REVIEW: Myeloid-Derived Suppressor Cells and Proinflammatory Cytokines as Targets for Cancer Therapy
K.-S. N. Atretkhany1,2 and M. S. Drutskaya1,2*
1Engelhardt Institute of Molecular Biology, 119991 Moscow, Russia; E-mail: marinadru@gmail.com2Lomonosov Moscow State University, Biological Faculty, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received July 5, 2016; Revision received July 9, 2016
Myeloid-derived suppressor cells represent a heterogeneous population of immature myeloid cells. Under normal conditions, these cells differentiate into macrophages, dendritic cells, and granulocytes. However, in pathological states such as inflammation, infection, or tumor growth, there is an arrest of their differentiation that results in the accumulation of immature myeloid cells in the organism. In addition, these cells acquire a suppressor phenotype, expressing anti-inflammatory cytokines and reactive oxygen and nitrogen species, and suppress T-cell immune response. Myeloid-derived suppressor cells (MDSC) contribute to cancerogenesis by forming a favorable microenvironment for tumor growth. Proinflammatory cytokines, secreted by tumor cells and the tumor microenvironment, induce angiogenesis and metastasis and promote tumor growth. They also provide signals necessary for survival, accumulation, and function of MDSC. Understanding the mechanisms of myeloid suppressor cell development and the use of proinflammatory cytokine inhibitors may prove beneficial for tumor therapy.
KEY WORDS: MDSC, TNF, IL-6, IL-1, tumor microenvironment, tumor-associated inflammationDOI: 10.1134/S0006297916110055
Abbreviations: Arg1, arginase 1; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; IDO, indolamine-2,3-dioxygenase; IL, interleukin; iNOS, inducible NO-synthase; M-CSF, monocyte-macrophage colony-stimulating factor; MDSC, myeloid-derived suppressor cells; ROS, reactive oxygen species; sTNF, soluble form of TNF; TCR, T-cell receptor; TGF-β, transforming growth factor beta; tmTNF, transmembrane TNF; TNF, tumor necrosis factor; TNFR, tumor necrosis factor receptor; Treg, regulatory T cells; VEGF, vascular endothelial growth factor.
Chronic inflammation is accompanied by increased expression of
proinflammatory cytokines that modulate immune response, chemokines
that attract immune cells to the inflammatory site, and growth factors
necessary for the local development and differentiation of immune
cells. Inflammation that contributes to tumor growth may be caused by
chronic infections and autoimmune processes, and also be provoked by
environmental factors or the use of chemotherapy. Furthermore, local
chronic inflammation that promotes tumor growth can be induced by the
cancer cells themselves [1]. When dysregulation of
the immune response and the induction of chronic inflammation occur,
activation of signaling cascades and transcription factors can give a
selective advantage to cancer cells. It is known that such
transcriptional programs can lead to induction of angiogenesis,
degradation of extracellular matrix proteins, tumor niche remodeling
and tumor cell proliferation through regulation of genes involved in
these processes. Active forms of nitrogen and oxygen that are produced
during inflammation may not only suppress the antitumor immune
response, but also promote genomic instability, such as mutations or
epigenetic changes. Furthermore, during inflammation signaling cascades
responsible for antiapoptotic protein expression are activated, which
provides resistance for cancer cells to apoptosis [2]. Finally, there is increasing evidence that chronic
inflammation is often accompanied by the accumulation of various
myeloid cells, which also may contribute to tumor growth [3].
MYELOID-DERIVED SUPPRESSOR CELLS
Myeloid-derived suppressor cells (MDSC) represent a heterogeneous population of immature neutrophils, monocytes, and dendritic cells capable of suppressing the immune response, including immune response against tumors [4]. MDSC are characterized by co-expression of myeloid surface markers CD11b and GR-1. Antibodies against GR-1 usually recognize one epitope on two structurally similar molecules – Ly6G and Ly6C. Using antibodies specific to these individual markers, two subpopulations of MDSC were identified: granulocyte fraction CD11b+Ly6G+Ly6Clow, characterized by high production of reactive oxygen species (ROS) and a low level of nitric oxide (NO), and monocyte fraction CD11b+Ly6G–Ly6Chi, producing large amounts of NO [5]. Both populations express arginase (Arg1). In humans, MDSC are characterized by LIN–HLA–DR–CD33+CD11b+ expression profile, but they may also express additional markers depending of the type of cancer. Thus, it was shown that the monocyte fraction expresses CD14, and the granulocytic fraction expresses CD15 [6]. The accumulation of MDSC was first discovered in patients with tumors, but later expansion of this myeloid cell population with suppressor functions was also reported in several experimental models of carcinogenesis in mice [7-9].
Accumulation and composition of MDSC is mainly influenced by the tumor microenvironment (Fig. 1). Cells of the tumor microenvironment secrete various factors that can be divided into two groups according to their functions [10]. The first group of factors, such as granulocyte-macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF), monocyte-macrophage colony-stimulating factor (M-CSF), vascular endothelial growth factor (VEGF), and IL-13 is responsible for myelopoiesis and suppression of differentiation of immature myeloid cells into terminally differentiated cells. The second group includes various proinflammatory factors such as TNF, IL-1β, IL-6, S100A8, and S100A9 secreted by the tumor cells, as well as IFN-γ, IL-4, IL-10, and IL-13 secreted by tumor-associated T-cells that affect differentiation of myeloid progenitors and promote suppressor activity of MDSC [3]. In addition, various important chemokines, such as CCL2, CXCL12, CXCL15, are necessary for attraction of myeloid cells to the tumor site [11-13].
Fig. 1. Chronic inflammation, MDSC, and cancer. Expansion of MDSC mediated by tumor microenvironment. The tumor microenvironment produces proinflammatory cytokines and growth factors that affect immature myeloid cells (IMC) formed from a common myeloid progenitor (CMP). Pro- and anti-inflammatory cytokines and growth factors such as IL-1β, IL-6, IL-10, G-SCF, M-CSF, and VEGF lead to activation of STAT3 that promotes the survival and proliferation of the cells through upregulation of antiapoptotic and cell cycle proteins and also drives the expression of S100A8 and S100A9 antimicrobial peptides [30, 31]. The transcription factor STAT1, activated by IFN-γ and IL-1β, is involved in the increase of iNOS and Arg1 gene expression [32], and another transcription factor of this family – STAT6 – activates the expression NOX2 and Arg1 [33]. STAT5 and STAT3, activated by GM-CSF, enhance the expression of antiapoptotic proteins [34] and IDO [18], respectively.
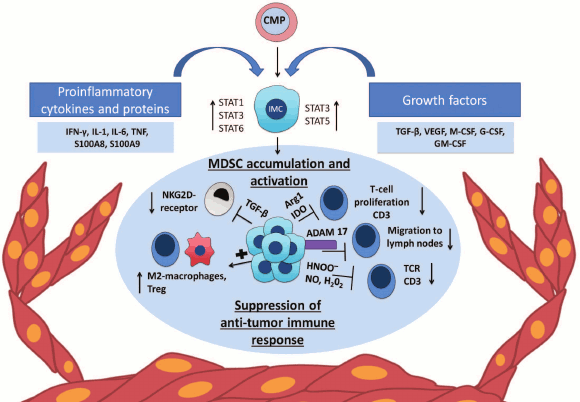
FUNCTIONS OF MYELOID SUPPRESSOR CELLS
MDSC suppress immune response through cell-to-cell interactions via molecules synthesized on the cell surface, as well as via short-lived mediators (Fig. 1) [14]. First, MDSC affect the metabolism of arginine and tryptophan, which leads to inhibition of T-cell antitumor immune response. The enzyme iNOS (inducible Nitric Oxide Synthase) converts arginine into citrulline and NO, while Arg1 converts it into ornithine and urea. Thus, elevated concentrations of these enzymes lead to a shortage of arginine at the site of inflammation [3, 15]. In turn, lack of arginine decreases T-cell proliferation and suppresses the expression of CD3ζ-chains of T-cell receptor (TCR) [16]. A similar mechanism of MDSC-mediated T-cell suppression is STAT3-dependent activation of indolamine-2,3-dioxygenase (IDO), which leads to tryptophan deficiency [17, 18]. It is remarkable that IDO, produced by tumor cells, contributes to attraction and activation of MDSC with participation of regulatory T-cells (Treg) [19]. Second, MDSC produce reactive oxygen and nitrogen species by NADPH oxidase and iNOS that supports the inflammatory microenvironment. Furthermore, peroxynitrite, which is formed by chemical reaction between superoxide anion-radical and nitric oxide [20], is involved in the nitration and nitrosylation of various amino acids that results in their decrease at the site of inflammation. Moreover, peroxynitrite modifies TCR-complex components, i.e. directly alters its structure, and thereby affects the interaction of T-cell receptor with the MHC (major histocompatibility complex) molecules, which ultimately leads to inhibition of specific T-cell response to tumor antigens [21]. ROS inhibit expression of CD3ζ-chains of TCR and various cytokines [22]. It was also shown that NO blocks signaling from IL-2 and inhibits T-cell proliferation [23].
In addition, MDSC can affect the migration of T-cells. MDSC express ADAM17 on their surface, which due to its protease activity is able to cleave the external domain of CD62L. CD62L is an important adhesion molecule that allows naïve lymphocytes to locate to the lymph nodes, where they may differentiate into activated T-cells in the presence of antigens. In addition, CD62L retains naive lymphocytes at the site of inflammation, e.g. in the tumor microenvironment, where their activation also occurs [24]. Thus, by removing CD62L, MDSC inhibit activation of naive T-cells and prevent their relocation to the site of inflammation [25]. MDSC produce reactive nitrogen species, leading to nitrosylation of CCL2 chemokine, modification of which prevents the infiltration of CD8+ T-cells in tumor tissues [26]. In addition, MDSC express galectin-9 (Gal-9) on their surface, which is a ligand for TIM-3. TIM-3, in turn, is expressed on IFN-γ-producing CD4+ and CD8+ T-cells and ensures their negative regulation. The interaction of TIM-3 with Gal-9 results in the death of T-cells and the expansion of myeloid suppressor cells [27]. Finally, MDSC inhibit NK-cell functions. In experimental models of carcinogenesis, it has been shown that inhibition of NK-cell functions correlates with expansion of MDSC. It was shown that MDSC reduce NK-cell cytotoxicity, IFN-γ production, as well as expression of NKG2D through the membrane-bound TGF-β1 [28].
Another mechanism of tumor development is associated with the induction and expansion of Treg. In several animal models, it has been shown that Treg can be activated by MDSC both in vitro and in vivo. MDSC, stimulated with IFN-γ, express large amounts of IL-10 and TGF-β, which are important for the induction and activation of Treg. Later, the same authors have shown that IFN-γ regulates CD40 expression on MDSC and thus affects intercellular interactions between CD40+ MDSC and CD40L+ T-cells in the presence of IL-10 and TGF-β, which leads to the expansion and activation of Treg [29].
Thus, innate and adaptive immune cells closely interact with tumor cells and form tumor microenvironment that may contribute to tumor progression. Myeloid cells are an important component of the tumor microenvironment. They may support tumor survival, suppress antitumor immune response, and lead to angiogenesis, metastasis, and further expansion of the tumor niche through induction of various signaling pathways (Fig. 1).
ROLE OF INFLAMMATION IN CANCER
Inflammation is one of the hallmarks of cancer [35] and is mediated by the complex work of proinflammatory cytokines, chemokines, and growth factors [1]. One of the most studied proinflammatory cytokines is tumor necrosis factor (TNF). It is a multifunctional cytokine whose physiological effects are associated with immunoregulation, protection from infections, and control of inflammation [36]. The role of TNF in the development of tumors is controversial. On one hand, TNF was discovered due to its antitumor activity [37]. However, there is evidence that TNF also promotes tumor development. For example, TNF-deficient ovarian cancer cell line was characterized as less invasive, with high levels of cell death and decreased expression of proinflammatory mediators such as VEGF, IL-6, and CCL-2 [38]. TNF deficient mice were resistant to the development of papillomas and squamous cell carcinoma in an experimental model of chemically induced skin cancer [39]. Mice deficient in TNFRI, exposed to azoxymethane and sodium dextran sulfate, had less severe symptoms of colon neoplasia – reduced number of macrophages and neutrophils and less damage of the intestinal mucosa [40]. In another study, the role of TNF was shown in a model of hepatocellular carcinoma, where inhibition of this cytokine resulted in suppression of tumor growth [41]. TNF is expressed as a homotrimer and exists in soluble and membrane-bound forms. TACE enzyme (TNF-α converting enzyme, also known as ADAM17) converts transmembrane TNF (tmTNF) into the soluble form (sTNF) by proteolytic cleavage [42]. When TNF interacts with TNFRI, the signaling part of the receptor recruits molecules that lead to different signals: apoptosis, necrosis, proliferation, activation of NF-κB [43]. Such complexity in TNF signaling most likely can be explained by the presence of several receptors that differ from each other by the expression pattern and the type of transmitted signals, ability to transmit these signals in both soluble and membrane-bound forms, as well as the presence of another ligand – lymphotoxin α binding to the same receptors [43]. Thus, expression of numerous genes is activated by NF-κB including IL-6, IL-8, IL-18, chemokines, cyclooxygenases, and other mediators of inflammation. Interaction of TNFRII with tmTNF also induces the signaling cascades resulting in activation of NF-κB [44]. Induction of this transcription factor leads to the development of tumors by stimulating the expression of proinflammatory cytokines and adhesion molecules. Furthermore, NF-κB is essential for cell proliferation and activation of cell cycle factors such as Cyclin-D1 and c-Myc, and antiapoptotic proteins – c-FLIP, BCL-2, and cIAPs. The interaction of tmTNF, expressed on tumor cells, with receptors leads to reverse signal transmission that ensures the survival of tumor cells by constitutive expression of NF-κB, whereas the immune cells, when interacting with tmTNF on the surface of tumor cells, obtain cell death signal through TNFRI [45].
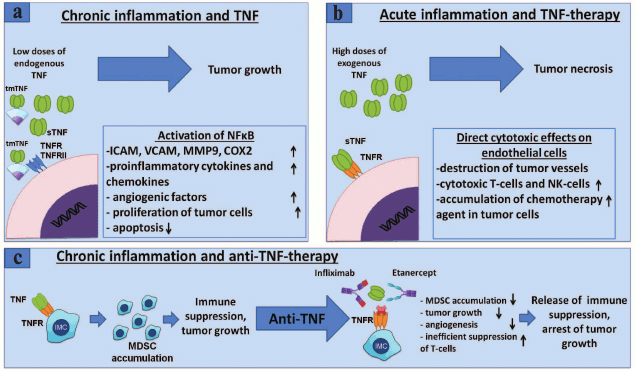
Thus, local effects of endogenous TNF produced by tumor cells or cells of the tumor microenvironment lead to survival and proliferation of cancer cells by NF-κB activation, whereas antitumor effect of exogenous TNF is caused by activation of cell death that occurs not because of TNF direct action on tumor cells, but due to its effect on blood vessels, which in turn are parts of a tumor microenvironment and are necessary for tumor growth [46]. Thus, the role of TNF in tumor development is ambiguous and depends largely on the surrounding inflammatory context, source of cells that produce TNF, its form (soluble or membrane-bound), and cells with which it interacts (Fig. 2).
Fig. 2. Role of TNF in cancer and MDSC development – anti-TNF-therapy. a) TNF promotes tumor growth. Binding of TNF to its receptors on endothelial cells or fibroblasts leads to NF-κB activation with subsequent expression of chemokines, adhesion molecules, growth factors, and proinflammatory cytokines that promote angiogenesis, inflammation, and recruitment of immune cells to the site of inflammation [81]. TNF through the same receptors activates tumor cell proliferation and induction of VEGF and metalloproteinase required for tumor angiogenesis and tumor niche remodeling [82]. b) Antitumor effect of TNF. High doses of exogenous TNF lead to tumor necrosis [83]. However, this effect is not caused by a direct cytotoxic effect on tumor cells, but rather on the endothelium of blood vessels, which results in the destruction of blood vessels associated with the tumor, and tumor regression [84]. In clinical tests, it has been shown that TNF monotherapy may be ineffective, but in conjunction with chemotherapy, local TNF concentration increases due to increased permeability of blood vessels and is used in the treatment of melanomas and sarcomas in isolated limb perfusion technique [85]. c) TNF and other proinflammatory cytokines secreted by tumor microenvironment lead to chronic inflammation associated with the tumor [2]. In chronic inflammation, immature myeloid cells acquire immunosuppressive phenotype. It has been shown that TNF is required for the development and activation of myeloid suppressor cells [64, 86, 87]. Neutralization of TNF with etanercept or infliximab results in a decrease of the tumor growth and MDSC accumulation, suppression of the immune response of T-cells, as well as a possible decrease in metastasis.
IL-6 is a proinflammatory cytokine with a wide range of immunoregulatory properties. It binds to the soluble or transmembrane form of the IL-6R receptor and through interaction with gp130 subunit activates JAK/STAT, MAPK, and PI3K signaling pathways. An important transcription factor that is activated by the interaction of IL-6 with its receptor is STAT3. This transcription factor contributes to the activation of genes involved in control of cell division and apoptosis, such as Bcl-2, Bcl-xL, Mcl-1, Cyclin D, c-Myc, HSP70, and survivin, which results in proliferation and survival of tumor cells and cells of the tumor microenvironment [47, 48]. The tumor microenvironment cells, such as macrophages, dendritic cells, T cells, and epithelial cells, produce IL-6 and contribute to cancer development [49]. Furthermore, tumor cells themselves are capable of producing IL-6, stimulating autocrine STAT3 activation and proliferation of tumor cells [50]. Elevated level of IL-6 is associated with cancers such as lymphomas and breast, lung, and stomach cancer. In addition, the IL6/JAK/STAT3 signaling pathway has been implicated in the development of colorectal cancer [49, 51]. Finally, IL-6 promotes angiogenesis by increasing the expression of VEGF, an important vascular endothelial growth factor, necessary for the growth of solid tumors [52].
The role of IL-1 in cancer development, as well as that of TNF, is not completely understood. On one hand, IL-1 is involved in the development of malignant neoplasms and metastasis via MyD88-dependent activation of NF-κB [53]. However, it activates effector mechanisms that are involved in the elimination of tumors [54]. Overexpression of IL-1 is associated with a variety of cancers, such as breast, lung, and colon cancer [55, 56]. IL-1α and IL-1β are products of distinct genes, with different processing and secretion; however, they have similar biological activity due to binding to the same receptor and MyD88-dependent activation of NF-κB [57, 58]. Voronov et al. [58] showed that solid tumors did not develop in IL-1α/IL-1β knockout mice, whereas wild-type mice died because of metastasis to the lungs. In addition, IL-1 may induce expression of matrix metalloproteinase 1 (MMP1) in bone marrow stromal cells in melanoma patients, and increase MMP1 activity which leads to tumor niche remodeling and metastasis. IL-1 induces the production of angiogenic growth factors and proteins in cell lines of colorectal cancer [59]. In a Myc-dependent mouse model of β-cell pancreatic islet carcinogenesis, activation of Myc induces production of IL-1β, which contributes to angiogenesis [60]. The tumor microenvironment secretes IL-1, which interacts with IL-1R on tumor cells leading to activation of angiogenic factors and adhesion molecules and promotes tumor invasion and metastasis [58, 61].
ROLE OF INFLAMMATORY CYTOKINES IN DEVELOPMENT OF MDSC
Proinflammatory cytokines secreted by tumor and tumor microenvironment inhibit terminal differentiation of myeloid cells and promote the expansion and activation of MDSC. Recent studies have shown that TNF is important for the development and activation of MDSC. For example, Zhao et al. found that transplanted tumor cell lines are often rejected in TNF-deficient mice, and tumor regression correlates with delayed dynamics of MDSC accumulation [62]. Reduction in the number of MDSC was due to their impaired survival through downregulation of antiapoptotic protein c-FLIP in the absence of TNF signaling. Furthermore, in TNF receptor deficient mice, it was shown that TNFRII, rather than TNFRI, is important for MDSC survival by activation of antiapoptotic protein expression [62]. Another group of researchers also confirmed the importance of the signaling cascade mediated by TNFRII in MDSC regulation. Thus, MDSC derived from mice deficient in TNFRII are characterized by reduced production of NO required for MDSC suppressor activity, and IL-6, which activates STAT3 through interaction with gp130 [63].
Another group of investigators demonstrated that TNF inhibits differentiation and enhances suppressive properties of MDSC in an experimental model of chronic inflammation. In this model, mice deficient in TNF were injected with BCG bacteria killed by heat, thus inducing chronic inflammation. After that, accumulation of myeloid cells was evaluated and their suppressive properties in the absence of TNF were studied. It was found that in spite of the increased expression of certain inflammatory cytokines, mice with complete inactivation of TNF were characterized by significant reduction in MDSC in the spleen. In addition, genetic inactivation of TNF was associated with a reduction in MDSC suppressor activity [64]. TNF-deficient MDSC did not inhibit T-cell response and were characterized by reduced expression of s100A8, s100A9, and their receptor RAGE, which is necessary for the activation and expansion of MDSC. This work showed that TNF inhibits further differentiation of immature myeloid cells in vitro, while in the absence of TNF their differentiation toward CD11c+ and F4/80+ cells occurred, which prevented MDSC accumulation.
Interaction of IL-1 with IL-1R leads to the activation of NF-κB via the MyD88-dependent pathway. In addition, NF-κB regulates the expression of suppressor factors IL-10 and Arg1, as well as expression of antiapoptotic proteins necessary for the survival of MDSC [15]. In 4T1 tumor model, overexpressing IL-1β NK-cells are functionally inhibited. Overexpression of IL-1β leads to expansion of granulocytic MDSC subpopulation, phenotypically characterized as CD11b+Ly6G+Ly6C–, which inhibits the expression of NKG2D receptor on NK-cells [65]. Overexpression of IL-1β by gastric parietal cells contributes to spontaneous development of stomach inflammation and cancer and is characterized by NF-κB-dependent activation of MDSC [53].
Overexpression of IL-6 is also typical for malignant neoplasms. In an experimental model of esophageal cancer in mice and in patients with esophagus squamous cell carcinoma, a positive correlation of IL-6 expression and the accumulation of MDSC was found. In vitro culturing of blood leukocytes with IL-6 leads to expansion of MDSC with distinctive for this cell population elevated arginase 1 and ROS levels. It is known that IL-6 leads to phosphorylation of STAT3, which is an important transcriptional factor for activation of genes responsible for differentiation and MDSC engraftment to the site of inflammation [66]. In an experimental model of breast cancer, a role of IL-6 in recruitment of MDSC has also been shown. In addition, MDSC themselves expressed IL-6 and increased the concentration of soluble IL-6R through the regulation of ADAM17 protease [67]. Collectively, proinflammatory cytokines regulate MDSC functions by activating downstream signaling pathways: JAK/STAT, NF-κB, COX-2, and PGE2 [31].
Thus, proinflammatory cytokines are important factors in the development and functioning of MDSC and tumor microenvironment. Based on these results, we can suggest that blocking of TNF, IL-1, and IL-6 for possible MDSC inhibition may be an important adjunct to cancer therapy.
ANTI-CYTOKINE THERAPY AND MDSC
Systemic anti-cytokine therapy is widely used in the treatment of various autoimmune diseases [68]. Currently, five blockers of TNF are approved for clinical use: infliximab, adalimumab, golimumab, certolizumab, and etanercept. These blockers have differences in structure, mechanism of inhibition and the effective dosage. They bind to and neutralize sTNF with comparable efficiency, although it was previously shown that etanercept blocks tmTNF less effectively [69]. Furthermore, etanercept binds the soluble form of lymphotoxin, thereby systemically inhibiting not only TNF-mediated functions, but also functions of soluble LTα. Not surprisingly, because of their structural differences, these drugs are used with varying effectiveness in the treatment of autoimmune diseases [70, 71]. The use of TNF inhibitors may have clinical significance in tumor therapy. For example, in experiments with mice, humanized for the TNF gene [72], it was found that anti-cytokine therapy with etanercept or infliximab reduces the growth of transplantable tumors. Furthermore, the use of TNF inhibitors was associated with reduced accumulation of MDSC and their decreased suppressive activity [73] (Fig. 2). In an adenocarcinoma model, it was shown that neutralization of TNF by etanercept or infliximab reduces tumor growth and metastasis [74]. Finally, in clinical trials, 60% of patients with kidney carcinoma showed prolonged stabilization of the disease during anti-TNF-therapy [75, 76].
The use of IL-6/IL-6R blockers was also investigated in several tumor models and in clinical trials. It was shown that monoclonal antibodies to IL-6R suppress tumor growth and MDSC accumulation at the tumor site [77]. Siltuksimab – an inhibitor of human IL-6 – has been approved for the treatment of lymphoproliferative syndrome [78]. Finally, clinical trial of IL-1 blockers demonstrated their effectiveness in patients with colorectal cancer and non-small cell lung cancer [79]. Therapy with anakinra, an antagonist of IL-1 receptor, in combination with dexamethasone in multiple myeloma patients also led to a stabilization of the disease [80].
The main problem, however, with anti-cytokine therapy are its systemic effects, because such drugs nonspecifically inhibit cytokines that perform both physiological and pathological functions. It is not surprising that systemic anti-cytokine therapy has several side effects. Now increasing attention is given to development of more specific inhibitors of proinflammatory cytokines for clinical use. In particular, a potential target for the combined treatment of tumors and autoimmune diseases, for which a pathological role of MDSC was shown, can be anti-cytokine cell-targeted therapy with bispecific antibodies, which inhibit proinflammatory cytokines in a particular cellular source. Therefore, the study of the effect of blocking TNF, IL-1, and IL-6 in the development of MDSC may be clinically relevant and important for understanding of molecular mechanisms of their regulation.
During the development of malignant tumors, there is an imbalance between the signals of cell death and survival, proliferation and termination of division, increased production of proinflammatory cytokines and chemokines, that recruits various immune cells, which in turn are suppressed by the tumor microenvironment and become part of it, and contribute to further growth of the tumor. There is a serious rearrangement of metabolism in tumor cells, which allows cells to proliferate and survive. Furthermore, immature myeloid cells rapidly accumulate, become immunosuppressive and inhibit antitumor immune response during tumor growth and inflammation. These immature cells become part of the tumor microenvironment together with tumor-associated fibroblasts, macrophages, neutrophils, and dendritic cells that actively infiltrate the tumor and protect it from the action of the immune system.
Various approaches to cancer therapy aimed at regulation of metabolism, inhibition of growth factors, activation of antitumor immune response, inhibition of cell proliferation or changing the epigenetic status of cells are now being developed. Proinflammatory cytokines provide signals for MDSC survival, differentiation and maintenance. Understanding the mechanisms of myeloid suppressor cell development and the use of proinflammatory cytokine inhibitors may be beneficial for tumor therapy.
For example, the use of specific inhibitors aimed at suppression of cytokine production by a specific cell type may be more advantageous than systemic therapy and may have less pronounced side effects on the body.
Acknowledgements
The authors express their special appreciation to Dr. R. I. Ataullakhanov and Dr. S. A. Nedospasov for criticism and discussion of the project, to V. S. Gogoleva for technical assistance, and to I. Sivova for help with translation.
The work was supported by the Russian Science Foundation (project No. 14-25-00160).
REFERENCES
1.Grivennikov, S. I., Greten, F. R., and Karin, M.
(2010) Immunity, inflammation, and cancer, Cell, 140,
883-899.
2.Colotta, F., Allavena, P., Sica, A., Garlanda, C.,
and Mantovani, A. (2009) Cancer-related inflammation, the seventh
hallmark of cancer: links to genetic instability,
Carcinogenesis, 30, 1073-1081.
3.Gabrilovich, D. I., Ostrand-Rosenberg, S., and
Bronte, V. (2012) Coordinated regulation of myeloid cells by tumours,
Nat. Rev. Immunol., 12, 253-268.
4.Ostrand-Rosenberg, S., and Sinha, P. (2009)
Myeloid-derived suppressor cells: linking inflammation and cancer,
J. Immunol., 182, 4499-4506.
5.Ponomarev, A. V. (2016) Myeloid supressor cells:
general properties, Immunologiya, 37, 47-50.
6.Trikha, P., and Carson, W. E., 3rd. (2014)
Signaling pathways involved in MDSC regulation, Biochim. Biophys.
Acta, 1846, 55-65.
7.Centuori, S. M., Trad, M., LaCasse, C. J.,
Alizadeh, D., Larmonier, C. B., Hanke, N. T., Kartchner, J.,
Janikashvili, N., Bonnotte, B., Larmonier, N., and Katsanis, E. (2012)
Myeloid-derived suppressor cells from tumor-bearing mice impair
TGF-β-induced differentiation of
CD4+CD25+FoxP3+ Tregs from
CD4+CD25–FoxP3– T-cells,
J. Leukoc. Biol., 92, 987-997.
8.Younos, I. H., Dafferner, A. J., Gulen, D.,
Britton, H. C., and Talmadge, J. E. (2012) Tumor regulation of
myeloid-derived suppressor cell proliferation and trafficking, Int.
Immunopharmacol., 13, 245-256.
9.Wang, L., Chang, E. W., Wong, S. C., Ong, S. M.,
Chong, D. Q., and Ling, K. L. (2013) Increased myeloid-derived
suppressor cells in gastric cancer correlate with cancer stage and
plasma S100A8/A9 proinflammatory proteins, J. Immunol.,
190, 794-804.
10.Gabrilovich, D. I., and Nagaraj, S. (2009)
Myeloid-derived-suppressor cells as regulators of the immune system,
Nat. Rev. Immunol., 9, 162-174.
11.Qian, B. Z., Li, J., Zhang, H., Kitamura, T.,
Zhang, J., Campion, L. R., Kaiser, E. A., Snyder, L. A., and Pollard,
J. W. (2011) CCL2 recruits inflammatory monocytes to facilitate
breast-tumour metastasis, Nature, 475, 222-225.
12.Allavena, P., Sica, A., Solinas, G., Porta, C.,
and Mantovani, A. (2008) The inflammatory micro-environment in tumor
progression: the role of tumor-associated macrophages, Crit. Rev.
Oncol. Hematol., 66, 1-9.
13.Draghiciu, O., Lubbers, J., Nijman, H. W., and
Daemen, T. (2015) Myeloid derived suppressor cells – an overview
of combat strategies to increase immunotherapy efficacy,
Oncoimmunology, 4, e954829.
14.Kumar, V., Patel, S., Tcyganov, E., and
Gabrilovich, D. I. (2016) The nature of myeloid-derived suppressor
cells in the tumor microenvironment, Trends Immunol., 37,
208-220.
15.Martino, A., Badell, E., Abadie, V., Balloy, V.,
Chignard, M., Mistou, M.-Y., Combadiere, B., Combadiere, C., and
Winter, N. (2010) Mycobacterium bovis bacillus
Calmette–Guerin vaccination mobilizes innate myeloid-derived
suppressor cells restraining in vivo T-cell priming via
IL-1R-dependent nitric oxide production, J. Immunol.,
184, 2038-2047.
16.Rodriguez, P. C., Quiceno, D. G., and Ochoa, A.
C. (2007) L-arginine availability regulates T-lymphocyte cell-cycle
progression, Blood, 109, 1568-1573.
17.Medzhitov, R., Shevach, E. M., Trinchieri, G.,
Mellor, A. L., Munn, D. H., Gordon, S., Libby, P., Hansson, G. K.,
Shortman, K., Dong, C., Gabrilovich, D., Gabrysova, L., Howes, A., and
O’Garra, A. (2011) Highlights of 10 years of immunology in
Nature Reviews Immunology, Nat. Rev. Immunol., 11,
693-702.
18.Yu, J., Wang, Y., Yan, F., Zhang, P., Li, H.,
Zhao, H., Yan, C., Yan, F., and Ren, X. (2014) Noncanonical NF-κB
activation mediates STAT3-stimulated IDO upregulation in
myeloid-derived suppressor cells in breast cancer, J. Immunol.,
193, 2574-2586.
19.Holmgaard, R. B., Zamarin, D., Li, Y., Gasmi, B.,
Munn, D. H., Allison, J. P., Merghoub, T., and Wolchok, J. D. (2015)
Tumor-expressed IDO recruits and activates MDSCs in a Treg-dependent
manner, Cell Rep., 13, 412-424.
20.Pacher, P., Beckman, J. S., and Liaudet, L.
(2007) Nitric oxide and peroxynitrite in health and disease,
Physiol. Rev., 87, 315-424.
21.Nagaraj, S., Gupta, K., Pisarev, V., Kinarsky,
L., Sherman, S., Kang, L., Herber, D. L., Schneck, J., and Gabrilovich,
D. I. (2007) Altered recognition of antigen is a mechanism of
CD8+ T-cell tolerance in cancer, Nat. Med.,
13, 828-835.
22.Schmielau, J., and Finn, O. J. (2001) Activated
granulocytes and granulocyte-derived hydrogen peroxide are the
underlying mechanism of suppression of T-cell function in advanced
cancer patients, Cancer Res., 61, 4756-4760.
23.Mazzoni, A., Bronte, V., Visintin, A., Spitzer,
J. H., Apolloni, E., Serafini, P., Zanovello, P., and Segal, D. M.
(2002) Myeloid suppressor lines inhibit T-cell responses by an
no-dependent mechanism, J. Immunol., 168, 689-695.
24.Khan, A. I., Landis, R. C., and Malhotra, R.
(2003) L-Selectin ligands in lymphoid tissues and models of
inflammation, Inflammation, 27, 265-280.
25.Hanson, E. M., Clements, V. K., Sinha, P.,
Ilkovitch, D., and Ostrand-Rosenberg, S. (2009) Myeloid-derived
suppressor cells down-regulate L-selectin expression on CD4+
and CD8+ T-cells, J. Immunol., 183,
937-944.
26.Molon, B., Ugel, S., Del Pozzo, F., Soldani, C.,
Zilio, S., Avella, D., De Palma, A., Mauri, P., Monegal, A., Rescigno,
M., Savino, B., Colombo, P., Jonjic, N., Pecanic, S., Lazzarato, L.,
Fruttero, R., Gasco, A., Bronte, V., and Viola, A. (2011) Chemokine
nitration prevents intratumoral infiltration of antigen-specific
T-cells, J. Exp. Med., 208, 1949-1962.
27.Sakuishi, K., Jayaraman, P., Behar, S. M.,
Anderson, A. C., and Kuchroo, V. K. (2011) Emerging Tim-3 functions in
antimicrobial and tumor immunity, Trends Immunol., 32,
345-349.
28.Li, H., Han, Y., Guo, Q., Zhang, M., and Cao, X.
(2009) Cancer-expanded myeloid-derived suppressor cells induce anergy
of NK-cells through membrane-bound TGF-β1, J. Immunol.,
182, 240-249.
29.Pan, P. Y., Ma, G., Weber, K. J., Ozao-Choy, J.,
Wang, G., Yin, B., Divino, C. M., and Chen, S. H. (2010) Immune
stimulatory receptor CD40 is required for T-cell suppression and
T-regulatory cell activation mediated by myeloid-derived suppressor
cells in cancer, Cancer Res., 70, 99-108.
30.Cheng, P., Corzo, C. A., Luetteke, N., Yu, B.,
Nagaraj, S., Bui, M. M., Ortiz, M., Nacken, W., Sorg, C., Vogl, T.,
Roth, J., and Gabrilovich, D. I. (2008) Inhibition of dendritic cell
differentiation and accumulation of myeloid-derived suppressor cells in
cancer is regulated by S100A9 protein, J. Exp. Med., 205,
2235-2249.
31.Botta, C., Gulla, A., Correale, P., Tagliaferri,
P., and Tassone, P. (2014) Myeloid-derived suppressor cells in multiple
myeloma: pre-clinical research and translational opportunities,
Front. Oncol., 4, 348.
32.Hix, L. M., Karavitis, J., Khan, M. W., Shi, Y.
H., Khazaie, K., and Zhang, M. (2013) Tumor STAT1 transcription factor
activity enhances breast tumor growth and immune suppression mediated
by myeloid-derived suppressor cells, J. Biol. Chem., 288,
11676-11688.
33.Bronte, V., Serafini, P., De Santo, C., Marigo,
I., Tosello, V., Mazzoni, A., Segal, D. M., Staib, C., Lowel, M.,
Sutter, G., Colombo, M. P., and Zanovello, P. (2003) IL-4-induced
arginase 1 suppresses alloreactive T-cells in tumor-bearing mice, J.
Immunol., 170, 270-278.
34.Kieslinger, M., Woldman, I., Moriggl, R.,
Hofmann, J., Marine, J. C., Ihle, J. N., Beug, H., and Decker, T.
(2000) Antiapoptotic activity of Stat5 required during terminal stages
of myeloid differentiation, Genes Dev., 14, 232-244.
35.Hanahan, D., and Weinberg, R. A. (2011) Hallmarks
of cancer: the next generation, Cell, 144, 646-674.
36.Drutskaya, M. S., Efimov, G. A., Kruglov, A. A.,
Kuprash, D. V., and Nedospasov, S. A. (2010) Tumor necrosis factor,
lymphotoxin and cancer, IUBMB Life, 62, 283-289.
37.Coley, W. B. (1894) Treatment of inoperable
malignant tumors with the toxines of erysipelas and the Bacillus
prodigiosus, Am. J. Med. Sci., 108, 50-66.
38.Kulbe, H., Thompson, R., Wilson, J. L., Robinson,
S., Hagemann, T., Fatah, R., Gould, D., Ayhan, A., and Balkwill, F.
(2007) The inflammatory cytokine tumor necrosis factor-α
generates an autocrine tumor-promoting network in epithelial ovarian
cancer cells, Cancer Res., 67, 585-592.
39.Moore, R. J., Owens, D. M., Stamp, G., Arnott,
C., Burke, F., East, N., Holdsworth, H., Turner, L., Rollins, B.,
Pasparakis, M., Kollias, G., and Balkwill, F. (1999) Mice deficient in
tumor necrosis factor-α are resistant to skin carcinogenesis,
Nat. Med., 5, 828-831.
40.Popivanova, B. K., Kitamura, K., Wu, Y., Kondo,
T., Kagaya, T., Kaneko, S., Oshima, M., Fujii, C., and Mukaida, N.
(2008) Blocking TNF-α in mice reduces colorectal carcinogenesis
associated with chronic colitis, J. Clin. Invest., 118,
560-570.
41.Pikarsky, E., Porat, R. M., Stein, I.,
Abramovitch, R., Amit, S., Kasem, S., Gutkovich-Pyest, E.,
Urieli-Shoval, S., Galun, E., and Ben-Neriah, Y. (2004) NF-κB
functions as a tumour promoter in inflammation-associated cancer,
Nature, 431, 461-466.
42.Mohan, M. J., Seaton, T., Mitchell, J., Howe, A.,
Blackburn, K., Burkhart, W., Moyer, M., Patel, I., Waitt, G. M.,
Becherer, J. D., Moss, M. L., and Milla, M. E. (2002) The tumor
necrosis factor-alpha converting enzyme (TACE): a unique
metalloproteinase with highly defined substrate selectivity,
Biochemistry, 41, 9462-9469.
43.Bauer, J., Namineni, S., Reisinger, F., Zoller,
J., Yuan, D., and Heikenwalder, M. (2012) Lymphotoxin, NF-κB, and
cancer: the dark side of cytokines, Dig. Dis., 30,
453-468.
44.Zhang, H., Yan, D., Shi, X., Liang, H., Pang, Y.,
Qin, N., Chen, H., Wang, J., Yin, B., Jiang, X., Feng, W., Zhang, W.,
Zhou, M., and Li, Z. (2008) Transmembrane TNF-α mediates
“forward” and “reverse” signaling, inducing
cell death or survival via the NF-κB pathway in Raji Burkitt
lymphoma cells, J. Leukoc. Biol., 84, 789-797.
45.Aggarwal, B. B., Gupta, S. C., and Kim, J. H.
(2012) Historical perspectives on tumor necrosis factor and its
superfamily: 25 years later, a golden journey, Blood,
119, 651-665.
46.Havell, E. A., Fiers, W., and North, R. J. (1988)
The antitumor function of tumor necrosis factor (TNF). I. Therapeutic
action of TNF against an established murine sarcoma is indirect,
immunologically dependent, and limited by severe toxicity, J. Exp.
Med., 167, 1067-1085.
47.Puthier, D., Derenne, S., Barille, S., Moreau,
P., Harousseau, J. L., Bataille, R., and Amiot, M. (1999) Mcl-1 and
Bcl-xL are co-regulated by IL-6 in human myeloma cells, Br. J.
Haematol., 107, 392-395.
48.Spets, H., Stromberg, T., Georgii-Hemming, P.,
Siljason, J., Nilsson, K., and Jernberg-Wiklund, H. (2002) Expression
of the bcl-2 family of pro- and anti-apoptotic genes in multiple
myeloma and normal plasma cells: regulation during
interleukin-6(IL-6)-induced growth and survival, Eur. J.
Haematol., 69, 76-89.
49.Neurath, M. F., and Finotto, S. (2011) IL-6
signaling in autoimmunity, chronic inflammation and
inflammation-associated cancer, Cytokine Growth Factor Rev.,
22, 83-89.
50.Suematsu, S., Matsusaka, T., Matsuda, T., Ohno,
S., Miyazaki, J., Yamamura, K., Hirano, T., and Kishimoto, T. (1992)
Generation of plasmacytomas with the chromosomal translocation t(12;15)
in interleukin 6 transgenic mice, Proc. Natl. Acad. Sci. USA,
89, 232-235.
51.Grivennikov, S. I., and Karin, M. (2011)
Inflammatory cytokines in cancer: tumour necrosis factor and
interleukin 6 take the stage, Ann. Rheum. Dis., 70
(Suppl. 1), i104-108.
52.Huang, S. P., Wu, M. S., Shun, C. T., Wang, H.
P., Lin, M. T., Kuo, M. L., and Lin, J. T. (2004) Interleukin-6
increases vascular endothelial growth factor and angiogenesis in
gastric carcinoma, J. Biomed. Sci., 11, 517-527.
53.Tu, S., Bhagat, G., Cui, G., Takaishi, S.,
Kurt-Jones, E. A., Rickman, B., Betz, K. S., Penz-Oesterreicher, M.,
Bjorkdahl, O., Fox, J. G., and Wang, T. C. (2008) Overexpression of
interleukin-1β induces gastric inflammation and cancer and
mobilizes myeloid-derived suppressor cells in mice, Cancer Cell,
14, 408-419.
54.Apte, R. N., and Voronov, E. (2008) Is
interleukin-1 a good or bad “guy” in tumor immunobiology
and immunotherapy? Immunol. Rev., 222, 222-241.
55.Lewis, A. M., Varghese, S., Xu, H., and
Alexander, H. R. (2006) Interleukin-1 and cancer progression: the
emerging role of interleukin-1 receptor antagonist as a novel
therapeutic agent in cancer treatment, J. Transl. Med.,
4, 48.
56.Apte, R. N., Dotan, S., Elkabets, M., White, M.
R., Reich, E., Carmi, Y., Song, X., Dvozkin, T., Krelin, Y., and
Voronov, E. (2006) The involvement of IL-1 in tumorigenesis, tumor
invasiveness, metastasis and tumor–host interactions, Cancer
Metastasis Rev., 25, 387-408.
57.Dinarello, C. A. (1996) Biologic basis for
interleukin-1 in disease, Blood, 87, 2095-2147.
58.Voronov, E., Shouval, D. S., Krelin, Y., Cagnano,
E., Benharroch, D., Iwakura, Y., Dinarello, C. A., and Apte, R. N.
(2003) IL-1 is required for tumor invasiveness and angiogenesis,
Proc. Natl. Acad. Sci. USA, 100, 2645-2650.
59.Konishi, N., Miki, C., Yoshida, T., Tanaka, K.,
Toiyama, Y., and Kusunoki, M. (2005) Interleukin-1 receptor antagonist
inhibits the expression of vascular endothelial growth factor in
colorectal carcinoma, Oncology, 68, 138-145.
60.Shchors, K., Shchors, E., Rostker, F., Lawlor, E.
R., Brown-Swigart, L., and Evan, G. I. (2006) The Myc-dependent
angiogenic switch in tumors is mediated by interleukin 1β,
Genes Dev., 20, 2527-2538.
61.Sawai, H., Funahashi, H., Yamamoto, M., Okada,
Y., Hayakawa, T., Tanaka, M., Takeyama, H., and Manabe, T. (2003)
Interleukin-1α enhances integrin
α6β1 expression and metastatic
capability of human pancreatic cancer, Oncology, 65,
167-173.
62.Zhao, X., Rong, L., Zhao, X., Li, X., Liu, X.,
Deng, J., Wu, H., Xu, X., Erben, U., Wu, P., Syrbe, U., Sieper, J., and
Qin, Z. (2012) TNF signaling drives myeloid-derived suppressor cell
accumulation, J. Clin. Invest., 122, 4094-4104.
63.Sander, L. E., Sackett, S. D., Dierssen, U.,
Beraza, N., Linke, R. P., Muller, M., Blander, J. M., Tacke, F., and
Trautwein, C. (2010) Hepatic acute-phase proteins control innate immune
responses during infection by promoting myeloid-derived suppressor cell
function, J. Exp. Med., 207, 1453-1464.
64.Sade-Feldman, M., Kanterman, J., Ish-Shalom, E.,
Elnekave, M., Horwitz, E., and Baniyash, M. (2013) Tumor necrosis
factor-α blocks differentiation and enhances suppressive activity
of immature myeloid cells during chronic inflammation, Immunity,
38, 541-554.
65.Elkabets, M., Ribeiro, V. S., Dinarello, C. A.,
Ostrand-Rosenberg, S., Di Santo, J. P., Apte, R. N., and Vosshenrich,
C. A. (2010) IL-1β regulates a novel myeloid-derived suppressor
cell subset that impairs NK cell development and function, Eur. J.
Immunol., 40, 3347-3357.
66.Chen, M. F., Kuan, F. C., Yen, T. C., Lu, M. S.,
Lin, P. Y., Chung, Y. H., Chen, W. C., and Lee, K. D. (2014)
IL-6-stimulated
CD11b+CD14+HLA-DR–
myeloid-derived suppressor cells, are associated with progression and
poor prognosis in squamous cell carcinoma of the esophagus,
Oncotarget, 5, 8716-8728.
67.Oh, K., Lee, O. Y., Shon, S. Y., Nam, O., Ryu, P.
M., Seo, M. W., and Lee, D. S. (2013) A mutual activation loop between
breast cancer cells and myeloid-derived suppressor cells facilitates
spontaneous metastasis through IL-6 trans-signaling in a murine model,
Breast Cancer Res., 15, R79.
68.Pereira, R., Lago, P., Faria, R., and Torres, T.
(2015) Safety of anti-TNF therapies in immune-mediated inflammatory
diseases: focus on infections and malignancy, Drug Dev. Res.,
76, 419-427.
69.Van Hauwermeiren, F., Vandenbroucke, R. E., and
Libert, C. (2011) Treatment of TNF mediated diseases by selective
inhibition of soluble TNF or TNFR1, Cytokine Growth Factor Rev.,
22, 311-319.
70.Kaymakcalan, Z., Sakorafas, P., Bose, S.,
Scesney, S., Xiong, L., Hanzatian, D. K., Salfeld, J., and Sasso, E. H.
(2009) Comparisons of affinities, avidities, and complement activation
of adalimumab, infliximab, and etanercept in binding to soluble and
membrane tumor necrosis factor, Clin. Immunol., 131,
308-316.
71.Furst, D. E., Wallis, R., Broder, M., and
Beenhouwer, D. O. (2006) Tumor necrosis factor antagonists: different
kinetics and/or mechanisms of action may explain differences in the
risk for developing granulomatous infection, Semin. Arthritis
Rheum., 36, 159-167.
72.Efimov, G. A., Kruglov, A. A., Khlopchatnikova,
Z. V., Rozov, F. N., Mokhonov, V. V., Rose-John, S., Scheller, J.,
Gordon, S., Stacey, M., Drutskaya, M. S., Tillib, S. V., and
Nedospasov, S. A. (2016) Cell-type-restricted anti-cytokine therapy:
TNF inhibition from one pathogenic source, Proc. Natl. Acad. Sci.
USA, 113, 3006-3011.
73.Atretkhany, K. S., Nosenko, M. A., Gogoleva, V.
S., Zvartsev, R. V., Qin, Z., Nedospasov, S. A., and Drutskaya, M. S.
(2016) TNF neutralization results in the delay of transplantable tumor
growth and reduced MDSC accumulation, Front. Immunol., 7,
147.
74.Egberts, J. H., Cloosters, V., Noack, A.,
Schniewind, B., Thon, L., Klose, S., Kettler, B., von Forstner, C.,
Kneitz, C., Tepel, J., Adam, D., Wajant, H., Kalthoff, H., and
Trauzold, A. (2008) Anti-tumor necrosis factor therapy inhibits
pancreatic tumor growth and metastasis, Cancer Res., 68,
1443-1450.
75.Harrison, M. L., Obermueller, E., Maisey, N. R.,
Hoare, S., Edmonds, K., Li, N. F., Chao, D., Hall, K., Lee, C.,
Timotheadou, E., Charles, K., Ahern, R., King, D. M., Eisen, T.,
Corringham, R., DeWitte, M., Balkwill, F., and Gore, M. (2007) Tumor
necrosis factor α as a new target for renal cell carcinoma: two
sequential phase II trials of infliximab at standard and high dose,
J. Clin. Oncol., 25, 4542-4549.
76.Larkin, J. M., Ferguson, T. R., Pickering, L. M.,
Edmonds, K., James, M. G., Thomas, K., Banerji, U., Berns, B., De Boer,
C., and Gore, M. E. (2010) A phase I/II trial of sorafenib and
infliximab in advanced renal cell carcinoma, Br. J. Cancer,
103, 1149-1153.
77.Sumida, K., Wakita, D., Narita, Y., Masuko, K.,
Terada, S., Watanabe, K., Satoh, T., Kitamura, H., and Nishimura, T.
(2012) Anti-IL-6 receptor mAb eliminates myeloid-derived suppressor
cells and inhibits tumor growth by enhancing T-cell responses, Eur.
J. Immunol., 42, 2060-2072.
78.Kurzrock, R., Voorhees, P. M., Casper, C.,
Furman, R. R., Fayad, L., Lonial, S., Borghaei, H., Jagannath, S.,
Sokol, L., Usmani, S. Z., Van de Velde, H., Qin, X., Puchalski, T. A.,
Hall, B., Reddy, M., Qi, M., and Van Rhee, F. (2013) A phase I,
open-label study of siltuximab, an anti-IL-6 monoclonal antibody, in
patients with B-cell non-Hodgkin lymphoma, multiple myeloma, or
Castleman disease, Clin. Cancer Res., 19, 3659-3670.
79.Hong, D. S., Hui, D., Bruera, E., Janku, F.,
Naing, A., Falchook, G. S., Piha-Paul, S., Wheler, J. J., Fu, S.,
Tsimberidou, A. M., Stecher, M., Mohanty, P., Simard, J., and Kurzrock,
R. (2014) MABp1, a first-in-class true human antibody targeting
interleukin-1α in refractory cancers: an open-label, phase 1
dose-escalation and expansion study, Lancet Oncol., 15,
656-666.
80.Lust, J. A., Lacy, M. Q., Zeldenrust, S. R.,
Dispenzieri, A., Gertz, M. A., Witzig, T. E., Kumar, S., Hayman, S. R.,
Russell, S. J., Buadi, F. K., Geyer, S. M., Campbell, M. E., Kyle, R.
A., Rajkumar, S. V., Greipp, P. R., Kline, M. P., Xiong, Y.,
Moon-Tasson, L. L., and Donovan, K. A. (2009) Induction of a chronic
disease state in patients with smoldering or indolent multiple myeloma
by targeting interleukin 1β-induced interleukin 6 production and
the myeloma proliferative component, Mayo Clin. Proc.,
84, 114-122.
81.Stuelten, C. H., DaCosta Byfield, S., Arany, P.
R., Karpova, T. S., Stetler-Stevenson, W. G., and Roberts, A. B. (2005)
Breast cancer cells induce stromal fibroblasts to express MMP-9 via
secretion of TNF-α and TGF-β, J. Cell Sci.,
118, 2143-2153.
82.Szlosarek, P., Charles, K. A., and Balkwill, F.
R. (2006) Tumour necrosis factor-α as a tumour promoter, Eur.
J. Cancer, 42, 745-750.
83.Carswell, E. A., Old, L. J., Kassel, R. L.,
Green, S., Fiore, N., and Williamson, B. (1975) An endotoxin-induced
serum factor that causes necrosis of tumors, Proc. Natl. Acad. Sci.
USA, 72, 3666-3670.
84.Stoelcker, B., Ruhland, B., Hehlgans, T.,
Bluethmann, H., Luther, T., and Mannel, D. N. (2000) Tumor necrosis
factor induces tumor necrosis via tumor necrosis factor receptor type
1-expressing endothelial cells of the tumor vasculature, Am. J.
Pathol., 156, 1171-1176.
85.Grunhagen, D. J., De Wilt, J. H., Graveland, W.
J., Verhoef, C., Van Geel, A. N., and Eggermont, A. M. (2006) Outcome
and prognostic factor analysis of 217 consecutive isolated limb
perfusions with tumor necrosis factor-α and melphalan for
limb-threatening soft tissue sarcoma, Cancer, 106,
1776-1784.
86.Hu, X., Li, B., Li, X., Zhao, X., Wan, L., Lin,
G., Yu, M., Wang, J., Jiang, X., Feng, W., Qin, Z., Yin, B., and Li, Z.
(2014) Transmembrane TNF-α promotes suppressive activities of
myeloid-derived suppressor cells via TNFR2, J. Immunol.,
192, 1320-1331.
87.Polz, J., Remke, A., Weber, S., Schmidt, D.,
Weber-Steffens, D., Pietryga-Krieger, A., Muller, N., Ritter, U.,
Mostbock, S., and Mannel, D. N. (2014) Myeloid suppressor cells require
membrane TNFR2 expression for suppressive activity, Immun. Inflamm.
Dis., 2, 121-130.