 production occurs, which triggers expression of IL-22 by ILC3s. It is suggested that this positive feedback loop between ILC3s and DC is required for providing efficient protection against a pathogen [107]. The scheme of interactions of ILC3s, MNP, and epithelial cells upon damage of intestinal epithelium as well as control mechanisms with participation of LTβR is shown in Fig. 2.
Fig. 2. Proposed mechanism of interaction of ILC3, mononuclear phagocytes (MNP), and epithelium upon mucosal damage. Epithelium damage results in penetration of pathogenic microorganisms deep inside tissues and MyD88-mediated signaling, which activates expression of NF-κB-dependent genes. Production of IL-23 by activated MNP (mainly by dendritic cells) stimulates production of IL-22 by ILC3s. In response to IL-22, epithelial cells proliferate and express antimicrobial peptides RegIIIγ and RegIIIβ, while goblet cells extensively excrete mucin. The described feedback loop between MNP and ILC3s, as well as LTβR-mediated interaction with epithelial cells, controls activity of pathogen at early stages of infection. Production of IL-12 and IL-18 by dendritic cells can cause differentiation of RORγt+ ILC3 into pathogenic T-bet+ ILC1 IFNγ-producing cells, which can aggravate inflammation.
ROLE OF LT AND TNF IN IgA PRODUCTION IN INTESTINE AND IN
REGULATION OF COMMENSAL MICROBIOTA
The gastrointestinal tract is densely populated by a variable bacterial community comprising about 100 trillion microbes of 500-1000 species [111]. Symbiotic relationships are established between the host organism and commensal microbiota. This facilitates development of the immune system and maintenance of normal physiology [112]. Constant interaction between intestinal microbes and the host immune system provides stable immune response that includes tolerance to food antigens and commensal microorganisms, and efficient response to pathogens [113]. In the gut, the immune system is comprised of Peyer’s patches, mesenteric lymph nodes, isolated lymphoid follicles, and diffused lymphoid tissue associated with epithelium and lamina propria [10, 114]. Homeostasis between intestinal microbiota and host tissues is maintained by several mechanisms including secretion of IgA antibodies, which are able to block attachment of pathogens to the epithelium and neutralize toxins [115, 116]. Transformation of В cells localized in Peyer’s patches or in the lamina propria from IgM-producing cells into IgA-producing plasma cells may occur by T cell-dependent or T cell-independent mechanisms [116-119] shown in Fig. 3.
Fig. 3. Mechanisms of induction of IgA production in the intestine. a) T cell-dependent IgA induction in Peyer’s patches. The single-layer epithelium associated with underlying B cell follicle (follicle-associated epithelium) contains approximately 10% of specialized microfold cells (M cells), which are able to capture antigens from the intestinal lumen and pass them to migrating dendritic cells (CD103+ DC). Local nonmigrating macrophage-like CX3CR1+ DCs can also directly capture antigens from the intestinal lumen by their long projections. Further, CD103+ DCs migrate into interfollicular region of Peyer’s patches for contact interactions and activation of naïve CD4+ T cells. Then, CD4+ T cells differentiate into follicular helper T cells (TFH) under the action of retinoic acid (RA), TGFβ, IL-10, and thymic stromal lymphopoietin (TSLP) produced by epithelial cells and DCs. TFH interact with antigen-specific B cells at the T–B region boundary and then migrate into B cell follicles following the CXCL13 gradient produced by follicular dendritic cells (FDC). CD40L, IL-21, and TGFβ produced by TFH activate B cells and induce expression of activation-induced cytidine deaminase (AID), which is required for switching the immunoglobulin classes (CSR) to IgA. TGFβ, IL-10, RA, and nitric oxide (NO) produced by FDC and DC facilitate this process. In germinal centers, В cells increase expression of α4β7 and CCR9 receptors in response to RA and migrate to the lamina propria upon interaction with high endothelial venules expressing MAdCAM-1. Further, B cells differentiate into plasmablasts and secrete polymeric IgA. During transfer through the epithelial cell layer, polymeric IgA is converted into secretory IgA (sIgA), which interacts with commensal microorganisms and neutralizes pathogens. LT with RORγt+ ILC3 (LTi cells) are required for Peyer’s patch development during embryogenesis. b) IgA induction in isolated lymphoid follicles (ILF) and in the lamina propria. CX3CR1+ DC capture antigen with transepithelial dendrites. CD103+ DC can capture antigen from goblet cells. In ILF, DCs can also capture antigen from М cells. DCs are activated by signals from toll-like receptors (TLRs) and by TSLP and RA, produced by epithelial cells, and present antigen to B cells in ILF or in the lamina propria. B cells receive IgA-inducing signals from TNF superfamily cytokines – B cell activating factor of the TNF family (BAFF) and proliferation-inducing ligand (APRIL) – and from TGFβ and NO, which are produced by DCs and stromal cells. The latter are activated in turn by LT and TNF produced by RORγt+ ILC3 cells. Interaction of BAFF and APRIL with BAFF-R, B-cell maturation antigen (BCMA), and transmembrane activator and CAML interactor (TACI) on В cells triggers expression of AID and formation of IgA-producing В cells within a lymphoid follicle. These В cells further differentiate into IgA-producing plasma cells in the lamina propria. As a result, special environment is created in the lamina propria, which is suitable for plasma cell survival. LT from RORγt+ ILC3 is required for postnatal development of ILF. Membrane LT (LTα1β2) from RORγt+ ILC3 is required for T cell-independent induction of IgA production in the lamina propria by activating DCs. Soluble LT (LTα3) homotrimer secreted by RORγt+ ILC is required for T cell-dependent IgA induction via T cell migration to the lamina propria.
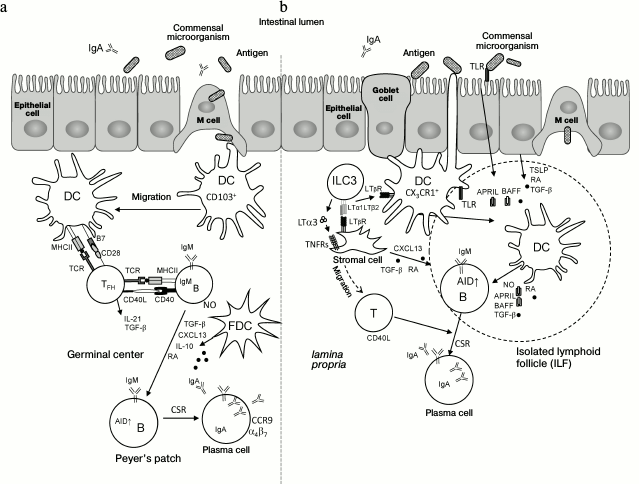
The T cell-dependent IgA production pathway takes place mainly in Peyer’s patches and depends on germinal center formation and interactions of B cells with follicular helper T cells (TFH) [120]. Lymphoid patch of the cecum also participates in production of IgA [121]. T cell-independent IgA production pathway occurs predominantly in isolated lymphoid follicles (ILF) and in own proper mucous plate (lamina propria) [122, 123]. LTβR is required for development of gut-associated lymphoid tissue (GALT), including Peyer’s patches, ILF, and mesenteric lymph nodes [10, 124].
Formation of Peyer’s patches, ILF, and mesenteric lymph nodes requires expression of LT on the surface of lymphoid tissue inducer cells (LTi), which interact with LTβR expressing stromal cells during embryogenesis and trigger the development of lymph nodes and Peyer’s patches [10, 125, 126]. TNF also participates in the development and maintenance of lymphoid organs in the intestine [18, 127], though to a lesser extent compared to LT. Therefore, in contrast to LTβR–/– mice, TNF–/– and TNFR–/– mice develop mesenteric lymph nodes, ILF, and, in some strains, Peyer’s patches [10, 127]. Twenty-five percent of LTβ–/– mice develop mesenteric lymph nodes, which display a disturbed microstructure [10, 127]. Production of IgA in mice with a deletion of the membrane lymphotoxin LTβ2LTα1 (LTβ–/– mice) and in LTβR–/– mice is sharply decreased, in contrast to slight reduction of IgA production in TNFR1–/– and TNFR2–/– mice [16, 128].
It is worth mentioning that the defect in IgA production in the gut of LTβR–/– and LTβ–/– mice cannot be fully explained by the defects in GALT development. In fact, recent studies have shown that in mice with deficiency of LTβ2LTα1 on RORγt+ ILC3, IgA levels in the intestine remains normal, despite the absence of GALT [16, 128]. Detailed experiments have demonstrated that LTα1β2 from RORγt+ ILC3 is required for the T cell-independent IgA induction in the lamina propria via DC activation. At the same time, soluble LT homotrimer (LTα3) secreted by RORγt+ ILC is required for T cell-dependent induction of IgA in the lamina propria by promoting T cell migration to the gut [16, 128] (see also Fig. 3).
Thus, members of the TNF superfamily play a key role in formation of isolated lymphoid follicles, and IgA production in the intestinal tissues, which is required for control of commensal microbiota, maintaining its proper composition and timely elimination of pathogens and toxins neutralization.
APPLICATION OF TNF BLOCKERS IN THERAPY OF INFLAMMATORY BOWEL
DISEASE
Patients with inflammatory bowel disease (IBD) suffer from chronic pains, bloody diarrhea, and weight loss. Such IBD-associated syndromes as skin inflammation and joint pain are also frequent [27, 129]. Two main forms of IBD are distinguished that differ in localization, etiology and symptoms: Crohn’s disease and ulcerative colitis [27, 30-32, 130]. However, the pathogenesis of these diseases has a common pattern and consists in a combination of genetic susceptibility and certain environmental factors affecting the patient directly or indirectly involving their microbiota [27, 34, 131]. In a general case, impairment of the mucosal barrier function results in penetration of commensal microorganisms into tissues and activation of immune response. At this stage, genetic susceptibility to development of inadequate reaction to environmental factors may cause formation of continuous uncontrolled inflammation of the intestinal mucosa. An aberrant immune response includes activation of both innate and adaptive arms of the immune system with participation of proinflammatory cytokines, chemokines, and transcription factors. Blocking the inflammatory process may inhibit disease development [35, 104]. Potentially, any molecule involved into initiation or progression of the disease may be considered as a target for therapy. Hypothetically, an ideal IBD treatment should induce and maintain continuous remission, which maximally reduces risk of complicated abdominal surgery. For a long time glucocorticosteroids, derivatives of 5-aminosalicylic acid and immunosuppressant azathioprine possessing wide spectrum of contraindications and side effects were used as primary therapy for IBD [132, 133]. Emergence of TNF antagonists in the medical arsenal was a valuable addition to the existing methods for IBD therapy [133].
Increased levels of TNF in mucosa [134, 135], serum [134], and in stool [136] were found in IBD patients in comparison with healthy donors. Initially, TNF antagonists were introduced into broad practice during treatment of intestinal inflammation mainly as a second line therapy for patients who did not respond to conservative treatment. However, efficiency of TNF blockers as a first line therapy was soon shown [19, 137]. TNF antagonists have been approved by the Food and Drug Administration of USA (FDA) for IBD treatment: infliximab, adalimumab, golimumab, and certolizumab pegol [133, 138-140]. Their characteristics are given in Table 2. These drugs are also used for IBD treatment in Russia [31, 141].
Table 2. Characteristics of monoclonal
anti-TNF antibodies approved by FDA (United States Food & Drug
Administration) for IBD therapy
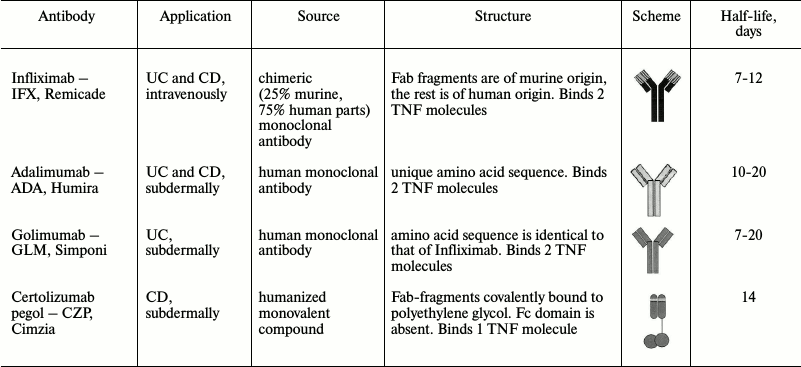
The listed inhibitors are monoclonal anti-TNF IgG antibodies consisting of Fab-fragment, which binds antigen, and Fc-domain (or substituting molecule) connected to the Fab-fragment by a hinge linker [142].
All the monoclonal anti-TNF antibodies were designed for inhibition of TNF signaling. However, they have different efficiency, mechanism of action, contraindications, and side effects [140, 143, 144]. Though TNF inhibitors have revolutionized therapy of colitis, one third of patients do not respond to such treatment upon initial drug administration (primary non-responders), and up to 50% of patients who take anti-TNF antibodies eventually stop responding (secondary non-responders) [19, 145]. Therefore, understanding the mechanisms of the blocker action becomes essential for developing a successful therapeutic strategy.
In a number of in vitro studies, it was shown that differences in clinical efficacy of antibody might be related to its different affinity and avidity to soluble and membrane-bound TNF [146-148]. All TNF inhibitors based on monoclonal antibodies design efficiently bind and neutralize soluble TNF [146, 147, 149]. However, the ability of these blockers to neutralize membrane TNF showed contradictory results [146-150]. It is generally accepted though that these TNF inhibitors bind membrane fraction with lower affinity compared to the soluble TNF. As a result, efficiency of an inhibitor in the context of membrane TNF blocking depends not only on properties of the inhibitor itself, but also on levels of soluble TNF, which “competes” more successfully for antagonist binding [140]. This should be considered both by researchers when choosing the experimental colitis model and by physicians for selection of individual IBD therapy. It should be noted that in experimental colitis models in mice, selective inhibition of soluble TNF was sufficient to reduce inflammation. However, blocking both soluble and membrane-bound forms of TNF in mice was more efficient and accompanied by continuous stable remission [151].
In contrast to the above-mentioned TNF inhibitors, etanercept blocker (Enbrel) predominantly binds soluble TNF and is successfully used for treatment of rheumatoid arthritis. However, it was inefficient in treatment of IBD [152, 153]. Etanercept is an all-human chimeric protein consisting of two soluble TNFR2 moieties bound to the Fc-fragment of IgG1. It was suggested that etanercept has lower binding activity because of increased dissociation rate (lower stability) of the complex with soluble and membrane-bound TNF [148]. Etanercept is also known to have lower ability to induce apoptosis in T cells, in contrast to adalimumab and infliximab, which may be a reason for low efficiency of etanercept in IBD therapy [19, 154]. Another potential explanation for limited efficiency of etanercept in IBD may be linked with its ability to block soluble LTα3 homotrimer [138], which is required for production of IgA and maintaining intestinal homeostasis [16].
The widespread use of anti-TNF therapy has led to an increase in number of registered systemic side effects, especially linked with infections, and inflammation in the skin and joints [138, 155]. One of the serious complications of anti-TNF therapy is activation of latent tuberculosis infection [156, 157]. Prognosis and risk management is complicated because in a number of cases it is hard to distinguish between side effects of anti-TNF therapy and non-intestinal manifestations of IBD. Taking into account that TNF blockers represent a fundamentally new class of therapeutic biological agents, fears about their long-term application safety required careful monitoring and fixation of all side effect cases [158, 159].
TNF antagonists are also efficient in treatment of severe forms of psoriasis [160]. However, continuous use of such therapy has revealed paradoxically high frequency of psoriasis development as a side effect [161]. Furthermore, cancellation of TNF blockers stopped the development psoriasis [162]. The mechanism of this reaction is not fully understood. Disruption of the balance between TNF and IFNα could be one of the reasons for development of psoriasis during anti-TNF therapy. It was shown that TNF blocks differentiation of plasmacytoid DCs (an IFNα source) from premature CD34+ hematopoietic precursor cells in vitro. On the other hand, TNF inhibits secretion of IFNα by plasmacytoid DCs. Hence, neutralization of endogenous TNF increases the amount of secreted IFNα, which plays an important role in the psoriasis induction [163].
Beside psoriasis, anti-TNF therapy may cause such side effect as TNF alpha antagonist-induced lupus-like syndrome (TAILS) [164]. The etiology of this disease is not determined, although there are a number of hypotheses. According to one, anti-TNF therapy provokes massive apoptosis of proinflammatory immune cells accompanied by the release of DNA and other autoantigens, which results in antibody production and development of the lupus-like syndrome [165]. It should be noted that this mechanism is not applicable for the lupus-like syndrome caused by a course of etanercept, which does not induce apoptosis. A second hypothesis connects the disease pathogenesis with autoantibodies produced by continuously activated B cells, which are forced to handle frequent infections during immunosuppressive anti-TNF therapy [166]. According to another hypothesis, anti-TNF therapy activates humoral autoimmunity, thus shifting the Th1-Th2 helper responses [167]. A number of studies have demonstrated that application of TNF inhibitors might increase the risk of squamous cell carcinoma of the skin [168, 169]. On the other hand, emergence of skin cancer may be related to side effects of psoriasis and IBD therapy using ultraviolet А, psoralen [170], thiopurine [171], and other immunosuppressants.
Local inflammation at the injection site may be attributed to the side effects of anti-TNF therapy. A Danish research group measured levels of IgG and IgE antibodies against infliximab in the serum of patients after acute reaction to drug administration. They found that the presence of severe infusion reactions correlates with high levels of IgG, but not IgE antibodies. Thus, such a side effect is not a true anaphylactic reaction [172]. Finally, it was shown that the risk of bacterial, viral, and fungal infections is increased in patients with rheumatoid arthritis and IBD undergoing anti-TNF therapy [173, 174].
At present, blocking endogenous TNF is the most efficient IBD therapy. However, lack of a clear understanding of the antagonist action mechanisms and severe side effects determine an urgent need for further studies of TNF biology. Use of new mouse models such as “humanized” mice expressing human TNF genes as well as mice with tissue-specific TNF expression might be a valuable tool for studying the role of TNF in pathogenesis of intestinal inflammation and design of more efficient therapy for IBD [20, 175]. Other biologicals for IBD treatment are also being extensively developed and tested [25, 176, 177]. Similar to TNF, LT also plays a key role in regulation of the intestinal inflammation, but use of selective modulators of this cytokine in the IBD therapy is still poorly studied and requires additional research.
Acknowledgements
We thank Pavel Matveev for assistance in preparation of the figures.
This work was supported by the grant of the President of the Russian Federation for support of leading scientific schools of the Russian Federation (NSH-10014.2016.4), by the Crohn and Colitis Foundation of America (grant No. 294083), and by Trudeau Institute.
REFERENCES
1.Locksley, R. M., Killeen, N., and Lenardo, M. J.
(2001) The TNF and TNF receptor superfamilies: integrating mammalian
biology, Cell, 104, 487-501.
2.Ward-Kavanagh, L. K., Lin, W. W., Sedy, J. R., and
Ware, C. F. (2016) The TNF receptor superfamily in co-stimulating and
co-inhibitory responses, Immunity, 44, 1005-1019.
3.Ware, C. F. (2005) Network communications:
lymphotoxins, LIGHT, and TNF, Annu. Rev. Immunol., 23,
787-819.
4.Croft, M., Benedict, C. A., and Ware, C. F. (2013)
Clinical targeting of the TNF and TNFR superfamilies, Nat. Rev. Drug
Discov., 12, 147-168.
5.Browning, J. L., and French, L. E. (2002)
Visualization of lymphotoxin-beta and lymphotoxin-beta receptor
expression in mouse embryos, J. Immunol., 168,
5079-5087.
6.Fu, Y. X., and Chaplin, D. D. (1999) Development
and maturation of secondary lymphoid tissues, Annu. Rev.
Immunol., 17, 399-433.
7.Sudhamsu, J., Yin, J., Chiang, E. Y., Starovasnik,
M. A., Grogan, J. L., and Hymowitz, S. G. (2013) Dimerization of
LTbetaR by LTalpha1beta2 is necessary and sufficient for signal
transduction, Proc. Natl. Acad. Sci. USA, 110,
19896-19901.
8.Faustman, D., and Davis, M. (2010) TNF receptor 2
pathway: drug target for autoimmune diseases, Nat. Rev. Drug
Discov., 9, 482-493.
9.Steinberg, M. W., Cheung, T. C., and Ware, C. F.
(2011) The signaling networks of the herpesvirus entry mediator
(TNFRSF14) in immune regulation, Immunol. Rev., 244,
169-187.
10.Randall, T. D., Carragher, D. M., and
Rangel-Moreno, J. (2008) Development of secondary lymphoid organs,
Annu. Rev. Immunol., 26, 627-650.
11.McCarthy, D. D., Summers-Deluca, L., Vu, F.,
Chiu, S., Gao, Y., and Gommerman, J. L. (2006) The lymphotoxin pathway:
beyond lymph node development, Immunol. Res., 35,
41-54.
12.Lo, J. C., Wang, Y., Tumanov, A. V., Bamji, M.,
Yao, Z., Reardon, C. A., Getz, G. S., and Fu, Y. X. (2007) Lymphotoxin
beta receptor-dependent control of lipid homeostasis, Science,
316, 285-288.
13.Tumanov, A. V., Christiansen, P. A., and Fu,
Y.-X. (2007) The role of lymphotoxin receptor signaling in diseases,
Curr. Mol. Med., 7, 567-578.
14.Haybaeck, J., Zeller, N., Wolf, M. J., Weber, A.,
Wagner, U., Kurrer, M. O., Bremer, J., Iezzi, G., Graf, R., Clavien, P.
A., Thimme, R., Blum, H., Nedospasov, S. A., Zatloukal, K., Ramzan, M.,
Ciesek, S., Pietschmann, T., Marche, P. N., Karin, M., Kopf, M.,
Browning, J. L., Aguzzi, A., and Heikenwalder, M. (2009) A
lymphotoxin-driven pathway to hepatocellular carcinoma, Cancer
Cell, 16, 295-308.
15.Upadhyay, V., and Fu, Y. X. (2013) Lymphotoxin
signalling in immune homeostasis and the control of microorganisms,
Nat. Rev. Immunol., 13, 270-279.
16.Kruglov, A. A., Grivennikov, S. I., Kuprash, D.
V., Winsauer, C., Prepens, S., Seleznik, G. M., Eberl, G., Littman, D.
R., Heikenwalder, M., Tumanov, A. V., and Nedospasov, S. A. (2013)
Nonredundant function of soluble LTalpha3 produced by innate lymphoid
cells in intestinal homeostasis, Science, 342,
1243-1246.
17.Macho-Fernandez, E., Koroleva, E. P., Spencer, C.
M., Tighe, M., Torrado, E., Cooper, A. M., Fu, Y. X., and Tumanov, A.
V. (2015) Lymphotoxin beta receptor signaling limits mucosal damage
through driving IL-23 production by epithelial cells, Mucosal
Immunol., 8, 403-413.
18.Tumanov, A. V., Grivennikov, S. I., Kruglov, A.
A., Shebzukhov, Y. V., Koroleva, E. P., Piao, Y., Cui, C. Y., Kuprash,
D. V., and Nedospasov, S. A. (2010) Cellular source and molecular form
of TNF specify its distinct functions in organization of secondary
lymphoid organs, Blood, 116, 3456-3464.
19.Nielsen, O. H., and Ainsworth, M. A. (2013) Tumor
necrosis factor inhibitors for inflammatory bowel disease, New Engl.
J. Med., 369, 754-762.
20.Winsauer, C., Kruglov, A. A., Chashchina, A. A.,
Drutskaya, M. S., and Nedospasov, S. A. (2014) Cellular sources of
pathogenic and protective TNF and experimental strategies based on
utilization of TNF humanized mice, Cytokine Growth Factor Rev.,
25, 115-123.
21.Dothel, G., Vasina, V., Barbara, G., and De
Ponti, F. (2013) Animal models of chemically induced intestinal
inflammation: predictivity and ethical issues, Pharmacol. Ther.,
139, 71-86.
22.DeVoss, J., and Diehl, L. (2014) Murine models of
inflammatory bowel disease (IBD): challenges of modeling human disease,
Toxicol. Pathol., 42, 99-110.
23.Jiminez, J. A., Uwiera, T. C., Douglas Inglis,
G., and Uwiera, R. R. (2015) Animal models to study acute and chronic
intestinal inflammation in mammals, Gut Pathog., 7,
29.
24.Mizoguchi, A., Takeuchi, T., Himuro, H., Okada,
T., and Mizoguchi, E. (2016) Genetically engineered mouse models for
studying inflammatory bowel disease, J. Pathol., 238,
205-219.
25.Danese, S., Fiocchi, C., and Panes, J. (2016)
Drug development in IBD: from novel target identification to early
clinical trials, Gut, 65, 1233-1239.
26.Jurjus, A. R., Khoury, N. N., and Reimund, J.-M.
(2004) Animal models of inflammatory bowel disease, J. Pharmacol.
Toxicol. Methods, 50, 81-92.
27.Vorobyov, G. I., and Khalif, I. L. (eds.) (2008)
Nonspecific Inflammatory Bowel Disease [in Russian], Miklosh,
Moscow.
28.Baumgart, D. C., and Sandborn, W. J. (2012)
Crohn’s disease, Lancet, 380, 1590-1605.
29.Danese, S., and Fiocchi, C. (2011) Ulcerative
colitis, New Engl. J. Med., 365, 1713-1725.
30.Zimmerman, Ya. S., Zimmerman, I. Ya., and
Tretyakova, Yu. I. (2013) Ulcerative colitis and Crohn’s disease:
modern concept. Part 1. Definition, terminology, occurrence, etiology
and pathogenеsis, diagnostics, complications, classification,
Klin. Med., 11, 27-33.
31.Zimmerman, Ya. S., Zimmerman, I. Ya., and
Tretyakova, Yu. I. (2013) Ulcerative colitis and Crohn’s disease:
modern concept. Part 2. Diagnostics and differentiation therapy,
Klin. Med., 12, 9-16.
32.Kaser, A., Zeissig, S., and Blumberg, R. S.
(2010) Inflammatory bowel disease, Annu. Rev. Immunol.,
28, 573-621.
33.Kaplan, G. G. (2015) The global burden of IBD:
from 2015 to 2025, Nat. Rev. Gastroenterol. Hepatol., 12,
720-727.
34.Wallace, K. L., Zheng, L. B., Kanazawa, Y., and
Shih, D. Q. (2014) Immunopathology of inflammatory bowel disease,
World J. Gastroenterol., 20, 6-21.
35.Neurath, M. F. (2014) Cytokines in inflammatory
bowel disease, Nat. Rev. Immunol., 14, 329-342.
36.Dalal, S. R., and Chang, E. B. (2014) The
microbial basis of inflammatory bowel diseases, The J. Clin.
Invest., 124, 4190-4196.
37.Khor, B., Gardet, A., and Xavier, R. J. (2011)
Genetics and pathogenesis of inflammatory bowel disease, Nature,
474, 307-317.
38.Sheehan, D., Moran, C., and Shanahan, F. (2015)
The microbiota in inflammatory bowel disease, J. Gastroenterol.,
50, 495-507.
39.Wirtz, S., Neufert, C., Weigmann, B., and
Neurath, M. F. (2007) Chemically induced mouse models of intestinal
inflammation, Nat. Protoc., 2, 541-546.
40.Low, D., Nguyen, D. D., and Mizoguchi, E. (2013)
Animal models of ulcerative colitis and their application in drug
research, Drug Des. Devel. Ther., 7, 1341-1357.
41.Okayasu, I., Hatakeyama, S., Yamada, M., Ohkusa,
T., Inagaki, Y., and Nakaya, R. (1990) A novel method in the induction
of reliable experimental acute and chronic ulcerative colitis in mice,
Gastroenterology, 98, 694-702.
42.Pers, M., and Cerar, A. (2012) Dextran sodium
sulphate colitis mouse model: traps and tricks, J. Biomed.
Biotechnol., 2012, 718617.
43.Chassaing, B., Aitken, J. D., Malleshappa, M.,
and Vijay-Kumar, M. (2014) Dextran sulfate sodium (DSS)-induced colitis
in mice, Curr. Protoc. Immunol., 104, Unit 15.25.
44.Te Velde, A. A., Verstege, M. I., and Hommes, D.
W. (2006) Critical appraisal of the current practice in murine
TNBS-induced colitis, Inflamm. Bowel Dis., 12,
995-999.
45.Motavallian-Naeini, A., Andalib, S., Rabbani, M.,
Mahzouni, P., Afsharipour, M., and Minaiyan, M. (2012) Validation and
optimization of experimental colitis induction in rats using
2,4,6-trinitrobenzene sulfonic acid, Res. Pharm. Sci., 7,
159-169.
46.Ko, J.-K.-S., Lam, F.-Y.-L., and Cheung, A.-P.-L.
(2005) Amelioration of experimental colitis by Astragalus
membranaceus through anti-oxidation and inhibition of adhesion
molecule synthesis, World J. Gastroenterol., 11,
5787-5794.
47.Heller, F., Fuss, I. J., Nieuwenhuis, E. E.,
Blumberg, R. S., and Strober, W. (2002) Oxazolone colitis, a Th2
colitis model resembling ulcerative colitis, is mediated by
IL-13-producing NK-T cells, Immunity, 17, 629-638.
48.Collins, J. W., Keeney, K. M., Crepin, V. F.,
Rathinam, V. A., Fitzgerald, K. A., Finlay, B. B., and Frankel, G.
(2014) Citrobacter rodentium: infection, inflammation and the
microbiota, Nat. Rev. Microbiol., 12, 612-623.
49.Koroleva, E. P., Halperin, S., Gubernatorova, E.
O., Macho-Fernandez, E., Spencer, C. M., and Tumanov, A. V. (2015)
Citrobacter rodentium-induced colitis: a robust model to study
mucosal immune responses in the gut, J. Immunol. Methods,
421, 61-72.
50.Rivera-Nieves, J., Bamias, G., Vidrich, A.,
Marini, M., Pizarro, T. T., McDuffie, M. J., Moskaluk, C. A., Cohn, S.
M., and Cominelli, F. (2003) Emergence of perianal fistulizing disease
in the SAMP1/YitFc mouse, a spontaneous model of chronic ileitis,
Gastroenterology, 124, 972-982.
51.Matsumoto, S., Okabe, Y., Setoyama, H., Takayama,
K., Ohtsuka, J., Funahashi, H., Imaoka, A., Okada, Y., and Umesaki, Y.
(1998) Inflammatory bowel disease-like enteritis and caecitis in a
senescence accelerated mouse P1/Yit strain, Gut, 43,
71-78.
52.Pizarro, T. T., Arseneau, K. O., Bamias, G., and
Cominelli, F. (2003) Mouse models for the study of Crohn’s
disease, Trends Mol. Med., 9, 218-222.
53.Kuhn, R., Lohler, J., Rennick, D., Rajewsky, K.,
and Muller, W. (1993) Interleukin-10-deficient mice develop chronic
enterocolitis, Cell, 75, 263-274.
54.Keubler, L. M., Buettner, M., Hager, C., and
Bleich, A. (2015) A multihit model: colitis lessons from the
interleukin-10-deficient mouse, Inflamm. Bowel Dis., 21,
1967-1975.
55.Kontoyiannis, D., Boulougouris, G., Manoloukos,
M., Armaka, M., Apostolaki, M., Pizarro, T., Kotlyarov, A., Forster,
I., Flavell, R., Gaestel, M., Tsichlis, P., Cominelli, F., and Kollias,
G. (2002) Genetic dissection of the cellular pathways and signaling
mechanisms in modeled tumor necrosis factor-induced Crohn’s-like
inflammatory bowel disease, J. Exp. Med., 196,
1563-1574.
56.Leach, M. W., Bean, A. G., Mauze, S., Coffman, R.
L., and Powrie, F. (1996) Inflammatory bowel disease in C.B-17 scid
mice reconstituted with the CD45RBhigh subset of
CD4+ T-cells, Am. J. Pathol., 148,
1503-1515.
57.Ostanin, D. V., Bao, J., Koboziev, I., Gray, L.,
Robinson-Jackson, S. A., Kosloski-Davidson, M., Price, V. H., and
Grisham, M. B. (2009) T-cell transfer model of chronic colitis:
concepts, considerations, and tricks of the trade, Am. J. Physiol.
Gastrointest. Liver Physiol., 296, G135-146.
58.Sadlack, B., Merz, H., Schorle, H., Schimpl, A.,
Feller, A. C., and Horak, I. (1993) Ulcerative colitis-like disease in
mice with a disrupted interleukin-2 gene, Cell, 75,
253-261.
59.Baumgart, D. C., Olivier, W. A., Reya, T.,
Peritt, D., Rombeau, J. L., and Carding, S. R. (1998) Mechanisms of
intestinal epithelial cell injury and colitis in interleukin 2
(IL2)-deficient mice, Cell Immunol., 187, 52-66.
60.Lee, E. G., Boone, D. L., Chai, S., Libby, S. L.,
Chien, M., Lodolce, J. P., and Ma, A. (2000) Failure to regulate
TNF-induced NF-kappaB and cell death responses in A20-deficient mice,
Science, 289, 2350-2354.
61.Hammer, G. E., Turer, E. E., Taylor, K. E., Fang,
C. J., Advincula, R., Oshima, S., Barrera, J., Huang, E. J., Hou, B.,
Malynn, B. A., Reizis, B., DeFranco, A., Criswell, L. A., Nakamura, M.
C., and Ma, A. (2011) Expression of A20 by dendritic cells preserves
immune homeostasis and prevents colitis and spondyloarthritis, Nat.
Immunol., 12, 1184-1193.
62.Mombaerts, P., Mizoguchi, E., Grusby, M. J.,
Glimcher, L. H., Bhan, A. K., and Tonegawa, S. (1993) Spontaneous
development of inflammatory bowel disease in T-cell receptor mutant
mice, Cell, 75, 274-282.
63.Nagatani, K., Wang, S., Llado, V., Lau, C. W.,
Li, Z., Mizoguchi, A., Nagler, C. R., Shibata, Y., Reinecker, H.-C.,
Mora, J. R., and Mizoguchi, E. (2012) Chitin microparticles for the
control of intestinal inflammation, Inflamm. Bowel Dis.,
18, 1698-1710.
64.Nenci, A., Becker, C., Wullaert, A., Gareus, R.,
Van Loo, G., Danese, S., Huth, M., Nikolaev, A., Neufert, C., Madison,
B., Gumucio, D., Neurath, M. F., and Pasparakis, M. (2007) Epithelial
NEMO links innate immunity to chronic intestinal inflammation,
Nature, 446, 557-561.
65.Watanabe, M., Ueno, Y., Yajima, T., Okamoto, S.,
Hayashi, T., Yamazaki, M., Iwao, Y., Ishii, H., Habu, S., Uehira, M.,
Nishimoto, H., Ishikawa, H., Hata, J., and Hibi, T. (1998) Interleukin
7 transgenic mice develop chronic colitis with decreased interleukin 7
protein accumulation in the colonic mucosa, J. Exp. Med.,
187, 389-402.
66.Rath, H. C., Wilson, K. H., and Sartor, R. B.
(1999) Differential induction of colitis and gastritis in HLA-B27
transgenic rats selectively colonized with Bacteroides
vulgatus or Escherichia coli, Infect. Immun.,
67, 2969-2974.
67.Rath, H. C. (2002) Role of commensal bacteria in
chronic experimental colitis: lessons from the HLA-B27 transgenic rat,
Pathobiology, 70, 131-138.
68.Peloquin, J. M., and Nguyen, D. D. (2013) The
microbiota and inflammatory bowel disease: insights from animal models,
Anaerobe, 24, 102-106.
69.Gkouskou, K. K., Deligianni, C., Tsatsanis, C.,
and Eliopoulos, A. G. (2014) The gut microbiota in mouse models of
inflammatory bowel disease, Front. Cell. Infect. Microbiol.,
4, 28.
70.Eri, R., McGuckin, M. A., and Wadley, R. (2012) T
cell transfer model of colitis: a great tool to assess the contribution
of T cells in chronic intestinal inflammation, Methods Mol.
Biol., 844, 261-275.
71.Shouval, D. S., Ouahed, J., Biswas, A., Goettel,
J. A., Horwitz, B. H., Klein, C., Muise, A. M., and Snapper, S. B.
(2014) Interleukin 10 receptor signaling: master regulator of
intestinal mucosal homeostasis in mice and humans, Adv.
Immunol., 122, 177-210.
72.Koboziev, I., Jones-Hall, Y., Valentine, J. F.,
Webb, C. R., Furr, K. L., and Grisham, M. B. (2015) Use of humanized
mice to study the pathogenesis of autoimmune and inflammatory diseases,
Inflamm. Bowel Dis., 21, 1652-1673.
73.Sonnenberg, G. F., and Artis, D. (2015) Innate
lymphoid cells in the initiation, regulation and resolution of
inflammation, Nat. Med., 21, 698-708.
74.Eberl, G., Colonna, M., Di Santo, J. P., and
McKenzie, A. N. (2015) Innate lymphoid cells: a new paradigm in
immunology, Science, 348, aaa6566.
75.Spits, H., and Cupedo, T. (2012) Innate lymphoid
cells: emerging insights in development, lineage relationships, and
function, Annu. Rev. Immunol., 30, 647-675.
76.Diefenbach, A., Colonna, M., and Koyasu, S.
(2014) Development, differentiation, and diversity of innate lymphoid
cells, Immunity, 41, 354-365.
77.Vivier, E., Van de Pavert, S. A., Cooper, M. D.,
and Belz, G. T. (2016) The evolution of innate lymphoid cells, Nat.
Immunol., 17, 790-794.
78.Spits, H., Artis, D., Colonna, M., Diefenbach,
A., Di Santo, J. P., Eberl, G., Koyasu, S., Locksley, R. M., McKenzie,
A. N., Mebius, R. E., Powrie, F., and Vivier, E. (2013) Innate lymphoid
cells – a proposal for uniform nomenclature, Nat. Rev.
Immunol., 13, 145-149.
79.McKenzie, A. N., Spits, H., and Eberl, G. (2014)
Innate lymphoid cells in inflammation and immunity, Immunity,
41, 366-374.
80.Klose, C. S., and Artis, D. (2016) Innate
lymphoid cells as regulators of immunity, inflammation and tissue
homeostasis, Nat. Immunol., 17, 765-774.
81.Wang, Y., Koroleva, E. P., Kruglov, A. A.,
Kuprash, D. V., Nedospasov, S. A., Fu, Y. X., and Tumanov, A. V. (2010)
Lymphotoxin beta receptor signaling in intestinal epithelial cells
orchestrates innate immune responses against mucosal bacterial
infection, Immunity, 32, 403-413.
82.Sawa, S., Lochner, M., Satoh-Takayama, N.,
Dulauroy, S., Berard, M., Kleinschek, M., Cua, D., Di Santo, J. P., and
Eberl, G. (2011) RORgammat(+) innate lymphoid cells regulate intestinal
homeostasis by integrating negative signals from the symbiotic
microbiota, Nat. Immunol., 12, 320-326.
83.Eberl, G. (2012) Development and evolution of
RORgammat+ cells in a microbe’s world, Immunol.
Rev., 245, 177-188.
84.Sonnenberg, G. F., Monticelli, L. A., Alenghat,
T., Fung, T. C., Hutnick, N. A., Kunisawa, J., Shibata, N., Grunberg,
S., Sinha, R., Zahm, A. M., Tardif, M. R., Sathaliyawala, T., Kubota,
M., Farber, D. L., Collman, R. G., Shaked, A., Fouser, L. A., Weiner,
D. B., Tessier, P. A., Friedman, J. R., Kiyono, H., Bushman, F. D.,
Chang, K. M., and Artis, D. (2012) Innate lymphoid cells promote
anatomical containment of lymphoid-resident commensal bacteria,
Science, 336, 1321-1325.
85.Hepworth, M. R., Fung, T. C., Masur, S. H.,
Kelsen, J. R., McConnell, F. M., Dubrot, J., Withers, D. R., Hugues,
S., Farrar, M. A., Reith, W., Eberl, G., Baldassano, R. N., Laufer, T.
M., Elson, C. O., and Sonnenberg, G. F. (2015) Immune tolerance. Group
3 innate lymphoid cells mediate intestinal selection of commensal
bacteria-specific CD4(+) T-cells, Science, 348,
1031-1035.
86.Goc, J., Hepworth, M. R., and Sonnenberg, G. F.
(2016) Group 3 innate lymphoid cells: regulating host–commensal
bacteria interactions in inflammation and cancer, Int. Immunol.,
28, 43-52.
87.Cording, S., Medvedovic, J., Aychek, T., and
Eberl, G. (2016) Innate lymphoid cells in defense, immunopathology and
immunotherapy, Nat. Immunol., 17, 755-757.
88.Zheng, Y., Valdez, P. A., Danilenko, D. M., Hu,
Y., Sa, S. M., Gong, Q., Abbas, A. R., Modrusan, Z., Ghilardi, N., De
Sauvage, F. J., and Ouyang, W. (2008) Interleukin-22 mediates early
host defense against attaching and effacing bacterial pathogens,
Nat. Med., 14, 282-289.
89.Hernandez, P. P., Mahlakoiv, T., Yang, I.,
Schwierzeck, V., Nguyen, N., Guendel, F., Gronke, K., Ryffel, B.,
Holscher, C., Dumoutier, L., Renauld, J. C., Suerbaum, S., Staeheli,
P., and Diefenbach, A. (2015) Interferon-lambda and interleukin 22 act
synergistically for the induction of interferon-stimulated genes and
control of rotavirus infection, Nat. Immunol., 16,
698-707.
90.Vonarbourg, C., Mortha, A., Bui, V. L.,
Hernandez, P. P., Kiss, E. A., Hoyler, T., Flach, M., Bengsch, B.,
Thimme, R., Holscher, C., Honig, M., Pannicke, U., Schwarz, K., Ware,
C. F., Finke, D., and Diefenbach, A. (2010) Regulated expression of
nuclear receptor RORγt confers distinct functional fates to NK
cell receptor-expressing RORγt(+) innate lymphocytes,
Immunity, 33, 736-751.
91.Fuchs, A., Vermi, W., Lee, J. S., Lonardi, S.,
Gilfillan, S., Newberry, R. D., Cella, M., and Colonna, M. (2013)
Intraepithelial type 1 innate lymphoid cells are a unique subset of
IL-12- and IL-15-responsive IFN-gamma-producing cells, Immunity,
38, 769-781.
92.Bernink, J. H., Peters, C. P., Munneke, M., Te
Velde, A. A., Meijer, S. L., Weijer, K., Hreggvidsdottir, H. S.,
Heinsbroek, S. E., Legrand, N., Buskens, C. J., Bemelman, W. A.,
Mjosberg, J. M., and Spits, H. (2013) Human type 1 innate lymphoid
cells accumulate in inflamed mucosal tissues, Nat. Immunol.,
14, 221-229.
93.Bernink, J. H., Krabbendam, L., Germar, K., De
Jong, E., Gronke, K., Kofoed-Nielsen, M., Munneke, J. M., Hazenberg, M.
D., Villaudy, J., Buskens, C. J., Bemelman, W. A., Diefenbach, A.,
Blom, B., and Spits, H. (2015) Interleukin-12 and -23 control
plasticity of CD127(+) group 1 and group 3 innate lymphoid cells in the
intestinal lamina propria, Immunity, 43, 146-160.
94.Spencer, S. P., Wilhelm, C., Yang, Q., Hall, J.
A., Bouladoux, N., Boyd, A., Nutman, T. B., Urban, J. F., Jr., Wang,
J., Ramalingam, T. R., Bhandoola, A., Wynn, T. A., and Belkaid, Y.
(2014) Adaptation of innate lymphoid cells to a micronutrient
deficiency promotes type 2 barrier immunity, Science,
343, 432-437.
95.Zook, E. C., and Kee, B. L. (2016) Development of
innate lymphoid cells, Nat. Immunol., 17, 775-782.
96.Koues, O. I., Collins, P. L., Cella, M.,
Robinette, M. L., Porter, S. I., Pyfrom, S. C., Payton, J. E., Colonna,
M., and Oltz, E. M. (2016) Distinct gene regulatory pathways for human
innate versus adaptive lymphoid cells, Cell, 165,
1134-1146.
97.Sonnenberg, G. F. (2016) Transcriptionally
defining ILC heterogeneity in humans, Nat. Immunol., 17,
351-352.
98.Magri, G., Miyajima, M., Bascones, S., Mortha,
A., Puga, I., Cassis, L., Barra, C. M., Comerma, L., Chudnovskiy, A.,
Gentile, M., Llige, D., Cols, M., Serrano, S., Arostegui, J. I., Juan,
M., Yague, J., Merad, M., Fagarasan, S., and Cerutti, A. (2014) Innate
lymphoid cells integrate stromal and immunological signals to enhance
antibody production by splenic marginal zone B cells, Nat.
Immunol., 15, 354-364.
99.Magri, G., and Cerutti, A. (2015) Role of group 3
innate lymphoid cells in antibody production, Curr. Opin.
Immunol., 33, 36-42.
100.Hanash, A. M., Dudakov, J. A., Hua, G.,
O’Connor, M. H., Young, L. F., Singer, N. V., West, M. L., Jenq,
R. R., Holland, A. M., Kappel, L. W., Ghosh, A., Tsai, J. J., Rao, U.
K., Yim, N. L., Smith, O. M., Velardi, E., Hawryluk, E. B., Murphy, G.
F., Liu, C., Fouser, L. A., Kolesnick, R., Blazar, B. R., and Van den
Brink, M. R. (2012) Interleukin-22 protects intestinal stem cells from
immune-mediated tissue damage and regulates sensitivity to graft versus
host disease, Immunity, 37, 339-350.
101.Dudakov, J. A., Hanash, A. M., and Van den
Brink, M. R. (2015) Interleukin-22: immunobiology and pathology,
Annu. Rev. Immunol., 33, 747-785.
102.Buonocore, S., Ahern, P. P., Uhlig, H. H.,
Ivanov, I. I., Littman, D. R., Maloy, K. J., and Powrie, F. (2010)
Innate lymphoid cells drive interleukin-23-dependent innate intestinal
pathology, Nature, 464, 1371-1375.
103.Geremia, A., Arancibia-Carcamo, C. V., Fleming,
M. P., Rust, N., Singh, B., Mortensen, N. J., Travis, S. P., and
Powrie, F. (2011) IL-23-responsive innate lymphoid cells are increased
in inflammatory bowel disease, J. Exp. Med., 208,
1127-1133.
104.Geremia, A., Biancheri, P., Allan, P., Corazza,
G. R., and Di Sabatino, A. (2014) Innate and adaptive immunity in
inflammatory bowel disease, Autoimmun. Rev., 13,
3-10.
105.Withers, D. R., Hepworth, M. R., Wang, X.,
Mackley, E. C., Halford, E. E., Dutton, E. E., Marriott, C. L.,
Brucklacher-Waldert, V., Veldhoen, M., Kelsen, J., Baldassano, R. N.,
and Sonnenberg, G. F. (2016) Transient inhibition of ROR-gammat
therapeutically limits intestinal inflammation by reducing TH17 cells
and preserving group 3 innate lymphoid cells, Nat. Med.,
22, 319-323.
106.Goldberg, R., Prescott, N., Lord, G. M.,
MacDonald, T. T., and Powell, N. (2015) The unusual suspects –
innate lymphoid cells as novel therapeutic targets in IBD, Nat. Rev.
Gastroenterol. Hepatol., 12, 271-283.
107.Tumanov, A. V., Koroleva, E. P., Guo, X., Wang,
Y., Kruglov, A., Nedospasov, S., and Fu, Y. X. (2011) Lymphotoxin
controls the IL-22 protection pathway in gut innate lymphoid cells
during mucosal pathogen challenge, Cell Host Microbe, 10,
44-53.
108.Spahn, T. W., Maaser, C., Eckmann, L.,
Heidemann, J., Lugering, A., Newberry, R., Domschke, W., Herbst, H.,
and Kucharzik, T. (2004) The lymphotoxin-beta receptor is critical for
control of murine Citrobacter rodentium-induced colitis,
Gastroenterology, 127, 1463.
109.Cerovic, V., Bain, C. C., Mowat, A. M., and
Milling, S. W. (2014) Intestinal macrophages and dendritic cells:
what’s the difference? Trends Immunol., 35,
270-277.
110.Guilliams, M., Ginhoux, F., Jakubzick, C.,
Naik, S. H., Onai, N., Schraml, B. U., Segura, E., Tussiwand, R., and
Yona, S. (2014) Dendritic cells, monocytes and macrophages: a unified
nomenclature based on ontogeny, Nat. Rev. Immunol., 14,
571-578.
111.Backhed, F., Ley, R. E., Sonnenburg, J. L.,
Peterson, D. A., and Gordon, J. I. (2005) Host–bacterial
mutualism in the human intestine, Science, 307,
1915-1920.
112.Gensollen, T., Iyer, S. S., Kasper, D. L., and
Blumberg, R. S. (2016) How colonization by microbiota in early life
shapes the immune system, Science, 352, 539-544.
113.Caballero, S., and Pamer, E. G. (2015)
Microbiota-mediated inflammation and antimicrobial defense in the
intestine, Annu. Rev. Immunol., 33, 227-256.
114.Hamada, H., Hiroi, T., Nishiyama, Y.,
Takahashi, H., Masunaga, Y., Hachimura, S., Kaminogawa, S.,
Takahashi-Iwanaga, H., Iwanaga, T., Kiyono, H., Yamamoto, H., and
Ishikawa, H. (2002) Identification of multiple isolated lymphoid
follicles on the antimesenteric wall of the mouse small intestine,
J. Immunol., 168, 57-64.
115.Mantis, N. J., McGuinness, C. R., Sonuyi, O.,
Edwards, G., and Farrant, S. A. (2006) Immunoglobulin A antibodies
against ricin A and B subunits protect epithelial cells from ricin
intoxication, Infect. Immun., 74, 3455-3462.
116.Pabst, O. (2012) New concepts in the generation
and functions of IgA, Nat. Rev. Immunol., 12,
821-832.
117.Bemark, M., Boysen, P., and Lycke, N. Y. (2012)
Induction of gut IgA production through T cell-dependent and
T-cell-independent pathways, Ann. N. Y. Acad. Sci., 1247,
97-116.
118.Cerutti, A. (2008) The regulation of IgA class
switching, Nat. Rev. Immunol., 8, 421-434.
119.Gutzeit, C., Magri, G., and Cerutti, A. (2014)
Intestinal IgA production and its role in host–microbe
interaction, Immunol. Rev., 260, 76-85.
120.Fagarasan, S., Kawamoto, S., Kanagawa, O., and
Suzuki, K. (2010) Adaptive immune regulation in the gut:
T-cell-dependent and T-cell-independent IgA synthesis, Annu. Rev.
Immunol., 28, 243-273.
121.Masahata, K., Umemoto, E., Kayama, H., Kotani,
M., Nakamura, S., Kurakawa, T., Kikuta, J., Gotoh, K., Motooka, D.,
Sato, S., Higuchi, T., Baba, Y., Kurosaki, T., Kinoshita, M., Shimada,
Y., Kimura, T., Okumura, R., Takeda, A., Tajima, M., Yoshie, O.,
Fukuzawa, M., Kiyono, H., Fagarasan, S., Iida, T., Ishii, M., and
Takeda, K. (2014) Generation of colonic IgA-secreting cells in the
caecal patch, Nat. Commun., 5, 3704.
122.Macpherson, A. J., Gatto, D., Sainsbury, E.,
Harriman, G. R., Hengartner, H., and Zinkernagel, R. M. (2000) A
primitive T-cell-independent mechanism of intestinal mucosal IgA
responses to commensal bacteria, Science, 288,
2222-2226.
123.Mora, J. R., Iwata, M., Eksteen, B., Song, S.
Y., Junt, T., Senman, B., Otipoby, K. L., Yokota, A., Takeuchi, H.,
Ricciardi-Castagnoli, P., Rajewsky, K., Adams, D. H., and Von Andrian,
U. H. (2006) Generation of gut-homing IgA-secreting B cells by
intestinal dendritic cells, Science, 314, 1157-1160.
124.Lorenz, R. G., Chaplin, D. D., McDonald, K. G.,
McDonough, J. S., and Newberry, R. D. (2003) Isolated lymphoid follicle
formation is inducible and dependent upon lymphotoxin-sufficient B
lymphocytes, lymphotoxin beta receptor, and TNF receptor I function,
J. Immunol., 170, 5475-5482.
125.Van de Pavert, S. A., and Mebius, R. E. (2010)
New insights into the development of lymphoid tissues, Nat. Rev.
Immunol., 10, 664-674.
126.Bar-Ephraim, Y. E., and Mebius, R. E. (2016)
Innate lymphoid cells in secondary lymphoid organs, Immunol.
Rev., 271, 185-199.
127.Kuprash, D. V., Tumanov, A. V., Liepinsh, D.
J., Koroleva, E. P., Drutskaya, M. S., Kruglov, A. A., Shakhov, A. N.,
Southon, E., Murphy, W. J., Tessarollo, L., Grivennikov, S. I., and
Nedospasov, S. A. (2005) Novel tumor necrosis factor-knockout mice that
lack Peyer’s patches, Eur. J. Immunol., 35,
1592-1600.
128.Kang, H. S., Chin, R. K., Wang, Y., Yu, P.,
Wang, J., Newell, K. A., and Fu, Y. X. (2002) Signaling via LTbetaR on
the lamina propria stromal cells of the gut is required for IgA
production, Nat. Immunol., 3, 576-582.
129.Danese, S., and Peyrin-Biroulet, L. (2014) IBD
in 2013: enriching the therapeutic armamentarium for IBD, Nat. Rev.
Gastroenterol. Hepatol., 11, 84-86.
130.Rogler, G. (2015) Where are we heading to in
pharmacological IBD therapy? Pharmacol. Res., 100,
220-227.
131.Scharl, M., and Rogler, G. (2012) Inflammatory
bowel disease pathogenesis: what is new? Curr. Opin.
Gastroenterol., 28, 301-309.
132.Bosani, M., Ardizzone, S., and Porro, G. B.
(2009) Biologic targeting in the treatment of inflammatory bowel
diseases, Biologics, 3, 77-97.
133.Olesen, C. M., Coskun, M., Peyrin-Biroulet, L.,
and Nielsen, O. H. (2016) Mechanisms behind efficacy of tumor necrosis
factor inhibitors in inflammatory bowel diseases, Pharmacol.
Ther., 159, 110-119.
134.Murch, S. H., Lamkin, V. A., Savage, M. O.,
Walker-Smith, J. A., and MacDonald, T. T. (1991) Serum concentrations
of tumour necrosis factor alpha in childhood chronic inflammatory bowel
disease, Gut, 32, 913-917.
135.Breese, E. J., Michie, C. A., Nicholls, S. W.,
Murch, S. H., Williams, C. B., Domizio, P., Walker-Smith, J. A., and
MacDonald, T. T. (1994) Tumor necrosis factor alpha-producing cells in
the intestinal mucosa of children with inflammatory bowel disease,
Gastroenterology, 106, 1455-1466.
136.Nilsen, E. M., Johansen, F. E., Kvale, D.,
Krajci, P., and Brandtzaeg, P. (1999) Different regulatory pathways
employed in cytokine-enhanced expression of secretory component and
epithelial HLA class I genes, Eur. J. Immunol., 29,
168-179.
137.D’Haens, G. (2010) Anti-TNF treatment in
Crohn’s disease: toward tailored therapy? Am. J.
Gastroenterol., 105, 1140-1141.
138.Tracey, D., Klareskog, L., Sasso, E. H.,
Salfeld, J. G., and Tak, P. P. (2008) Tumor necrosis factor antagonist
mechanisms of action: a comprehensive review, Pharmacol. Ther.,
117, 244-279.
139.Ueda, N., Tsukamoto, H., Mitoma, H., Ayano, M.,
Tanaka, A., Ohta, S., Inoue, Y., Arinobu, Y., Niiro, H., Akashi, K.,
and Horiuchi, T. (2013) The cytotoxic effects of certolizumab pegol and
golimumab mediated by transmembrane tumor necrosis factor alpha,
Inflamm. Bowel Dis., 19, 1224-1231.
140.Slevin, S. M., and Egan, L. J. (2015) New
insights into the mechanisms of action of anti-tumor necrosis
factor-alpha monoclonal antibodies in inflammatory bowel disease,
Inflamm. Bowel Dis., 21, 2909-2920.
141.Shchigoleva, N. E., Matina, I. A., Ponomariova,
A. P., Karpina, L. M., and Bologov, A. A. (2010) Use of infliximab in
children with inflammatory bowel diseases, Pediatr. Pharmacol.,
7, 55-61.
142.Benucci, M., Saviola, G., Manfredi, M.,
Sarzi-Puttini, P., and Atzeni, F. (2012) Tumor necrosis factors
blocking agents: analogies and differences, Acta Biomed.,
83, 72-80.
143.Thorlund, K., Druyts, E., Toor, K., and Mills,
E. J. (2015) Comparative efficacy of golimumab, infliximab, and
adalimumab for moderately to severely active ulcerative colitis: a
network meta-analysis accounting for differences in trial designs,
Expert. Rev. Gastroenterol. Hepatol., 9, 693-700.
144.Peake, S. T., Bernardo, D., Mann, E. R.,
Al-Hassi, H. O., Knight, S. C., and Hart, A. L. (2013) Mechanisms of
action of anti-tumor necrosis factor alpha agents in Crohn’s
disease, Inflamm. Bowel Dis., 19, 1546-1555.
145.Steenholdt, C., Brynskov, J., Thomsen, O. O.,
Munck, L. K., Fallingborg, J., Christensen, L. A., Pedersen, G.,
Kjeldsen, J., Jacobsen, B. A., Oxholm, A. S., Kjellberg, J., Bendtzen,
K., and Ainsworth, M. A. (2014) Individualised therapy is more
cost-effective than dose intensification in patients with Crohn’s
disease who lose response to anti-TNF treatment: a randomised,
controlled trial, Gut, 63, 919-927.
146.Kaymakcalan, Z., Sakorafas, P., Bose, S.,
Scesney, S., Xiong, L., Hanzatian, D. K., Salfeld, J., and Sasso, E. H.
(2009) Comparisons of affinities, avidities, and complement activation
of adalimumab, infliximab, and etanercept in binding to soluble and
membrane tumor necrosis factor, Clin. Immunol., 131,
308-316.
147.Shealy, D. J., Cai, A., Staquet, K., Baker, A.,
Lacy, E. R., Johns, L., Vafa, O., Gunn, G., 3rd, Tam, S., Sague, S.,
Wang, D., Brigham-Burke, M., Dalmonte, P., Emmell, E., Pikounis, B.,
Bugelski, P. J., Zhou, H., Scallon, B. J., and Giles-Komar, J. (2010)
Characterization of golimumab, a human monoclonal antibody specific for
human tumor necrosis factor alpha, MAbs, 2, 428-439.
148.Scallon, B., Cai, A., Solowski, N., Rosenberg,
A., Song, X. Y., Shealy, D., and Wagner, C. (2002) Binding and
functional comparisons of two types of tumor necrosis factor
antagonists, J. Pharmacol. Exp. Ther., 301, 418-426.
149.Nesbitt, A., Fossati, G., Bergin, M., Stephens,
P., Stephens, S., Foulkes, R., Brown, D., Robinson, M., and Bourne, T.
(2007) Mechanism of action of certolizumab pegol (CDP870): in
vitro comparison with other anti-tumor necrosis factor alpha
agents, Inflamm. Bowel Dis., 13, 1323-1332.
150.Vos, A. C., Wildenberg, M. E., Arijs, I.,
Duijvestein, M., Verhaar, A. P., De Hertogh, G., Vermeire, S.,
Rutgeerts, P., Van den Brink, G. R., and Hommes, D. W. (2012)
Regulatory macrophages induced by infliximab are involved in healing
in vivo and in vitro, Inflamm. Bowel Dis.,
18, 401-408.
151.Perrier, C., De Hertogh, G., Cremer, J.,
Vermeire, S., Rutgeerts, P., Van Assche, G., Szymkowski, D. E., and
Ceuppens, J. L. (2013) Neutralization of membrane TNF, but not soluble
TNF, is crucial for the treatment of experimental colitis, Inflamm.
Bowel Dis., 19, 246-253.
152.Oikonomopoulos, A., Van Deen, W. K., and
Hommes, D. W. (2013) Anti-TNF antibodies in inflammatory bowel disease:
do we finally know how it works? Curr. Drug Targets, 14,
1421-1432.
153.Sandborn, W. J., Hanauer, S. B., Katz, S.,
Safdi, M., Wolf, D. G., Baerg, R. D., Tremaine, W. J., Johnson, T.,
Diehl, N. N., and Zinsmeister, A. R. (2001) Etanercept for active
Crohn’s disease: a randomized, double-blind, placebo-controlled
trial, Gastroenterology, 121, 1088-1094.
154.Van den Brande, J. M., Braat, H., Van den
Brink, G. R., Versteeg, H. H., Bauer, C. A., Hoedemaeker, I., Van
Montfrans, C., Hommes, D. W., Peppelenbosch, M. P., and Van Deventer,
S. J. (2003) Infliximab but not etanercept induces apoptosis in lamina
propria T-lymphocytes from patients with Crohn’s disease,
Gastroenterology, 124, 1774-1785.
155.Mocci, G., Marzo, M., Papa, A., Armuzzi, A.,
and Guidi, L. (2013) Dermatological adverse reactions during anti-TNF
treatments: focus on inflammatory bowel disease, J. Crohn’s
Colitis, 7, 769-779.
156.Targownik, L. E., and Bernstein, C. N. (2013)
Infectious and malignant complications of TNF inhibitor therapy in IBD,
Am. J. Gastroenterol., 108, 1835-1842, quiz 1843.
157.Cleynen, I., and Vermeire, S. (2012)
Paradoxical inflammation induced by anti-TNF agents in patients with
IBD, Nat. Rev. Gastroenterol. Hepatol., 9, 496-503.
158.Wiens, A., Venson, R., Correr, C. J., Otuki, M.
F., and Pontarolo, R. (2010) Meta-analysis of the efficacy and safety
of adalimumab, etanercept, and infliximab for the treatment of
rheumatoid arthritis, Pharmacotherapy, 30, 339-353.
159.Heldmann, F., Brandt, J., Van der
Horst-Bruinsma, I. E., Landewe, R., Sieper, J., Burmester, G. R., Van
den Bosch, F., De Vlam, K., Geusens, P., Gaston, H., Schewe, S.,
Appelboom, T., Emery, P., Dougados, M., Leirisalo-Repo, M., Breban, M.,
Listing, J., and Braun, J. (2011) The European ankylosing spondylitis
infliximab cohort (EASIC): a European multicentre study of long term
outcomes in patients with ankylosing spondylitis treated with
infliximab, Clin. Exp. Rheumatol., 29, 672-680.
160.Tzu, J., and Kerdel, F. (2008) From
conventional to cutting edge: the new era of biologics in treatment of
psoriasis, Dermatol. Ther., 21, 131-141.
161.Angelucci, E., Cocco, A., Viscido, A., Vernia,
P., and Caprilli, R. (2007) Another paradox in Crohn’s disease:
new onset of psoriasis in a patient receiving tumor necrosis
factor-alpha antagonist, Inflamm. Bowel Dis., 13,
1059-1061.
162.Cullen, G., Kroshinsky, D., Cheifetz, A. S.,
and Korzenik, J. R. (2011) Psoriasis associated with anti-tumour
necrosis factor therapy in inflammatory bowel disease: a new series and
a review of 120 cases from the literature, Aliment. Pharmacol.
Ther., 34, 1318-1327.
163.Palucka, A. K., Blanck, J. P., Bennett, L.,
Pascual, V., and Banchereau, J. (2005) Cross-regulation of TNF and
IFN-alpha in autoimmune diseases, Proc. Natl. Acad. Sci. USA,
102, 3372-3377.
164.Williams, V. L., and Cohen, P. R. (2011) TNF
alpha antagonist-induced lupus-like syndrome: report and review of the
literature with implications for treatment with alternative TNF alpha
antagonists, Int. J. Dermatol., 50, 619-625.
165.Bout-Tabaku, S., Rivas-Chacon, R., and
Restrepo, R. (2007) Systemic lupus erythematosus in a patient treated
with etanercept for polyarticular juvenile rheumatoid arthritis, J.
Rheumatol., 34, 2503-2504.
166.Ferraccioli, G., Mecchia, F., Di Poi, E., and
Fabris, M. (2002) Anticardiolipin antibodies in rheumatoid patients
treated with etanercept or conventional combination therapy: direct and
indirect evidence for a possible association with infections, Ann.
Rheum. Dis., 61, 358-361.
167.Via, C. S., Shustov, A., Rus, V., Lang, T.,
Nguyen, P., and Finkelman, F. D. (2001) In vivo neutralization
of TNF-alpha promotes humoral autoimmunity by preventing the induction
of CTL, J. Immunol., 167, 6821-6826.
168.Fidder, H., Schnitzler, F., Ferrante, M.,
Noman, M., Katsanos, K., Segaert, S., Henckaerts, L., Van Assche, G.,
Vermeire, S., and Rutgeerts, P. (2009) Long-term safety of infliximab
for the treatment of inflammatory bowel disease: a single-centre cohort
study, Gut, 58, 501-508.
169.Hochman, D., and Wolff, B. (2006) Risk of
serious infections and malignancies with anti-TNF antibody therapy in
rheumatoid arthritis, JAMA, 296, 2203; author reply
2203-2204.
170.Stern, R. S., Liebman, E. J., and Vakeva, L.
(1998) Oral psoralen and ultraviolet-A light (PUVA) treatment of
psoriasis and persistent risk of nonmelanoma skin cancer. PUVA
Follow-up Study, J. Natl. Cancer Inst., 90,
1278-1284.
171.Peyrin-Biroulet, L., Khosrotehrani, K., Carrat,
F., Bouvier, A. M., Chevaux, J. B., Simon, T., Carbonnel, F., Colombel,
J. F., Dupas, J. L., Godeberge, P., Hugot, J. P., Lemann, M., Nahon,
S., Sabate, J. M., Tucat, G., and Beaugerie, L. (2011) Increased risk
for nonmelanoma skin cancers in patients who receive thiopurines for
inflammatory bowel disease, Gastroenterology, 141,
1621-1628.
172.Steenholdt, C., Svenson, M., Bendtzen, K.,
Thomsen, O. O., Brynskov, J., and Ainsworth, M. A. (2011) Severe
infusion reactions to infliximab: aetiology, immunogenicity and risk
factors in patients with inflammatory bowel disease, Aliment.
Pharmacol. Ther., 34, 51-58.
173.Slifman, N. R., Gershon, S. K., Lee, J. H.,
Edwards, E. T., and Braun, M. M. (2003) Listeria monocytogenes
infection as a complication of treatment with tumor necrosis factor
alpha-neutralizing agents, Arthritis. Rheum., 48,
319-324.
174.Peyrin-Biroulet, L., Deltenre, P., De Suray,
N., Branche, J., Sandborn, W. J., and Colombel, J. F. (2008) Efficacy
and safety of tumor necrosis factor antagonists in Crohn’s
disease: meta-analysis of placebo-controlled trials, Clin.
Gastroenterol. Hepatol., 6, 644-653.
175.Shebzukhov, Y. V., Kuchmiy, A. A., Kruglov, A.
A., Zipp, F., Siffrin, V., and Nedospasov, S. A. (2014) Experimental
applications of TNF-reporter mice with far-red fluorescent label,
Methods Mol. Biol., 1155, 151-162.
176.Lowenberg, M., and D’Haens, G. (2015)
Next-generation therapeutics for IBD, Curr. Gastroenterol. Rep.,
17, 21.
177.Gomez-Gomez, G. J., Masedo, A., Yela, C.,
Martinez-Montiel Mdel, P., and Casis, B. (2015) Current stage in
inflammatory bowel disease: what is next? World J.
Gastroenterol., 21, 11282-11303.