Mitochondria-Targeted Antioxidant SkQR1 Reduces TNF-Induced Endothelial Permeability in vitro
I. I. Galkin1*, O. Yu. Pletjushkina1, R. A. Zinovkin1,2, V. V. Zakharova1, B. V. Chernyak1, and E. N. Popova1
1Lomonosov Moscow State University, Belozersky Institute of Physico-Chemical Biology, 119991 Moscow, Russia; E-mail: galkin.ivan.i@gmail.com2Lomonosov Moscow State University, Faculty of Biology, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received June 10, 2016; Revision received July 20, 2016
Prolonged or excessive increase in the circulatory level of proinflammatory tumor necrosis factor (TNF) leads to abnormal activation and subsequent damage to endothelium. TNF at high concentrations causes apoptosis of endothelial cells. Previously, using mitochondria-targeted antioxidants of SkQ family, we have shown that apoptosis of endothelial cells is dependent on the production of reactive oxygen species (ROS) in mitochondria (mito-ROS). Now we have found that TNF at low concentrations does not cause cell death but activates caspase-3 and caspase-dependent increase in endothelial permeability in vitro. This effect is probably due to the cleavage of β-catenin – an adherent junction protein localized in the cytoplasm. We have also shown that extracellular matrix metalloprotease 9 (MMP9) VE-cadherin shedding plays a major role in the TNF-induced endothelial permeability. The mechanisms of the caspase-3 and MMP9 activation are probably not related to each other since caspase inhibition did not affect VE-cadherin cleavage and MMP9 inhibition had no effect on the caspase-3 activation. Mitochondria-targeted antioxidant SkQR1 inhibited TNF-induced increase in endothelial permeability. SkQR1 also inhibited caspase-3 activation, β-catenin cleavage, and MMP9-dependent VE-cadherin shedding. The data suggest that mito-ROS are involved in the increase in endothelial permeability due to the activation of both caspase-dependent cleavage of intracellular proteins and of MMP9-dependent cleavage of the transmembrane cell-to-cell contact proteins.
KEY WORDS: endothelium, permeability, apoptosis, intercellular junctions, mitochondria-targeted antioxidants, inflammation, tumor necrosis factor (TNF)DOI: 10.1134/S0006297916100163
Abbreviations: Ac-DEVD-MCA, L-acetyl-Asp-Glu-Val-Asp-α-(4-methylcoumaryl-7-amide; DEVD, N-carbobenzyloxy-Asp(OMe)-Glu(OMe)-Val-Asp(OMe)-fluoromethyl ketone; MMP, matrix metalloprotease; NAC, N-acetyl-D-cysteine; PARP, poly(ADP-ribose) polymerase; ROS, reactive oxygen species; SkQ, conjugates of plastoquinone and penetrating cations; SkQ1, plastoquinonyl-10(6′-decyltriphenyl)phosphonium; SkQR1, 10-(6′-plastoquinonyl)decylrhodamine 19; TNF, tumor necrosis factor; ZVAD, N-benzyloxycarbonyl-Val-Ala-Asp-trifluoromethyl.
One of the most important functions performed by vascular endothelium is
the regulation of macromolecule and cell transport from the blood flow
to tissues. Endothelial barrier dysfunctions are typical for a number
of pathologies associated with acute and chronic inflammation [1-3]. They lead to edema, enhanced
transmigration of leukocytes to tissues, and subsequently to organ
damage [1, 2]. The injection of
recombinant Slit protein, which interacts with Robo 4 receptor and thus
stabilizes intercellular contacts, decreases organ damage and increases
survival of animals challenged by sepsis and viral infections [4]. Another way to lower endothelial permeability is
the inhibition of intercellular matrix metalloprotease 9 (MMP9) that
cleaves both glycocalyx and transmembrane intercellular junction
proteins, namely VE-cadherin, claudin-5, and others [5, 6]. Blood cells and
macromolecules migrate through the endothelium cell layer using either
(i) trans-cellular route through the body of the endothelial
cells by endocytosis and vesicular traffic, or (ii)
para-cellular through the gaps between nearby endothelial cells
formed due to the rearrangement of intercellular contacts [7]. These two modes are possibly linked to each other,
since many effectors of endothelial permeability stimulate both
vesicular transport and destruction of the integrity of intercellular
contacts [8]. Nevertheless, it is still unclear
whether these processes occur simultaneously in the same vessels or
they take place in various parts of the vascular system. The mechanisms
providing para-cellular transport are linked to destabilization
and rearrangement of protein complexes of the intercellular contacts
[7, 9, 10]. The most common mechanisms involve stretching of
nearby cells due to Rho/MLC1-dependent contraction of actomyosin
filaments attached to the contacts and dissociation of the protein
complexes of the intercellular contacts due to their phosphorylation
and/or degradation [7, 9, 10]. TNF is known to induce Rho-dependent
cytoskeleton rearrangement [11, 12], phosphorylation of adhesion junction proteins
[13], and their cleavage by caspases and
intercellular matrix metalloproteases (MMPs) [14].
Although mitochondria are not the main source of ATP in endothelial cells [15], they execute important signal functions under both normal and pathological conditions [16-18]. Mitochondria can influence intracellular signaling in multiple ways. The most studied are: release of proteins from the intermembrane space of mitochondria; regulation of ATP, ADP, and AMP levels; regulation of Ca2+ level in cytoplasm and regulation of ROS levels [16]. With the use of mitochondria-targeted antioxidants of the SkQ family, which are based on plastoquinone and penetrating cations, we recently showed that mitochondria-derived ROS (mito-ROS) participate in the induction of TNF-dependent endothelial permeability [19]. SkQ1 partially prevented the decrease in the intercellular contact VE-cadherin protein. TNF is known to induce shedding of the intercellular domain of this transmembrane protein via intercellular MMPs [14, 20]. On the other hand, we have shown that mito-ROS participate in signal transduction to caspases and in apoptotic stimulation driven by high doses of TNF [21]. Caspase activation may be caused not only by toxic but also by subtoxic concentrations of apoptotic inducers. Moreover, caspases can have non-apoptotic activities: induce cell proliferation, survival, and differentiation, regulate cell spreading and migration, cytokine maturation, and receptor internalization [22, 23]. There is no consensus on the role of caspases in the regulation of endothelial barrier function. There have been reports of involvement of caspases in the cytokine-induced permeability [24-26], and simultaneously that caspases are dispensable for the cytokine-induced alteration of endothelial barrier functions [27, 28].
In the current work, we estimated the contribution of caspase-dependent pathways of β-catenin cleavage and MMP9-dependent mechanisms of VE-cadherin shedding in TNF-induced endothelial permeability and studied the role of mito-ROS in these processes.
MATERIALS AND METHODS
Materials. SkQR1 was synthesized by G. A. Korshunova and N. V. Sumbatyan at the A. N. Belozersky Institute of Physico-Chemical Biology [29]; human recombinant TNFα was kindly procured by L. N. Shingarova (Institute of Bioorganic Chemistry, Moscow). All other reagents were from Sigma (USA). Laboratory plastic ware and Transwell® plates were from Costar (USA).
Cell cultures. Endothelial cell line EaHy926 (ATCC CRL-2922) [30] was cultured in DMEM medium (Dulbecco’s modified Eagle’s medium) (Gibco, USA) supplemented with 10% fetal bovine serum (FBS) (HyClone, USA) and with 100 µM hypoxanthine and 20 µM thymidine (HyClone). After cell attachment and spreading, the antioxidants (5 mM NAC, 200 µM Trolox, 20 nM SkQR1) were added to the cells (105 cells per ml) and incubated for 4 days. Then the medium was replaced by fresh medium containing 0.2% FBS. After 24 h, human recombinant TNF was added (concentrations and incubation conditions are indicated in the figure legends). For studies on caspase and MMP9 activities, total caspase inhibitor ZVAD (1 µM), caspase-3 inhibitor DEVD (1 µM), and MMP9 inhibitor GM6001 (Merck, USA) (25 µM) were added 30 min prior to TNF treatment.
Estimation of apoptosis. Two days after TNF treatment, the cells (including those that detached and emerged from the bottom of the dish) were collected, washed with cold PBS (2 mM KH2PO4, 10 mM Na2HPO4·7H2O, 137 mM NaCl, 2.7 mM KCl), fixed with ice-cold mixture of PBS–ethanol (1 : 2), and incubated overnight at 4°C. After this, the cells were washed with PBS and stained in PBS containing 30 µg/ml propidium iodide (MP Biomedicals, France) and 10 µg/ml RNase A (Fermentas, Lithuania) in the dark for 45 min at 37°C and then analyzed with a Beckman Coulter FC500 flow cytometer.
Estimation of caspase-3 activation. After incubation with the examined compounds and TNF, the cells were lysed with lysis buffer (10 mM Tris-HCl, 5 mM EDTA, 320 mM sucrose, 1% Triton X-100, 1 mM PMSF, 1 mM DTT) for 10 min at 4°C, then centrifuged at 14,000g for 5 min at 4°C. The supernatant was transferred into a 96-well plate. An equal volume of reaction buffer (100 mM HEPES, 10% sucrose, 0.2% CHAPS, 1 mM PMSF, 1 mM DTT, 100 µM caspase-3 fluorescent substrate AC-DEVD-MCA (Peptide Institute, Inc., Japan)) was added. The reaction was carried out for 1 h, 37°C. Caspase-3 activity was measured with a Fluoroskan Ascent plate fluorimeter at 355/460 nm (Thermo Fisher Scientific, USA).
Western blot analysis. Western blot analysis was performed as described previously [21]. The antibodies used were to human proteins: VE-cadherin (eBioscience, USA, 14-1449-82), β-catenin (BD Transduction Laboratories, C19220), GAPDH (G8795) and horseradish peroxidase-conjugated antibodies to rabbit Ig (A0545) and mouse Ig (A9917). A SuperSignal West Dura kit (Thermo Fisher Scientific) was used for HRP detection according to manufacturer’s instructions. Images were obtained and analyzed with a ChemiDoc™ MP System (Bio-Rad, USA) under ImageLab 5.2.1 (Bio-Rad) software.
In vitro endothelial permeability. The level of endothelial permeability was estimated as previously described by Chen et al. [31]. The cells were cultured up to 100% confluent monolayer in the upper chambers of Transwell® Plates (pore diameter 0.4 µm). After 24 h of incubation with TNF (10 ng/ml), a mixture of conjugates of 65-85 kDa dextran with TRITC (TRITC-dextran, 1 mg/ml) was added to the upper chamber. The amount of TRITC-dextran passed through the cell monolayer to the lower chamber was measured 4 h later with the plate fluorimeter at 485/590 nm.
Fluorescent microscopy. The cells were cultured on coverslips placed into the wells of a culture plate. After incubation with the examined compounds and TNF, the cells were fixed with 2% paraformaldehyde (10 min, 37°C) and permeabilized with 1% Triton X-100 (5 min, room temperature). The reaction with antibodies to VE-cadherin (eBioscience, 14-1449-82) and β-catenin (BD Transduction Laboratories, C19220) was carried for 12-15 h, 4°C. The reaction with antibodies to mouse IgG conjugated with Oregon Green 488 (Invitrogen, USA, O-6380) or Texas Red (Molecular Probes, USA, T-2767) fluorophores, along with Hoechst 33342 (5 µg/ml), was carried out for 30 min at room temperature. Images were obtained using an Axiovert 200M fluorescence microscope with AxioCAM HRM camera (Carl Zeiss, Germany). To estimate the effect of TNF on the number of attached cells, photographs of three cell fields were randomly taken and the cell nuclei were counted. A lens with 20× amplification was used; the average number of cells in a single field of view was 269. ImageJ 1.51d software was used for cell counting and fluorescence profiling.
Statistical analysis. Data are presented as mean ± standard deviation (SD). Statistical significance was calculated using the Mann–Whitney criteria.
RESULTS AND DISCUSSION
The response of endothelial cells to apoptosis induction varies in different cell models. In some cases, TNF alone can induce apoptosis [32], in other cases additional co-simulation is required by either inflammatory cytokines or hydrogen peroxide [33, 34]. We have shown previously that TNF at high concentrations (>10 ng/ml) induces apoptosis of more than 40% of EAhy926 endothelial cells [21] (Fig. 1, a, c, and d). TNF at 5 ng/ml did not induce apoptosis (Fig. 1, a, b, and d) but led to the remarkable changes in cell morphology. The cells became stretched; the adhesion contacts proteins VE-cadherin and β-catenin disappeared from the contacts; gaps appeared between the cells (Figs. 2 and 3). This TNF-dependent change in primary endothelial cell morphology was observed earlier, and it was accompanied by the suppression of cell proliferation and cell detachment from the surface [35]. Incubation of EAhy926 cells with TNF also led to a decrease in the number of attached cells by 24%. The number of cells in S and G2/M-phase increased after TNF incubation, probably because of cell cycle arrest (Fig. 1, b and c). Moreover, we observed three-fold increase in detached cells with non-fragmented nuclei. Large amounts of circulating endothelial cells are typical for various pathologies linked to systemic inflammation [36, 37]. The content of apoptotic cells among circulating endothelial cells is usually not high, though endothelial cell detachment may cause their apoptosis in vitro. This occurs due to the interruption of the survival signaling induced by the integrins and intercellular contacts. TNF treatment also promoted increase in endothelial permeability for macromolecular (30-75 kDa) dextran-TRITC conjugates (Fig. 4).
Fig. 1. TNF induces apoptosis (a-d) and caspase-3 activation (e) in EAhy926 endothelial cells. a-c) Typical results of cell cycle analysis; d) statistical analysis. Apoptosis was measured 48 h after TNF treatment; caspase-3 activity was measured 12-15 h after TNF treatment. n ≥ 5; ** p ≤ 0.01, *** p ≤ 0.001 for 5, 10, 50, and 100 ng/ml of TNF.
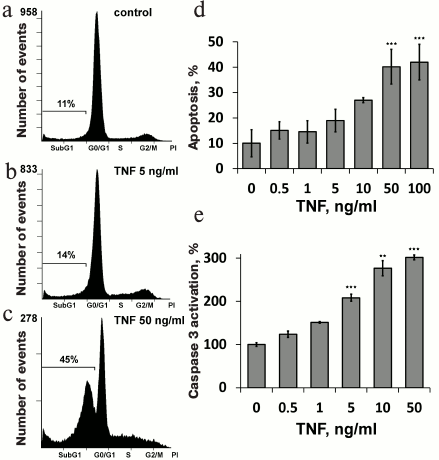
Fig. 2. Mitochondria-targeted antioxidant SkQR1 (20 nM), caspase inhibitor ZVAD (1 µM), and MMP9 inhibitor (MMP9i, 25 µM) prevent TNF-dependent (5 ng/ml, 24 h) VE-cadherin rearrangement in EAhy926 cells. Immunofluorescent staining of VE-cadherin is stained green, and cell nuclei are stained cyan (Hoechst 33342 staining). Bar size – 15 µm. Oregon Green 488 fluorescence profiles containing cross sections of the cells are shown for control and TNF-treated cells. Quantitative analysis of the distribution of VE-cadherin is shown.
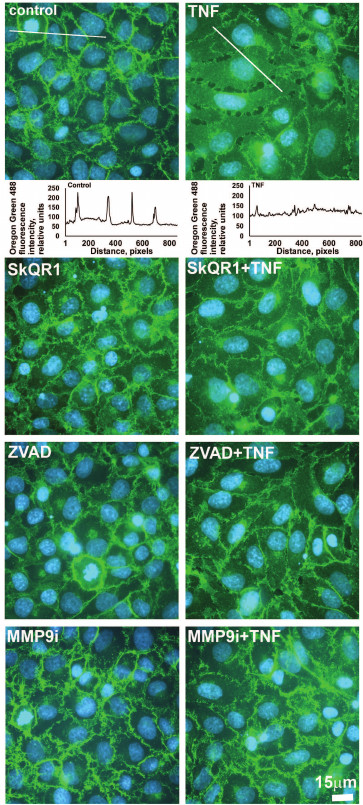
Fig. 3. Mitochondria-targeted antioxidant SkQR1 (20 nM), caspase inhibitor ZVAD (1 µM), and MMP9 inhibitor (MMP9i, 25 µM) prevent TNF-dependent (5 ng/ml, 24 h) β-catenin rearrangement in EAhy926 cells. β-Catenin is stained green (immunofluorescent staining, monoclonal antibodies); cell nuclei are stained cyan (Hoechst 33342 staining). Bar size – 15 µm. Oregon Green 488 fluorescence profiles containing cross sections of the cells, which are shown for control and TNF-treated cells. Quantitative analysis of the distribution of β-catenin is shown.
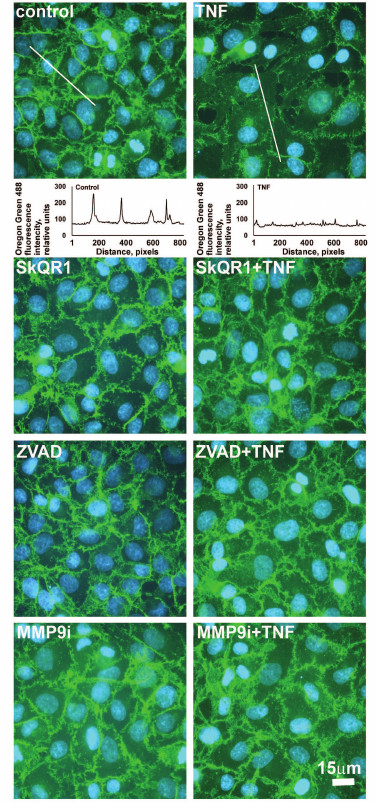
Fig. 4. Effect of antioxidants SkQR1 (20 nM) and Trolox (200 µM) and of the caspase inhibitor ZVAD (1 µM) and MMP9 inhibitor (25 µM) on TNF-dependent (5 ng/ml, 24 h) endothelial cell monolayer permeability to TRITC-dextran. n ≥ 5; ** p ≤ 0.01.
Low concentrations of TNF (<5 ng/ml) did not cause apoptosis of EAhy926 cells but activated caspase-3 (Fig. 1e). Inhibition of the caspases by total caspase inhibitor ZVAD led to small (10-15%) but statistically significant decrease in TNF-stimulated endothelial permeability (Fig. 4). Additionally, ZVAD and caspase-3 inhibitor DEVD prevented TNF-dependent disappearance of VE-cadherin and β-catenin from contact areas and prevented formation of gaps (Figs. 2 and 3, data from the experiments with ZVAD are shown). TNF-induced β-catenin cleavage in the EAhy926 cells was blocked by ZVAD (Fig. 5) in full accordance with published data [14]. β-Catenin cleavage was also decreased in the presence of DEVD (Fig. 5), but this effect was weaker compared to ZVAD treatment. This points out that not only caspase-3 but also other caspases may participate in β-catenin cleavage.
Fig. 5. Effect of antioxidants, caspase inhibitors ZVAD and DEVD (1 µM), and MMP9 inhibitor (25 µM) on TNF-dependent (5 ng/ml, 24 h) cleavage of β-catenin in EAhy926 cells. a, b) Representative Western blots; c) densitometric analysis of Western blots; n = 3; & p ≤ 0.01 for Control – TNF; * p ≤ 0.05; ** p ≤ 0.01 for TNF – antioxidants or inhibitors.
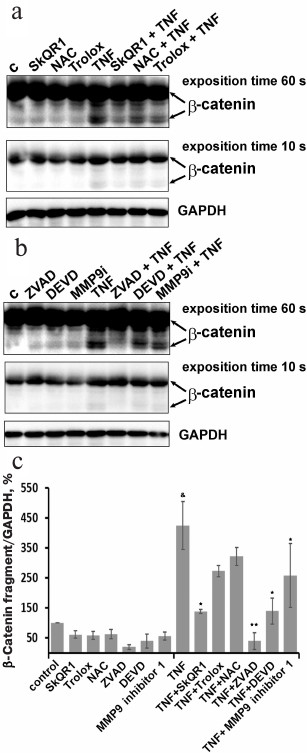
ZVAD completely inhibited TNF-induced decrease in the number of monolayer cells. The detachment of cells was probably promoted by β-catenin cleavage by caspases and subsequent disruption of intercellular contacts. Additionally, caspase-3 may inhibit cell proliferation by the cleavage of p27 and p21 [23]. Moreover, β-catenin regulates endothelial proliferation and cell survival [38]. β-Catenin cleavage by caspases in cancer cells leads to arrest of the cell cycle in the G2/M phase and apoptosis due to suppression of c-Myc expression and kinase Cdc2/cyclin B1 inactivation [39].
The role of caspases in the regulation of endothelial barrier function is not completely clear. The data are limited and contradictory [24-28]. Adhesion contact proteins β-catenin and plakoglobin were shown to be cleaved by caspases in TNF treated endothelial cells [14]. The data presented above show that the cleavage of cell proteins by caspases makes a small but statistically significant contribution to the TNF-dependent increase in permeability and disassembly of intercellular contacts in endothelium. It may be assumed that the cleavage of β-catenin and possibly other proteins of intercellular contacts by caspases plays an important role in stimulation of caspase-dependent permeability of endothelium.
Subtoxic TNF doses caused a decrease in total VE-cadherin content in EAhy926 cells (Fig. 6). The inhibition of caspases had no effect on this process, while MMP9 inhibition abrogated the effect of TNF (Fig. 6) and prevented disassembly of the intercellular contacts and gap formation (Figs. 2 and 3). It also completely blocked TNF-dependent increase in endothelial permeability for dextran-TRITC (Fig. 4). Thus, MMP9-driven shedding of VE-cadherin (and, presumably, other proteins) makes the most significant contribution to the TNF-dependent increase in permeability. The increase in endothelial permeability by shedding of transmembrane proteins including VE-cadherin, as well as glycocalyx proteins, has been reported for a number of pathologies associated both with chronic and acute inflammation [20, 40-42].
Fig. 6. Effect of antioxidants SkQR1 (20 nM) and Trolox (200 µM), caspase inhibitor ZVAD (1 µM), and MMP9 inhibitor (25 µM) on TNF-dependent (5 ng/ml, 48 h) VE-cadherin decrease in EAhy926 cells. a) Representative Western blots; b) densitometric analysis of Western blots. n = 5; && p ≤ 0.01 for Control – TNF; ** p ≤ 0.01 for TNF – antioxidants or inhibitors.
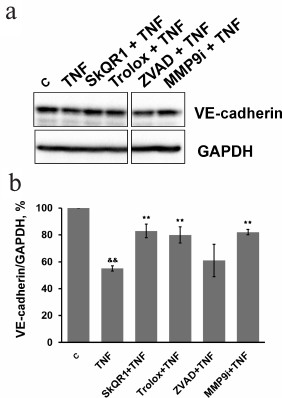
Inhibition of MMP9 substantially prevented the TNF-induced loss of adherent cells: the number of cells in vehicle control was taken as 100%, TNF incubation – 76%, TNF + MMP9 inhibitor – 93.4%. VE-cadherin is one of the key regulators of endothelial cell detachment from the substrate [36, 37], and its cleavage by MMP9 may facilitate this process. Moreover, MMP cleaves other intercellular matrix proteins – fibronectin, vitronectin, and collagen [5, 6] – that are also involved in the adhesion of endothelial cells [36, 37].
Caspases can regulate several signal transduction pathways [22, 23]. This regulation mainly occurs via NFκB transcription factor and mitogen-activated protein kinases (MAPK) [22, 23]. For example, caspase-dependent NFκB activation and expression of the adhesion molecules ICAM1 and VCAM1 were demonstrated in murine kidney endothelium [43]. TNF may induce MMP9 expression via NFκB, Sp-1, and AP-1 activation [44, 45]. One can assume that MMP9-dependent shedding of VE-cadherin is also connected with caspase activation. However, the inhibition of caspases with ZVAD did not prevent VE-cadherin cleavage (Fig. 6). In turn, inhibition of MMP9 did not affect caspase-3 activation (Fig. 7). Therefore, caspase-3 activation and MMP9-dependent shedding of VE-cadherin are regulated independently. At the same time, MMP9 inhibitor partially decreased caspase-dependent cleavage of β-catenin (Fig. 5). Destabilization of the protein complex that forms adhesion contacts due to cleavage of VE-cadherin possibly promotes β-catenin cleavage by caspases.
Fig. 7. Effect of mitochondria-targeted antioxidant SkQR1 (20 nM) and MMP9 inhibitor (25 µM) on TNF-dependent (5 ng/ml, 12-15 h) caspase-3 activation in EAhy926 cells. n = 5; && p ≤ 0.01 for Control – TNF; ** p ≤ 0.01 for TNF – SkQR1 or MMP9 inhibitor.
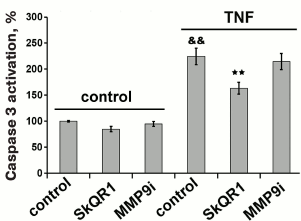
We have previously shown that apoptosis of EAhy926 cells induced by high TNF concentrations was prevented by classical antioxidants (NAC and Trolox) as well as mitochondria-targeted antioxidants, with SkQR1 being the most efficient [21]. The experiments with subtoxic TNF concentrations proved that SkQR1 (along with NAC and Trolox) prevented the disassembly of intercellular adhesion contacts and endothelial permeability caused by TNF (Figs. 2-4). Similar effects were earlier observed in experiments with another mitochondria-targeted antioxidant, SkQ1 [19]. SkQR1 inhibited caspase-3 activation caused by subtoxic TNF concentrations (Fig. 7) and completely abrogated caspase-dependent cleavage of β-catenin (Fig. 5). The effect of classical antioxidants was less pronounced (Fig. 5). Moreover, SkQR1 prevented the TNF-induced decrease in adherent cells: the number of cells in vehicle control was taken as 100%, TNF incubation – 76%, TNF + SkQR1 – 86%. We investigated the effect of SkQR1 on TNF-induced apoptosis and found that SkQR1 did not influence cleavage of caspase-8 and its substrate proapoptotic protein Bid, but it did inhibit cytochrome c release from mitochondria and cleavage (activation) of caspase-3 and its substrate PARP. It can be assumed that non-apoptotic subthreshold activation of caspase-3 also depends on mitochondrial cytochrome c release with the participation of mito-ROS. This assumption points to the important role of mitochondria in the non-apoptotic activation of caspases that plays a crucial role in the regulation of various physiological processes [22, 23].
It was shown recently that partial release of cytochrome c into the cytoplasm from a small population of mitochondria might lead to subliminal activation of caspases, which does not induce apoptosis but provokes genomic instability and cell transformation [46]. The authors observed partial release of cytochrome c when treating cells with ABT-737, which mimics the BH3 domain of the Bcl2 protein family, as well as at the conditions of limited tBid protein expression carrying BH3 domain only. We observed TNF-induced activation of exactly the same protein in our model, but it was not dependent on mito-ROS [21]. Thus, mito-ROS possibly play an important role in the non-apoptotic caspase activation by induction of partial release of cytochrome c from mitochondria.
Our experiments have demonstrated that the main impact on the TNF-dependent increase in endothelial permeability is made by MMP9-dependent shedding of transmembrane proteins of adhesion contacts, particularly VE-cadherin (Fig. 6). SkQR1 suppressed VE-cadherin cleavage (Fig. 6), and this effect was not linked to caspase inhibition (Fig. 7). NFκB is the most important transcription factor regulating TNF-induced MMP9 expression [44, 45]. We earlier showed that NFκB activation by TNF in endothelial cells depends on mito-ROS [47]. We can assume it is NFκB inhibition by SkQR1 that is responsible for VE-cadherin cleavage protection.
Our data show that mito-ROS play a crucial role in the TNF-induced increase in endothelial permeability. Mitochondria of endothelial cells regulate both caspase-dependent cleavage of intracellular proteins of adhesion contacts and MMP9-dependent shedding of transmembrane proteins. The mito-ROS may also promote endothelial cell detachment and/or decrease their proliferation, which also seems to be induced by caspase and MMP9 activation. Our data suggest that therapeutic effects of mitochondria-targeted antioxidants in various animal models are linked to the prevention of the increase in endothelial permeability during inflammation. This may help to extend possible applications of the mito-ROS scavengers.
Acknowledgements
We thank V. P. Skulachev for critical discussion of the manuscript and providing valuable suggestions.
This work was supported by the Russian Foundation for Basic Research (project No. 16-04-01074, immunofluorescent microscopy of intercellular contacts) and by the Russian Science Foundation (project No. 14-14-00055, other experiments).
REFERENCES
1.Aird, W. C. (2003) The role of the endothelium in
severe sepsis and multiple organ dysfunction syndrome, Blood,
101, 3765-3777.
2.Peters, K., Unger, R. E., Brunner, J., and
Kirkpatrick, C. J. (2003) Molecular basis of endothelial dysfunction in
sepsis, Cardiovasc. Res., 60, 49-57.
3.Pober, J. S., and Sessa, W. C. (2007) Evolving
functions of endothelial cells in inflammation, Nat. Rev.
Immunol., 7, 803-815.
4.London, N. R., Zhu, W., Bozza, F. A., Smith, M. C.,
Greif, D. M., Sorensen, L. K., Chen, L., Kaminoh, Y., Chan, A. C.,
Passi, S. F., Day, C. W., Barnard, D. L., Zimmerman, G. A., Krasnow, M.
A., and Li, D. Y. (2010) Targeting Robo4-dependent Slit signaling to
survive the cytokine storm in sepsis and influenza, Sci. Transl.
Med., 2, 23ra19.
5.Dubois, B., Starckx, S., Pagenstecher, A., Oord,
J., Arnold, B., and Opdenakker, G. (2002) Gelatinase B deficiency
protects against endotoxin shock, Eur. J. Immunol., 32,
2163-2171.
6.Mulivor, A. W., and Lipowsky, H. H. (2009)
Inhibition of glycan shedding and leukocyte-endothelial adhesion in
postcapillary venules by suppression of matrixmetalloprotease activity
with doxycycline, Microcirculation, 16, 657-666.
7.Komarova, Y., and Malik, A. B. (2010) Regulation of
endothelial permeability via paracellular and transcellular transport
pathways, Annu. Rev. Physiol., 72, 463-493.
8.Dejana, E. (2004) Endothelial cell–cell
junctions: happy together, Nat. Rev. Mol. Cell Biol., 5,
261-270.
9.Dejana, E., Orsenigo, F., and Lampugnani, M. G.
(2008) The role of adherens junctions and VE-cadherin in the control of
vascular permeability, J. Cell Sci., 121, 2115-2122.
10.Vestweber, D. (2008) VE-cadherin: the major
endothelial adhesion molecule controlling cellular junctions and blood
vessel formation, Arterioscler. Thromb. Vasc. Biol., 28,
223-232.
11.McKenzie, J. A., and Ridley, A. J. (2007) Roles
of Rho/ROCK and MLCK in TNF-alpha-induced changes in endothelial
morphology and permeability, J. Cell. Physiol., 213,
221-228.
12.Wojciak-Stothard, B., and Ridley, A. J. (2002)
Rho GTPases and the regulation of endothelial permeability, Vasc.
Pharmacol., 39, 187-199.
13.Angelini, D. J., Hyun, S. W., Grigoryev, D. N.,
Garg, P., Gong, P., Singh, I. S., Passaniti, A., Hasday, J. D., and
Goldblum, S. E. (2006) TNF-alpha increases tyrosine phosphorylation of
vascular endothelial cadherin and opens the paracellular pathway
through fyn activation in human lung endothelia, Am. J. Physiol.
Lung Cell. Mol. Physiol., 291, L1232-1245.
14.Herren, B., Levkau, B., Raines, E. W., and Ross,
R. (1998) Cleavage of beta-catenin and plakoglobin and shedding of
VE-cadherin during endothelial apoptosis: evidence for a role for
caspases and metalloproteinases, Mol. Biol. Cell, 9,
1589-1601.
15.Culic, O., Gruwel, M. L., and Schrader, J. (1997)
Energy turnover of vascular endothelial cells, Am. J. Physiol.,
273, C205-213.
16.Davidson, S. M., and Duchen, M. R. (2007)
Endothelial mitochondria: contributing to vascular function and
disease, Circ. Res., 100, 1128-1141.
17.Madamanchi, N. R., and Runge, M. S. (2007)
Mitochondrial dysfunction in atherosclerosis, Circ. Res.,
100, 460-473.
18.Schulz, E., Dopheide, J., Schuhmacher, S.,
Thomas, S. R., Chen, K., Daiber, A., Wenzel, P., Munzel, T., and
Keaney, J. F., Jr. (2008) Suppression of the JNK pathway by induction
of a metabolic stress response prevents vascular injury and
dysfunction, Circulation, 118, 1347-1357.
19.Demyanenko, I. A., Popova, E. N., Zakharova, V.
V., Ilyinskaya, O. P., Vasilieva, T. V., Romashchenko, V. P., Fedorov,
A. V., Manskikh, V. N., Skulachev, M. V., Zinovkin, R. A.,
Pletjushkina, O. Y., Skulachev, V. P., and Chernyak, B. V. (2015)
Mitochondria-targeted antioxidant SkQ1 improves impaired dermal wound
healing in old mice, Aging (Albany, N. Y.), 7,
475-485.
20.Sidibe, A., Mannic, T., Arboleas, M., Subileau,
M., Gulino-Debrac, D., Bouillet, L., Jan, M., Vandhuick, T., Le Loet,
X., Vittecoq, O., and Vilgrain, I. (2012) Soluble VE-cadherin in
rheumatoid arthritis patients correlates with disease activity:
evidence for tumor necrosis factor alpha-induced VE-cadherin cleavage,
Arthritis Rheum., 64, 77-87.
21.Galkin, I. I., Pletjushkina, O. Y., Zinovkin, R.
A., Zakharova, V. V., Birjukov, I. S., Chernyak, B. V., and Popova, E.
N. (2014) Mitochondria-targeted antioxidants prevent TNFα-induced
endothelial cell damage, Biochemistry (Moscow), 79,
124-130.
22.Algeciras-Schimnich, A., Barnhart, B. C., and
Peter, M. E. (2002) Apoptosis-independent functions of killer caspases,
Curr. Opin. Cell. Biol., 14, 721-726.
23.Shalini, S., Dorstyn, L., Dawar, S., and Kumar,
S. (2015) Old, new and emerging functions of caspases, Cell Death
Differ., 22, 526-539.
24.Lopez-Ramirez, M. A., Fischer, R.,
Torres-Badillo, C. C., Davies, H. A., Logan, K., Pfizenmaier, K., Male,
D. K., Sharrack, B., and Romero, I. A. (2012) Role of caspases in
cytokine-induced barrier breakdown in human brain endothelial cells,
J. Immunol., 189, 3130-3139.
25.Bannerman, D. D., Sathyamoorthy, M., and
Goldblum, S. E. (1998) Bacterial lipopolysaccharide disrupts
endothelial monolayer integrity and survival signaling events through
caspase cleavage of adherens junction proteins, J. Biol. Chem.,
273, 35371-35380.
26.Zehendner, C. M., Librizzi, L., De Curtis, M.,
Kuhlmann, C. R., and Luhmann, H. J. (2011) Caspase-3 contributes to
ZO-1 and Cl-5 tight-junction disruption in rapid anoxic neurovascular
unit damage, PLoS One, 6, e16760.
27.Bruewer, M., Luegering, A., Kucharzik, T.,
Parkos, C. A., Madara, J. L., Hopkins, A. M., and Nusrat, A. (2003)
Proinflammatory cytokines disrupt epithelial barrier function by
apoptosis-independent mechanisms, J. Immunol., 171,
6164-6172.
28.Petrache, I., Verin, A. D., Crow, M. T.,
Birukova, A., Liu, F., and Garcia, J. G. (2001) Differential effect of
MLC kinase in TNF-alpha-induced endothelial cell apoptosis and barrier
dysfunction, Am. J. Physiol. Lung Cell. Mol. Physiol.,
280, L1168-1178.
29.Antonenko, Y. N., Avetisyan, A. V., Bakeeva, L.
E., Chernyak, B. V., Chertkov, V. A., Domnina, L. V., Ivanova, O. Yu.,
Izyumov, D. S., Khailova, L. S., Klishin, S. S., Korshunova, G. A.,
Lyamzaev, K. G., Muntyan, M. S., Nepryakhina, O. K., Pashkovskaya, A.
A., Pletjushkina, O. Yu., Pustovidko, A. V., Roginsky, V. A.,
Rokitskaya, T. I., Ruuge, E. K., Saprunova, V. B., Severina, I. I.,
Simonyan, R. A., Skulachev, I. V., Skulachev, M. V., Sumbatyan, N. V.,
Sviryaeva, I. V., Tashlitsky, V. N., Vassiliev, J. M., Vyssokikh, M.
Yu., Yaguzhinsky, L. S., Zamyatnin, A. A., Jr., and Skulachev, V. P.
(2008) Mitochondria-targeted plastoquinone derivatives as tools to
interrupt execution of the aging program. 1. Cationic plastoquinone
derivatives: synthesis and in vitro studies, Biochemistry
(Moscow), 73, 1273-1287.
30.Edgell, C. J., McDonald, C. C., and Graham, J. B.
(1983) Permanent cell line expressing human factor VIII-related antigen
established by hybridization, Proc. Natl. Acad. Sci. USA,
80, 3734-3737.
31.Chen, C., Jamaluddin, M. S., Yan, S.,
Sheikh-Hamad, D., and Yao, Q. (2008) Human stanniocalcin-1 blocks
TNF-alpha-induced monolayer permeability in human coronary artery
endothelial cells, Arterioscler. Thromb. Vasc. Biol., 28,
906-912.
32.Polunovsky, V. A., Wendt, C. H., Ingbar, D. H.,
Peterson, M. S., and Bitterman, P. B. (1994) Induction of endothelial
cell apoptosis by TNF alpha: modulation by inhibitors of protein
synthesis, Exp. Cell Res., 214, 584-594.
33.Ohmori, Y., Schreiber, R. D., and Hamilton, T. A.
(1997) Synergy between interferon-gamma and tumor necrosis factor-alpha
in transcriptional activation is mediated by cooperation between signal
transducer and activator of transcription 1 and nuclear factor kappaB,
J. Biol. Chem., 272, 14899-14907.
34.Luo, T., and Xia, Z. (2006) A small dose of
hydrogen peroxide enhances tumor necrosis factor-alpha toxicity in
inducing human vascular endothelial cell apoptosis: reversal with
propofol, Anesth. Analg., 103, 110-116.
35.Stolpen, A. H., Guinan, E. C., Fiers, W., and
Pober, J. S. (1986) Recombinant tumor necrosis factor and immune
interferon act singly and in combination to reorganize human vascular
endothelial cell monolayers, Am. J. Pathol., 123,
16-24.
36.Dignat-George, F., and Sampol, J. (2000)
Circulating endothelial cells in vascular disorders: new insights into
an old concept, Eur. J. Haematol., 65, 215-220.
37.Blann, A. D., Woywodt, A., Bertolini, F., Bull,
T. M., Buyon, J. P., Clancy, R. M., Haubitz, M., Hebbel, R. P., Lip, G.
Y., Mancuso, P., Sampol, J., Solovey, A., and Dignat-George, F. (2005)
Circulating endothelial cells. Biomarker of vascular disease,
Thromb. Haemost., 93, 228-235.
38.Masckauchan, T. N., Shawber, C. J., Funahashi,
Y., Li, C. M., and Kitajewski, J. (2005) Wnt/beta-catenin signaling
induces proliferation, survival and interleukin-8 in human endothelial
cells, Angiogenesis, 8, 43-51.
39.Jaiswal, A. S., Marlow, B. P., Gupta, N., and
Narayan, S. (2002) Beta-catenin-mediated transactivation and cell-cell
adhesion pathways are important in curcumin (diferuylmethane)-induced
growth arrest and apoptosis in colon cancer cells, Oncogene,
21, 8414-8427.
40.Hermant, B., Bibert, S., Concord, E., Dublet, B.,
Weidenhaupt, M., Vernet, T., and Gulino-Debrac, D. (2003)
Identification of proteases involved in the proteolysis of vascular
endothelium cadherin during neutrophil transmigration, J. Biol.
Chem., 278, 14002-14012.
41.Navaratna, D., McGuire, P. G., Menicucci, G., and
Das, A. (2007) Proteolytic degradation of VE-cadherin alters the
blood-retinal barrier in diabetes, Diabetes, 56,
2380-2387.
42.Vilgrain, I., Sidibe, A., Polena, H., Cand, F.,
Mannic, T., Arboleas, M., Boccard, S., Baudet, A., Gulino-Debrac, D.,
Bouillet, L., Quesada, J. L., Mendoza, C., Lebas, J. F., Pelletier, L.,
and Berger, F. (2013) Evidence for post-translational processing of
vascular endothelial (VE)-cadherin in brain tumors: towards a candidate
biomarker, PLoS One, 8, e80056.
43.Wu, X., Guo, R., Chen, P., Wang, Q., and
Cunningham, P. N. (2009) TNF induces caspase-dependent inflammation in
renal endothelial cells through a Rho- and myosin light chain
kinase-dependent mechanism, Am. J. Physiol. Renal Physiol.,
297, F316-326.
44.Moon, S. K., Cha, B. Y., and Kim, C. H. (2004)
ERK1/2 mediates TNF-alpha-induced matrix metalloproteinase-9 expression
in human vascular smooth muscle cells via the regulation of NF-kappaB
and AP-1: involvement of the ras dependent pathway, J. Cell.
Physiol., 198, 417-427.
45.Huhtala, P., Chow, L. T., and Tryggvason, K.
(1990) Structure of the human type IV collagenase gene, J. Biol.
Chem., 265, 11077-11082.
46.Ichim, G., Lopez, J., Ahmed, S. U., Muthalagu,
N., Giampazolias, E., Delgado, M. E., Haller, M., Riley, J. S., Mason,
S. M., Athineos, D., Parsons, M. J., Van de Kooij, B., Bouchier-Hayes,
L., Chalmers, A. J., Rooswinkel, R. W., Oberst, A., Blyth, K., Rehm,
M., Murphy, D. J., and Tait, S. W. (2015) Limited mitochondrial
permeabilization causes DNA damage and genomic instability in the
absence of cell death, Mol. Cell, 57, 860-872.
47.Zinovkin, R. A., Romaschenko, V. P., Galkin, I.
I., Zakharova, V. V., Pletjushkina, O. Y., Chernyak, B. V., and Popova,
E. N. (2014) Role of mitochondrial reactive oxygen species in
age-related inflammatory activation of endothelium, Aging
(Albany, N. Y.), 6, 661-674.