Mechanisms of Inflammatory Injury of Renal Tubular Cells in a Cellular Model of Pyelonephritis
M. A. Morosanova1, E. Y. Plotnikov2*, L. D. Zorova3, I. B. Pevzner2, V. A. Popkov1, D. N. Silachev2, S. S. Jankauskas2, V. A. Babenko1, and D. B. Zorov2*
1Lomonosov Moscow State University, Faculty of Bioengineering and Bioinformatics, 119991 Moscow, Russia2Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; E-mail: plotnikov@genebee.msu.ru, zorov@genebee.msu.su
3Lomonosov Moscow State University, International Laser Center, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received April 29, 2016; Revision received July 5, 2016
Previously, we have assembled a cellular model of pyelonephritis which contains a primary culture of renal tubular epithelial cells, mononuclear leukocytes, and bacterial lysate or lipopolysaccharide. After cocultivation of renal cells with leukocytes and bacterial lysate, proinflammatory changes were observed in the renal cells, followed by nitrosative and oxidative stress and cell death. The interaction of bacterial antigens not only with leukocytes, but also with epithelial cells of the renal tubules, was partially mediated by signaling pathways involving Toll-like receptors (TLR2 and TLR4). Activation of these receptors led to increased levels of oxidative stress and synthesis of proinflammatory cytokines (TNF, IL-6, IL-1α) in the renal epithelium, while TLR4 blockade decreased the severity of these processes. Apart from the fact that activation of inflammatory signaling in response to bacterial antigens is observed directly in the renal cells, the presence of leukocytes significantly amplifies the inflammatory response as measured by the level of cytokines generated in the ensemble. In the presence of activated leukocytes, higher expression of TLR2 on the surface of renal cells was observed in response to exposure to bacterial components, which might explain the increased inflammatory response in the presence of leukocytes. The synthesis of IL-1α in the epithelial cells of the renal tubules in this inflammatory model leads to its accumulation in the nuclei, which has been reduced by the TLR4 antagonist polymyxin. TLR2 agonists also led to increased levels of IL-1α. The elevation in the content of IL-1α in nuclei was accompanied by increased acetylation of nuclear proteins, which has been reduced to control values after exposure to protective agents (Trolox, mitochondria-targeted antioxidant SkQR1 or LiCl). The high level of acetylation of histones is probably regulated by proinflammatory cytokines, and to some extent it is a marker of inflammation, which can indirectly be reduced by protective agents.
KEY WORDS: cytokines, acetylation, inflammation, pyelonephritisDOI: 10.1134/S000629791611002X
Abbreviations: DCF, 2,7-dihydrodichlorofluorescein; IL, interleukin; LPS, lipopolysaccharide; PBS, phosphate-buffered saline; ROS, reactive oxygen species; RTECs, renal tubular epithelial cells; TLR, Toll-like receptor; TNF, tumor necrosis factor.
Treatment of pyelonephritis is one of the main problems in urology. It
is complicated by the increasing need for pharmaceutical agents,
primarily antibiotics [1, 2].
This infectious disease that eventually results in renal failure is
mediated by bacteria penetrated in the kidney tissue, which may become
resistant to ongoing antibiotic therapy, therefore requiring use of new
approaches for treatment. Moreover, it is not always possible to use
antibiotics because they are not recommended for children, pregnant
women, individuals suffering from allergy, and, as mentioned above,
patients infected with antibiotic-resistant bacteria [3, 4]. Recently proposed
administration of mitochondria-targeted antioxidants in a combination
therapy of pyelonephritis [5] is within the
framework of contemporary treatment strategies that a priori
assume that oxidants of various origin may be involved in the
pathogenesis of the disease. It should be noted that the majority of
various kidney diseases is accompanied by oxidative stress occurring in
kidney tissues, including acute renal failure of various origin, e.g.,
ischemia-related diseases, ureteral obstruction, myoglobinuria (crush
syndrome), etc. [6-9]. Because
the major part of the examined renal pathologies may be, at least in
part, cured with mitochondria-targeted antioxidants [5-10], this implies that
mitochondria play an important role in pathogenesis of various renal
diseases [11]. However, in case of pyelonephritis,
according to the mode of action, positive effects of
mitochondria-targeted antioxidants might involve not only mitochondria,
but bacteria as well [5] (which are known to be
evolutionary ancestors of mitochondria [12]),
which invade kidney tissues during disease progression by ascending
along ureters from the bladder toward the kidneys.
Pyelonephritis is accompanied by injury of urinary tract mucosal layers and kidney parenchyma, mainly targeting interstitial tissues. During this disease, inflammation caused both by bacteria occupying renal tissue and their waste and decay products is considered as important component of pathological processes. In turn, developing inflammation most frequently is associated with oxidative stress.
Leukocyte infiltration as a response to bacterial invasion is the major effector in developing inflammation and subsequent destruction of kidney tissues [13]. As a natural response to pathogens, it does not affect surrounding tissues in a pathological way when reactive oxygen species (ROS) production is limited by phagocytic vesicles of leukocytes and macrophages. However, in case of extracellular release of ROS, they begin to damage surrounding cells, resulting in destruction of kidney tissues and subsequent organ failure. Unfortunately, in the majority of cases bacterial infection causes excessive activation of inflammatory response, so neutrophilic oxidative burst in response to pathogenic bacteria affects surrounding tissues [14]. Inflammatory response also includes release of locally acting cytokines, eicosanoids, activation of the complement system, and various damaging enzymes followed by a final ROS explosion [15]. The latter, in the “eukaryotic cell–bacterial cell–leukocyte” axis is mainly accomplished by leukocyte membrane NADPH oxidase [16], whose activity increases with progression of the disease at the site of infection. In addition, if at the onset of acute pyelonephritis there is a locally developed inflammation, then later, especially upon transition to a chronic phase, it may systemically spread and target the entire kidney, resulting in the death of a significant portion of its functionally active tissue [17]. Moreover, sepsis as a life-threatening condition as well as systemic inflammation of all tissues with subsequent propagation of toxins and bacteria over the body may develop as well.
Pyelonephritis is far from being the only renal disorder accompanied by inflammation; therefore, unveiling universal stages describing inflammatory processes in the kidneys is a strategically important task for therapy of renal pathology. In the current study, the goal was to explore the mechanism of inflammatory injury in renal tubular epithelial cells by simulating pyelonephritis in vitro. As a cellular model of pyelonephritis, renal tubular epithelial cells (RTECs) were cocultured with peripheral blood mononuclear cells supplemented with bacterial lysate [5].
Previously, using this model it has been found that 24-h co-culture of RTECs with leukocytes and bacterial lysate or lipopolysaccharide (LPS) resulted in increased fluorescence of 2,7-dihydrodichlorofluorescein (DCF), which pointed to enhanced ROS production in renal cells [5]. The broad-spectrum antioxidant Trolox, mitochondria-targeted antioxidants SkQ1 and SkQR1, as well as LiCl significantly lowered ROS levels. The following scheme of developing oxidative stress in RTECs was proposed: primary ROS are generated in leukocytes and RTECs in response to bacterial stimulation, and further ROS burst in the cells may be due to a self-amplified cascade of ROS production (so-called ROS-induced ROS release [18]). It should be noted that apart from increased ROS caused by added bacterial product, elevated levels of reactive nitrogen species occurred as well, and they were lowered also after using the same protective agents (antioxidants and LiCl). A remarkable death of both RTECs and leukocytes was noted in parallel with upregulated production of reactive oxygen and nitrogen species. We assumed that activation of pro-inflammatory signaling in response to bacterial invasion in the kidneys is an important pathogenic factor eventually responsible for death of renal cells.
MATERIALS AND METHODS
Primary rat renal tubular epithelial cell culture. Kidneys from 1-3-day-old rats were excised aseptically, chopped with scissors, and treated with 0.5% collagenase solution in PBS for 30 min, 37°C. After the cell suspension was centrifuged at 500g for 5 min, the pellet was resuspended in ~10 ml DMEM/F12 medium supplemented with 10% fetal bovine serum, and the cell suspension was left to sit for 2 min while bulk non-dissociated pieces sedimented, and the supernatant was transferred into another test tube. Then, 10 min later supernatant containing dissociated cells was decanted, and the pellet with renal tubules was resuspended in DMEM/F12 medium supplemented with 10% FBS to be seeded into 24-well plates or on cover slips placed into 35-mm Petri dishes. The renal epithelial cells were characterized by expression of Tamm–Horsfall protein (>90% positive cultured cells).
Bacterial culture and preparation of bacterial lysate. Escherichia coli (commensal strain 85 obtained from the bacterial collection of Lomonosov Moscow State University, Faculty of Biology, Department of Microbiology) bacteria were grown overnight in liquid culture medium containing 1% tryptone, 0.5% yeast extract, and 1% sodium chloride, pH 7.5. Culture medium containing ~109 CFU/ml was centrifuged at 2000g for 10 min to sediment the bacteria. Bacterial lysate was prepared by using 30 ml overnight bacterial culture diluted in 3 ml 0.9% NaCl solution. The final cell suspension was autoclaved for 1.5 h at 120°C for further use in RTEC culture.
Preparation of mononuclear leukocytes from peripheral blood. Peripheral blood was collected from the jugular vein of adult male rats into tubes containing 1000 U of heparin. Then, 5 ml of the heparinized blood was slowly layered onto 5 ml Ficoll-Urografin (density 1.077 g/cm3) followed by centrifugation for 30 min at 500g. This resulted in sedimentation of red blood cells, whereas the mononuclear cell fraction of ring-shaped peripheral blood leukocytes occupied the interphase on the Ficoll surface. Mononuclear leukocytes were transferred into another tube and centrifuged for 5 min at 500g. The remaining red blood cells were lysed by resuspending the cell pellet in 10 ml of cooled 0.88% NH4Cl solution and incubated for 10 min at 4°C, followed by centrifugation for 5 min at 500g. The final leukocyte pellet was resuspended in 5 ml DMEM/F12 culture medium followed by counting cells in a hemocytometer and diluting in DMEM/F12 medium at concentration ~105 cells/ml.
Cell coculture and treatment with protective substances. Two days after isolation of RTECs, the culture medium was exchanged for fresh DMEM/F12 medium supplemented with LiCl (9 mM), SkQ1 (10 nM), Trolox (100 µM), or SkQR1 (10 nM) and incubated for 2 h followed by adding ~105 mononuclear leukocytes/ml in DMEM/F12 medium together with bacterial lysate. All cells together with bacterial lysate were cocultured for 24 or 48 h. In control wells, DMEM/F12 medium was added instead of leukocytes and bacterial lysate.
Treatment with TLR4 antagonists and agonists and with TLR2 agonists. Instead of bacterial lysate, in some experiments RTECs were supplemented with 5 µg/ml lipopolysaccharide derived from E. coli (Sigma, USA), 100 µg/ml zymosan (Sigma), 10 µg/ml PAM3CSK4 (Sigma), or bacterial lysate at varying dilution degree (in phosphate buffer), per 100 µl/well and 400 µl/dish. To inhibit TLR4-mediated signaling, DMEM/F12 medium was partially replaced by a solution containing TLR4 antagonists in DMEM/F12 medium: 1 mg/ml polymyxin B (Sigma), 5 µg/ml LPS-RS (Sigma), per 100 µl/well and 400 µl/dish.
Measurement of ROS production. ROS production in the cells was estimated using the fluorescent probe DCF diacetate dissolved in bicarbonate-free DMEM/F12 medium to concentration 10 µM, supplemented to RTECs and incubated for 15 min at 37°C. The dye was washed out, followed by analysis of ROS production using a confocal laser scanning microscope (LSM510; Carl Zeiss Jena, Germany) through measuring DCF fluorescence intensity (excitation at 488 nm, emission at 505-530 nm), which is proportional to intracellular ROS levels [19]. In each experiment, the cells were used at the same seeding concentration. Mean DCF fluorescence intensity was calculated using confocal images for quantitative analysis. Randomly selected single confocal images revealing the highest number of cells in the focus were used for estimation (confocal slice thickness 1.5 µm, pinhole 150 µm, oil immersion objective Plan-Neofluar 63×/1.25 Oil). On average, 10 fields of view in three independent experiments were taken.
Measurement of TNF. Supernatant collected from 24-h cell cultures was centrifuged for 5 min at 1500g. The TNF concentration in the supernatant was estimated using a TNFα ELISA Ready-SET-Go! kit (eBioscience, USA).
RTECs immunocytochemistry staining. RTECs were fixed using 4% formalin solution in PBS for 30 min at 4°C, washed with PBS, permeabilized with 0.2% Triton X-100 in PBS for 60 min at 4°C, washed with PBS, and incubated in blocking solution containing 1% bovine serum albumin (BSA) in PBS for 60 min at 25°C. Then, the RTECs were incubated with primary antibodies for 2 h at 25°C at the following dilutions: 1 : 100 anti-Tamm–Horsfall protein (Millipore, USA); 1 : 200 anti-TLR2 (Abcam, Great Britain); 1 : 100 anti-IL-6 (Abcam); 1 : 100 anti-IL-1α (Thermo Fisher Scientific, USA), and 1 : 200 anti-acetyl lysine (Cell Signaling, USA). Then cells were washed with 1% BSA solution in PBS three times. The cells were then incubated with secondary antibodies for 1 h at 25°C (dilution 1 : 100), followed by three washouts in 1% BSA solution in PBS. Finally, the RTECs in plate wells were supplemented with 1% BSA in PBS, embedded in 50% glycerol solution in PBS, and mounted on microscopic slides.
Statistical analysis. Mean fluorescence intensity normalized per area covered by cells was calculated to estimate fluorescence intensity after immunocytochemical staining or experiments with DCF probe. The data was processed using STATISTICA 6.0 (StatSoft Inc., USA) and BioStat 4.03 (McGraw-Hill, USA) software and presented as mean ± SEM. Groups were compared using Student’s t-test.
RESULTS AND DISCUSSION
Interaction of bacterial ligands with Toll-like receptors on RTECs and mononuclear leukocytes. Using our in vitro pyelonephritis model, we pursue a goal to demonstrate the potential incidence of inflammatory response in the presence of bacterial products. It has been known that Toll-like receptors are responsible for recognition of various structurally conserved molecular patterns derived from both fragments of bacterial cell wall and proteins (coined as PAMPs – pathogen associated molecular patterns [20]) and host mitochondrial structures (coined as DAMPs – damage associated molecular patterns [21]). Since Toll-like receptors are known to be expressed on the surface of various renal cells [22] as well as leukocytes, it might happen that such receptors are also involved in our model during recognition primarily of bacterial ligands.
TLR4 as one of the crucial Toll-like receptors is involved in recognition of LPS residing in bacterial cell wall. The fact that in our model LPS induced oxidative stress [5] points to a potential role for TLR4 in this process. To test this, polymyxin B (an antibiotic that binds to LPS, thereby preventing its attachment to cognate receptor) known to inhibit signaling from TLR4 was used. In contrast, polymyxin B usage upregulated rather than lowered the intensity of oxidative stress (Fig. 1a). This effect may be due to its nephrotoxicity [23], as many nephrotoxic antibiotics provoke oxidative stress in RTECs. On the other hand, polymyxin successfully inhibited TNF production (Fig. 1b) in both the case of bacterial lysate as well as LPS. This suggests that LPS is the major active compound in the bacterial lysate, despite the other components contained. However, in the case of using bacterial lysate rather than pure LPS, the level of detected TNF was still higher than in control, as the other components in the bacterial lysate seemed to activate some receptors that were not affected by polymyxin. By hindering TLR4 activation, polymyxin reduces the activation of mononuclear leukocytes and RTECs, which is indicated by decreased TNF production. Due to this, oxidative stress in RTECs might decline as well, but it was not observed, perhaps due to polymyxin side effects such as toxicity to RTECs, although another yet unknown mechanism of ROS generation related to this agent might be involved. Since bacterial lysate is able to induce oxidative stress in RTECs, even without leukocytes (data not shown), most probably Toll-like receptors expressed by RTECs alone might be involved.
Fig. 1. Changes in fluorescence intensity of ROS-specific probe (a) and TNF production (b) in RTECs cocultured with mononuclear leukocytes (LC) supplemented with bacterial products (bacterial lysate, lipopolysaccharide (LPS)) with/without polymyxin inhibiting TLR4-mediated signal transduction. RTECs cocultured with leukocytes supplemented with bacterial products results in increased oxidative stress inside the cells (a) and simultaneously upregulates TNF production (b), whereas polymyxin lowers release of TNF into the medium, although oxidative stress was even increased. * p < 0.01; NS, not significant.
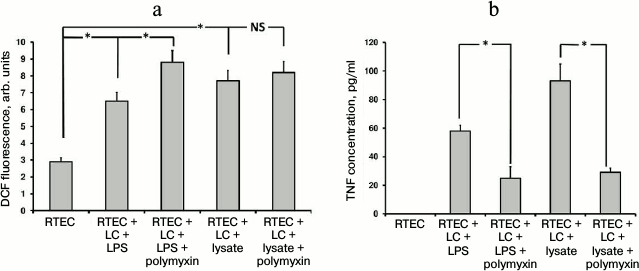
Indeed, adding LPS also results in oxidative stress inside RTECs (Fig. 1). However, like bacterial lysate, nephrotoxic activity of polymyxin results in even greater increase in ROS generation in RTECs.
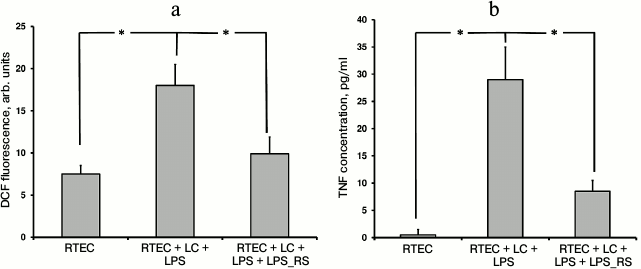
Usage of another TLR4 antagonist such as lipopolysaccharide derived from the nonpathogenic bacterium Rhodobacter sphaeroides (LPS_RS) decreases both TNF synthesis and level of oxidative stress (Fig. 2). This suggests an important role for TLR4 activation in mononuclear leukocytes and RTECs for developing inflammatory response.
Fig. 2. Changes in fluorescence intensity of ROS-specific probe in cells (a) and TNF concentration in the medium (b) after RTECs were cocultured with mononuclear leukocytes (LC) supplemented with lipopolysaccharide (LPS) with or without TLR4 antagonist LPS_RS. Both parameters characterizing magnitude of inflammatory response were decreased upon inhibiting binding of LPS to TLR4. * p < 0.01.
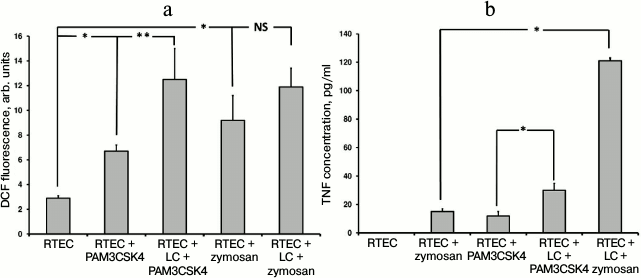
TLR2 is another well-characterized Toll-like receptor expressed in RTECs. Using specific agonists such as PAM3CSK4 (a peptide containing a fatty acid moiety) and zymosan (a component from yeast cell wall), it was found that the magnitude of oxidative stress in RTECs was increased with or without mononuclear leukocytes (Fig. 3a). Thus, when leukocytes were supplemented in the system a higher increase in ROS production in RTECs was observed compared to PAM3CSK4 or zymosan alone (PAM3CSK4 vs. zymosan: ROS production was significantly increased or tended to elevate, respectively). On the other hand, this may be explained by the fact that a greater cell number responds to receptor agonist because the receptor is expressed both in RTECs and in leukocytes. In addition, leukocytes might be activated more strongly, which is confirmed by a markedly higher TNF production (Fig. 3b). These data point to the involvement of RTEC’s TLR2 in recognizing cognate ligand, thereby allowing them to produce proinflammatory cytokine independently and taking part in the inflammatory response.
Fig. 3. Changes in fluorescence intensity of ROS-specific probe (a) in RTECs and TNF production (b) during incubation with TLR2 agonists (PAM3CSK4, zymosan) with or without mononuclear leukocytes (LC). Oxidative stress and inflammatory response were upregulated after incubating RTECs with TLR2 agonists, which were further increased by adding leukocytes into the system. * p < 0.01, ** p < 0.05; NS, not significant.
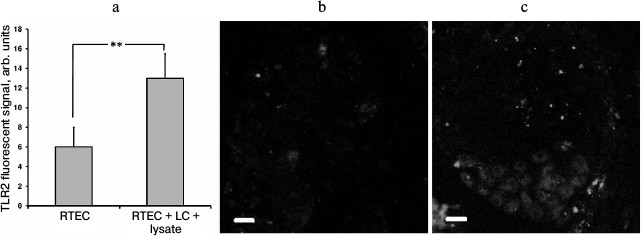
In our model, TLR2 expression in RTECs was upregulated in the presence of leukocytes activated by bacterial lysate (Fig. 4a), which occurred unevenly in different cells (Fig. 4c). This might be another explanation for enhanced inflammatory response observed in the presence of leukocytes. Apparently, interaction with leukocytes causes stronger involvement of RTECs in inflammatory response. Previously, emergence of surface TLR2 on nonimmune cells after exposure to an inflammatory milieu (cytokines, LPS) was demonstrated in endothelial cells [24], which were also shown to upregulate TLR2 expression in response to NADPH oxidase activation in LPS-primed neutrophils [25]. It seems that such interaction between various cell types may result in organization of a more efficient anti-bacterial defense.
Fig. 4. Upregulated TLR2 expression in RTECs cocultured with mononuclear leukocytes (LC) supplemented with bacterial lysate (a). TLR2 expression was measured as fluorescence intensity of cells immunocytochemically stained with FITC-labeled anti-TLR2 antibody (b, c: representative staining for TLR2 in control and experimental group, respectively); ** p < 0.05.
Thus, we conclude that in the proposed cellular model of pyelonephritis Toll-like receptors in leukocytes and RTECs are extensively involved in a developed inflammatory response. Activation of TLRs in leukocytes results in subsequently upregulated production of proinflammatory cytokines, but in the case of RTECs, their expression level becomes increased after exposure to the inflammatory milieu. Because RTECs contact foreign ligands prior to leukocytes after bacteria invade renal tissues, it was assumed [26] that it was RTECs that were responsible for the onset of developing renal inflammation upon bacterial invasion. Later, to develop peak inflammatory response both cell populations are required and must interact with each other.
Proinflammatory cytokines in the pyelonephritis model. There is a reason to believe that the two cell types interact with each other in the proposed model, and in the setting of inflammatory reaction, it may be mediated by release of proinflammatory cytokines. Damage of RTECs was detected after adding supernatant collected from mononuclear leukocytes after 24 h culture with bacterial lysate, and probably containing such cytokines. When RTECs were treated for 24 h with the supernatant, increase in ROS level from 4.5 ± 0.8 to 7.0 ± 0.8 arbitrary units (n = 5, p < 0.05) was observed.
In our model, we found that the level of TNF as one of the most examined proinflammatory cytokine was increased. Culture with bacterial lysate may let both leukocytes and RTECs synthesize and release TNF (control RTECs: TNF not detected, 24-h culture with bacterial lysate: concentration of TNF reached 6.3 ± 0.8 pg/ml; leukocytes cultured with bacterial lysate: 5.3 ± 0.7 pg/ml). A comparable amount of the cytokine detected in RTECs and leukocyte culture might be explained by longer survival of RTECs vs. leukocytes as well as their higher seeding concentration (leukocytes, 4·104; RTECs, 4·105). Thus, the overall TNF concentration measured 24 h after co-culturing RTECs and mononuclear leukocytes together with bacterial lysate was much higher than it was in individual cell cultures (co-culture RTECs + leukocytes added with bacterial lysate: 27 ± 1 pg/ml). This is consistent with the earlier assumption that the two cell populations should interact with each other to develop the most efficient inflammatory response.
By adding antioxidants or inhibitors of NO synthesis (Trolox, SkQR1, L-NAME, LiCl), there was virtually no effect on TNF production, thus suggesting that neither oxidative nor nitrosative stress were involved in this process. We demonstrate that its activation is caused by interaction of ligands with Toll-like receptors, while blocking relevant signaling pathways results in both lowered TNF production and decreased level of oxidative stress. Thus, the proinflammatory cytokine TNF may be one of causes, rather than a consequence, for developing oxidative and nitrosative stresses, so named above protective substances were unable to downregulate TNF concentration.
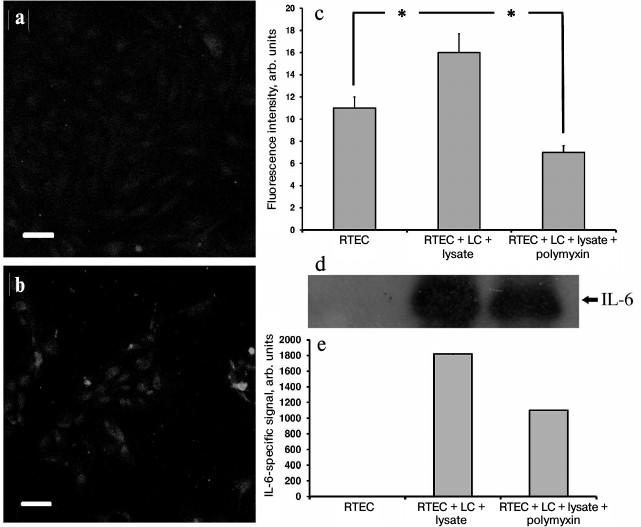
In addition, in our model a large amount of the proinflammatory cytokine IL-6 was found both inside RTECs (Fig. 5, a-c) and in the culture medium (Fig. 5, d and e), and polymyxin was able to lower IL-6 synthesis, thereby confirming the assumption that proinflammatory cytokines are produced in RTECs and mononuclear leukocytes resulting from activation of Toll-like receptors, primarily TLR4. Indeed, while culturing human renal epithelial cells with uropathogenic E. coli bacteria, a 20-fold increase in IL-6 production occurred [27]. Moreover, similar activation was observed by adding an NO donor. Because in our model RTECs are exposed to both bacterial substances and nitrosative stress, we assume that IL-6 is produced by both RTECs and leukocytes due to TLR activation, and possibly due to effects of NO.
Fig. 5. Upregulated IL-6 production in RTECs co-cultured with mononuclear leukocytes (LC) supplemented with bacterial lysate. a) Control group; b) RTECs co-cultured with mononuclear leukocytes supplemented with bacterial lysate for 24 h. Confocal microscopy after immunocytochemistry staining for IL-6; scale, 50 µm; c) amount of IL-6 in RTECs is increased after co-culture with leukocytes supplemented with bacterial lysate; polymyxin decreases the amount of IL-6 to the levels less than observed in the control group; * p < 0.01; d) cell culture immunoblot; e) densitometric analysis of immunoblot.
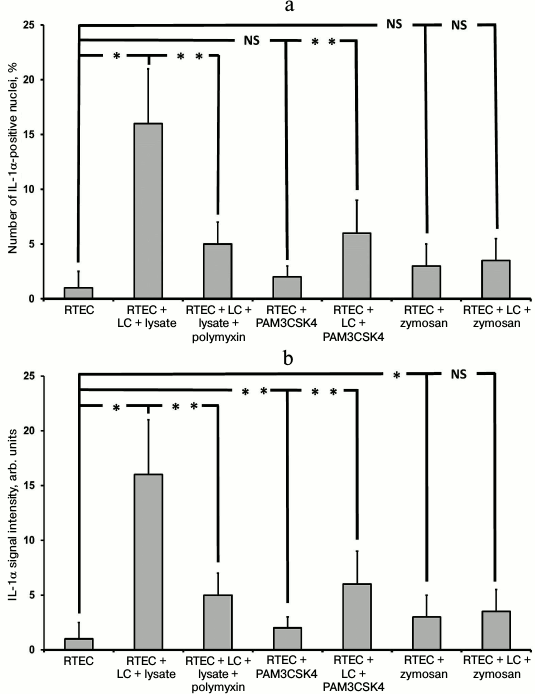
Immunocytochemical staining showed that after the modeling of inflammation, the signal of IL-1α was mainly within the nuclei of RTECs (not shown). In particular, IL-1α-positive nuclei were detected after RTECs were cocultured with leukocytes supplemented with bacterial lysate, whereas polymyxin decreased the percentage of IL-1-positive cells (Fig. 6a). In addition, TLR2 agonists also increased the amount of IL-1α within RTEC nuclei, which was even higher after co-culture with leukocytes. Thus, activation of Toll-like receptor 2 and 4 leads to upregulated synthesis of IL-1α and its accumulation in the cell nuclei.
Fig. 6. Detection of nuclear (a) and total (b) IL-1α in RTECs cocultured with mononuclear leukocytes (LC) and bacterial products (bacterial lysate, PAM3CSK4, zymosan) as well as with TLR4 antagonist polymyxin. TLR4 activation by bacterial lysate results in increased number of cells positively stained for nuclear IL-1α, whereas supplementation with the TLR4 antagonist polymyxin decreased the percentage of positive cells. Activation of TLR2 by its agonists also results in upregulated IL-1α synthesis, but to a lower extent. * p < 0.01, ** p < 0.05; NS, not significant.
The number of positively stained nuclei correlates with increased intensity of IL-1α-specific signal and points to generally elevated IL-1α synthesis (Fig. 6b). Perhaps, IL-1α found in the cytosol might then be transported into the nuclei or secreted into the culture medium. The former seems more plausible, as no significant increase in IL-1α concentration was detected in the medium. It is known that under stressed conditions full-size IL-1α is accumulated inside the cell nuclei, which is different from the excreted form by the N-terminal peptide containing the nuclear localization signal. One of the proposed functions for nuclear IL-1α is that it might interact with histone acetyltransferases resulting in histone acetylation and potentially activation of mRNA synthesis [28]. The support comes from the data showing that upon bacterial infection, including infection of renal tubular cells, nuclear IL-1α is involved in IL-8 production [29], and that the amount of acetylated proteins during oxidative stress and exposure to TNF elevates simultaneously with upregulated IL-8 production [30]. It is likely that increased synthesis and nuclear translocation of full-size IL-1α causing upregulation of histone acetyltransferase in the cells’ nucleus results in elevating the acetyltransferase activity, which in turn may lead to histone acetylation and transcription activation.
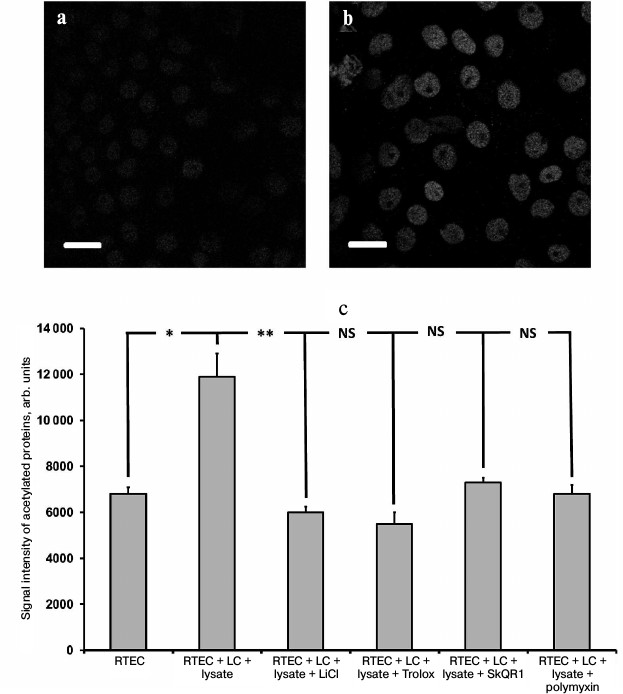
In support of this suggestion, in our inflammatory model we succeeded in detection of an increased amount of acetylated proteins in the RTECs nuclei (Fig. 7, a and b) as well as in finding a positive effect of protective compounds, which reduced the levels of histone acetylation (Fig. 7c). This effect might be mediated by downregulated production of nuclear IL-1α, as its level was also reduced after using such compounds (not shown). Polymyxin, which lowers the level of IL-1α, rather than oxidative stress, also reduces the intensity of histone acetylation. This implies that the presence of IL-1α in the nucleus can upregulate acetylation of nuclear proteins (possibly via increasing activity of histone acetyltransferase), and the level of oxidative stress per se plays no crucial role in this process. Protective compounds may also decrease the level of histone acetylation, perhaps by downregulation of the rate of IL-1α synthesis. It is likely that this effect is mediated by a suppressed activity of signaling ROS. Similar to hydrogen peroxide resulting in activated IL-8 production (likely via intracellular IL-1α) [30], ROS produced by cells in response to bacterial substances might also trigger IL-1α synthesis inside the RTECs. It can be assumed that both activation of Toll-like receptors and mere ROS may result in upregulation of proinflammatory cytokines produced in our model.
Fig. 7. Upregulated acetylation in RTECs nuclei. Immunocytochemical staining of RTECs with antibodies against acetylated proteins is shown: a) control group; b) RTECs cocultured with mononuclear leukocytes supplemented with bacterial lysate for 24 h; scale, 20 µm. Nuclear proteins, presumably histones, were mainly acetylated; c) mean fluorescence intensity of nuclei. RTECs cocultured with mononuclear leukocytes (LC) supplemented with bacterial lysate for 24 h were found to increase markedly the fluorescence intensity. Preincubation with protective compounds (Trolox, SkQR1, LiCl) or polymyxin decreased the level of fluorescence intensity to the control range. * p < 0.01, ** p < 0.05; NS, not significant.
Acknowledgements
This study was conducted with financial support by the Russian Science Foundation (project No. 14-15-00147).
REFERENCES
1.Prabhu, A., Taylor, P., Konecny, P., and Brown, M.
A. (2013) Pyelonephritis: what are the present day causative organisms
and antibiotic susceptibilities? Nephrology, 18,
463-467.
2.Meier, S., Weber, R., Zbinden, R., Ruef, C., and
Hasse, B. (2011) Extended-spectrum β-lactamase-producing
Gram-negative pathogens in community-acquired urinary tract infections:
an increasing challenge for antimicrobial therapy, Infection,
39, 333-340.
3.Shaikh, N., Ewing, A. L., Bhatnagar, S., and
Hoberman, A. (2010) Risk of renal scarring in children with a first
urinary tract infection: a systematic review, Pediatrics,
126, 1084-1091.
4.Smaill, F. M., and Vazquez, J. C. (2015)
Antibiotics for asymptomatic bacteriuria in pregnancy, Cochrane
Database Syst. Rev., 8, CD000490.
5.Plotnikov, E. Y., Morosanova, M. A., Pevzner, I.
B., Zorova, L. D., Manskikh, V. N., Pulkova, N. V., Galkina, S. I.,
Skulachev, V. P., and Zorov, D. B. (2013) Protective effect of
mitochondria-targeted antioxidants in an acute bacterial infection,
Proc. Natl. Acad. Sci. USA, 110, E3100-E3108.
6.Plotnikov, E. Y., Kazachenko, A. V., Vyssokikh, M.
Y., Vasileva, A. K., Tcvirkun, D. V., Isaev, N. K., Kirpatovsky, V. I.,
and Zorov, D. B. (2007) The role of mitochondria in oxidative and
nitrosative stress during ischemia/reperfusion in the rat kidney,
Kidney Int., 72, 1493-1502.
7.Plotnikov, E. Y., Chupyrkina, A. A., Jankauskas, S.
S., Pevzner, I. B., Silachev, D. N., Skulachev, V. P., and Zorov, D. B.
(2011) Mechanisms of nephroprotective effect of mitochondria-targeted
antioxidants under rhabdomyolysis and ischemia/reperfusion, Biochim.
Biophys. Acta, 1812, 77-86.
8.Jankauskas, S. S., Plotnikov, E. Y., Morosanova, M.
A., Pevzner, I. B., Zorova, L. D., Skulachev, V. P., and Zorov, D. B.
(2012) Mitochondria-targeted antioxidant SkQR1 ameliorates
gentamycin-induced renal failure and hearing loss, Biochemistry
(Moscow), 77, 666-670.
9.Kirpatovsky, V. I., Plotnikov, E. Y., Mudraya, I.
S., Golovanov, S. A., Drozhzheva, V. V., Khromov, R. A., Chernikov, D.
Y., Skulachev, V. P., and Zorov, D. B. (2013) Role of oxidative stress
and mitochondria in onset of urinary bladder dysfunction under acute
urine retention, Biochemistry (Moscow), 78, 542-548.
10.Plotnikov, E. Y., Silachev, D. N., Chupyrkina, A.
A., Danshina, M. I., Jankauskas, S. S., Morosanova, M. A., Stelmashook,
E. V., Vasileva, A. K., Goryacheva, E. S., Pirogov, Y. A., Isaev, N.
K., and Zorov, D. B. (2010) New-generation Skulachev ions exhibiting
nephroprotective and neuroprotective properties, Biochemistry
(Moscow), 75, 145-150.
11.Skulachev, M. V., Antonenko, Y. N., Anisimov, V.
N., Chernyak, B. V., Cherepanov, D. A., Chistyakov, V. A., Egorov, M.
V., Kolosova, N. G., Korshunova, G. A., Lyamzaev, K. G., Plotnikov, E.
Y., Roginsky, V. A., Savchenko, A. Y., Severina, I. I., Severin, F. F.,
Shkurat, T. P., Tashlitsky, V. N., Shidlovsky, K. M., Vyssokikh, M. Y.,
Zamyatnin, A. A., Jr., Zorov, D. B., and Skulachev, V. P. (2011)
Mitochondrial-targeted plastoquinone derivatives. Effect on senescence
and acute age-related pathologies, Curr. Drug Targets,
12, 800-826.
12.Zorov, D. B., Plotnikov, E. Y., Silachev, D. N.,
Zorova, L. D., Pevzner, I. B., Zorov, S. D., Babenko, V. A.,
Jankauskas, S. S., Popkov, V. A., and Savina, P. S. (2014) Microbiota
and mitobiota. Putting an equal sign between mitochondria and bacteria,
Biochemistry (Moscow), 79, 1017-1031.
13.Gupta, A., Sharma, S., Nain, C. K., Sharma, B.
K., and Ganguly, N. K. (1996) Reactive oxygen species-mediated tissue
injury in experimental ascending pyelonephritis, Kidney Int.,
49, 26-33.
14.Mundi, H., Bjorksten, B., Svanborg, C., Ohman,
L., and Dahlgren, C. (1991) Extracellular release of reactive oxygen
species from human neutrophils upon interaction with Escherichia
coli strains causing renal scarring, Infect. Immun.,
59, 4168-4172.
15.Nassar, G. M., and Badr, K. F. (1998) Novel
approaches to treatment of glomerulonephritis, J. Nephrol.,
11, 177-184.
16.Gupta, A., Sharma, N., Sharma, B. K., Sharma, S.,
and Ganguly, N. K. (1996) Oxygen-dependent and -independent mechanisms
of renal injury in experimental ascending pyelonephritis, FEMS
Immunol. Med. Microbiol., 13, 35-42.
17.Badr, K. F. (1997) Glomerulonephritis, Curr.
Opin. Nephrol. Hypertens., 6, 111-118.
18.Zorov, D. B., Filburn, C. R., Klotz, L.-O.,
Zweier, J. L., and Sollott, S. J. (2000) Reactive oxygen species
(ROS)-induced ROS release: a new phenomenon accompanying induction of
the mitochondrial permeability transition in cardiac myocytes, J.
Exp. Med., 192, 1001-1014.
19.Keston, A. S., and Brandt, R. (1965) The
fluorometric analysis of ultramicro quantities of hydrogen peroxide,
Anal. Biochem., 11, 1-5.
20.Janeway, C. A. (1989) Approaching the asymptote?
Evolution and revolution in immunology, Cold Spring Harb. Symp.
Quant. Biol., 54, 1-13.
21.Krysko, D. V., Agostinis, P., Krysko, O., Garg,
A. D., Bachert, C., Lambrecht, B. N., and Vandenabeele, P. (2011)
Emerging role of damage-associated molecular patterns derived from
mitochondria in inflammation, Trends Immunol., 32,
157-164.
22.Vandewalle, A. (2008) Toll-like receptors and
renal bacterial infections, Chang Gung Med. J., 31,
525-537.
23.Mingeot-Leclercq, M. P., Tulkens, P. M., Denamur,
S., Vaara, T., and Vaara, M. (2012) Novel polymyxin derivatives are
less cytotoxic than polymyxin B to renal proximal tubular cells,
Peptides, 35, 248-252.
24.Satta, N., Kruithof, E. K. O., Reber, G., and De
Moerloose, P. (2008) Induction of TLR2 expression by inflammatory
stimuli is required for endothelial cell responses to lipopeptides,
Mol. Immunol., 46, 145-157.
25.Fan, J., Frey, R. S., and Malik, A. B. (2003)
TLR4 signaling induces TLR2 expression in endothelial cells via
neutrophil NADPH oxidase, J. Clin. Invest., 112,
1234-1243.
26.Gribar, S. C., Anand, R. J., Sodhi, C. P., and
Hackam, D. J. (2008) The role of epithelial Toll-like receptor
signaling in the pathogenesis of intestinal inflammation, J. Leukoc.
Biol., 83, 493-498.
27.Demirel, I., Vumma, R., Mohlin, C., Svensson, L.,
Save, S., and Persson, K. (2012) Nitric oxide activates IL-6 production
and expression in human renal epithelial cells, Am. J. Nephrol.,
36, 524-530.
28.Buryskova, M., Pospisek, M., Grothey, A., Simmet,
T., and Burysek, L. (2004) Intracellular interleukin-1α
functionally interacts with histone acetyltransferase complexes, J.
Biol. Chem., 279, 4017-4026.
29.Cheng, W., Shivshankar, P., Zhong, Y., Chen, D.,
Li, Z., and Zhong, G. (2008) Intracellular interleukin-1α
mediates interleukin-8 production induced by Chlamydia
trachomatis infection via a mechanism independent of type I
interleukin-1 receptor, Infect. Immun., 76, 942-951.
30.Rahman, I., Gilmour, P. S., Jimenez, L. A., and
MacNee, W. (2002) Oxidative stress and TNF-alpha induce histone
acetylation and NF-kappaB/AP-1 activation in alveolar epithelial cells:
potential mechanism in gene transcription in lung inflammation, Mol.
Cell. Biochem., 234-235, 239-248.