Interaction of Chloramphenicol Tripeptide Analogs with Ribosomes
A. G. Tereshchenkov1, A. V. Shishkina2, V. N. Tashlitsky1, G. A. Korshunova2, A. A. Bogdanov1,2, and N. V. Sumbatyan1*
1Lomonosov Moscow State University, Faculty of Chemistry, 119991 Moscow, Russia; E-mail: sumbtyan@belozersky.msu.ru2Lomonosov Moscow State University, Belozersky Institute of Physico-Chemical Biology, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received September 24, 2015; Revision received November 18, 2015
Chloramphenicol amine peptide derivatives containing tripeptide fragments of regulatory “stop peptides” – MRL, IRA, IWP – were synthesized. The ability of the compounds to form ribosomal complexes was studied by displacement of the fluorescent erythromycin analog from its complex with E. coli ribosomes. It was found that peptide chloramphenicol analogs are able to bind to bacterial ribosomes. The dissociation constants were 4.3-10 µM, which is 100-fold lower than the corresponding values for chloramphenicol amine–ribosome complex. Interaction of the chloramphenicol peptide analogs with ribosomes was simulated by molecular docking, and the most probable contacts of “stop peptide” motifs with the elements of nascent peptide exit tunnel were identified.
KEY WORDS: ribosome, “stop peptides”, chloramphenicol, peptide derivatives, nascent peptide exit tunnelDOI: 10.1134/S000629791604009X
Abbreviations: Bhoc, N-benzhydryloxycarbonyl; Boc, tert-butyloxycarbonyl; BODIPY, (4,4-difluoro-4-bora-5,7-dimethyl)-3a,4a-diaza-s-indacene-3-pentanoic acid; Caeg, 3-(2-aminoethyl)-3-[2-(cytosin-1-yl)acetyl]glycine; DCC, 1,3-dicyclohexylcarbodiimide; DIPEA, diisopropylethylamine; Ery, erythromycin; Fmoc, fluorenylmethyloxycarbonyl; LC-MS, liquid chromatography-mass spectrometry; PNA, peptide-nucleic acids; PTC, peptidyl transferase center; RT, ribosomal tunnel; TFA, trifluoroacetic acid.
One of the important achievements of X-ray analysis of ribosome was the
proof of the existence of a tunnel located in its large subunit [1-3]. The start of the ribosome
tunnel (RT) overlaps with the ribosome peptidyl transferase center
(PTC). The main function of the RT is to ensure unhindered exit of
newly synthesized polypeptide chain from the ribosome during
translation and its delivery to the place where functionally complete
protein molecule formation begins [4-6]. The binding sites of many antibiotics are located
in the RT, and consequently, modification of rRNA nucleotide residues
and protein amino acid residues forming RT walls leads to bacterial
resistance to antibiotics [7, 8]. RT walls take part in amino acid sequence
monitoring of polypeptide chain moving along them and, in some cases,
polypeptides come into strong interactions with RT elements, which
results in translation arrest [4, 9]. The translation arrest is caused by the inhibition
of ribosome peptidyl transferase activity by relatively short segments
of the growing polypeptide chain called “stop peptides”,
which are usually encoded in the leader strands of the regulated genes
[9-12]. It appeared that RT
elements are able to interact not only with the nascent peptide chain,
but also with peptides coming from the outside. This was established by
X-ray and biochemical data for bacterial ribosomal complexes with
antimicrobial peptides, particularly oncocines. The structures of their
ribosomal complexes have recently been resolved by two scientific
groups [13, 14].
In recent years, the study of the ribosome and its complexes with antibiotics and peptides as ligands was studied using biochemical methods [15-19], X-ray analysis [13, 14], and cryoelectron microscopy [12, 20, 21]. It has been revealed that the RT is a dynamic functional system where specific interactions between the peptide chain and certain structural elements of the ribosome affect its functioning allosterically. One approach to the study of the principles of peptide chain recognition by the RT, as well as related mechanisms of translation regulation, is the creation of molecular probes based on ribosomal antibiotics, whose structure includes amino acid and peptide residues simulating the nascent polypeptide chain [19, 22-24].
In this work, novel peptide analogs of chloramphenicol modeling the 3′-terminal region of the peptidyl-tRNA were synthesized. The dissociation constants of the complexes of compounds obtained with E. coli ribosomes were measured by displacement of a fluorescent erythromycin analog from its ribosomal complexes, and molecular docking based modeling of these complexes was accomplished for determination of probable contacts of the peptides and RT. In the chloramphenicol analog structure, the dichloroacetic fragment is replaced by short regulatory “stop peptide” sequences (Fig. 1a), which will be oriented towards the RT when interacting with the ribosome (similarly to the growing polypeptide chain), with the amphenicol ring serving as an “anchor” for the molecule binding in the PTC A-site (Fig. 1b).
Fig. 1. a) Structures of chloramphenicol peptide analogs: AcMRL-CAM (I), AcIRA-CAM (II), AcIWP-CAM (III). b) Imposition of structures of E. coli ribosome complex with poly(Ala)-tRNA (PDB – 2WWQ) and ribosomal complex with AcIRA-CAM modeled by molecular docking. Peptidyl-tRNA and 23S rRNA are dark, and AcIRA-CAM is light colored.
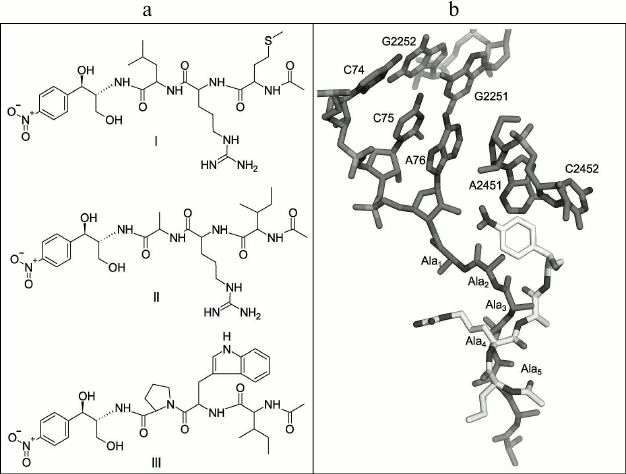
As the peptide fragments, short conserved peptide motifs were selected: MRL, IRA, IWP (Fig. 1a), that are met in the “stop peptide” C-terminal sequences, located at the PTC during translation (Table S1; see Supplement to this paper on the site of the journal (http://protein.bio.msu.ru/biokhimiya) and on Springer site (Link.springer.com)) [11, 18, 25].
In particular, the MRL-motif is found in sequences that when arranged in the ribosomal tunnel in the presence of antibiotics causes translational stalling of signal polypeptides encoded in the gene operon of the proteins responsible for resistance to macrolides [25]. The IRA motif is presented in the leader strand of SecM peptide that is responsible for regulation of SecA protein, playing an important role in E. coli protein secretion and membrane transport [26]. The IWP motif is characteristic for leader sequences regulating translation of E. coli proteins, including polyproline “stop peptides” defined by genetic selection [18].
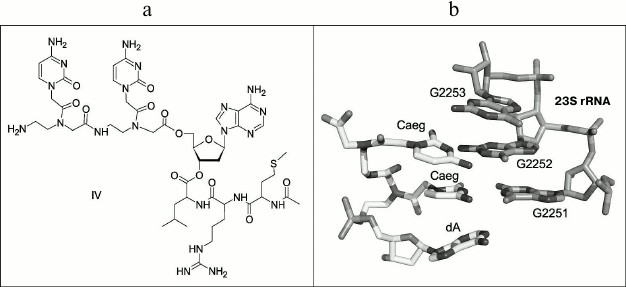
The conjugate containing peptide-nucleic acid (PNA) [27-29] in the 3′-terminal tRNA region and “stop peptide” MRL in the peptide part (Fig. 2a) was synthesized as another type of compound modeling peptidyl-tRNA for comparison. The conjugate of dipeptide-nucleotide, whose skeleton consists of N-(2-aminoethyl)-glycine moieties (aeg), with 2′-deoxyadenosine – CaegCaegdA – was designed as the PNA to address binding with ribosomal PTC. When interacting with the ribosome, the cytosine bases of this compound should be complementary to 23S rRNA nucleotide residues G2251 and G2252 that are used by peptidyl-tRNA for a specific binding with PTC P-site (Fig. 2b).
Fig. 2. a) Structure of peptidyl-tRNA analog: AcMetArgLeu-dACaegCaeg (IV). b) Static modeling of PNA CaegCaegdA interaction with PTC P-site of H. marismortui ribosome (PDB – 1VQ6).
MATERIALS AND METHODS
Chemicals used in this work were as follows: chloramphenicol from Sigma (China); BODIPY-OSu from Invitrogen (USA); amino acids, their derivatives, and reagents for peptide synthesis from Fluka (Switzerland), Merck (USA), and Reanal (Hungary); FmocCaeg(Bhoc)OH from Panagene (Korea). The fluorescent erythromycin derivative BODIPY-Ery was obtained previously [24]. The tripeptides AcMetArg(Pbf)LeuOH, AcIleArg(Pbf)AlaOH, and AcIleTrp(Boc)ProOH were synthesized and kindly provided by M. N. Zhmak (Department of Molecular Bases of Neurosignalization, Shemyakin–Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences).
TLC was performed on Kieselgel 60 F254 plates from Merck (Germany), and Silica gel 60 (0.063-0.200 and 0.040-0.063 mm) from Merck was used for column chromatography. The compounds containing UV-absorbing groups were detected with UV-cabinet Camag (England); the substances with free or Boc-protected amino groups were found by ninhydrin reactive; Sakaguchi and Ehrlich reagents were used for detection of compounds containing arginine with free guanidine group and tryptophan correspondingly.
UV absorption spectra were registered using a Cary 50 Bio spectrophotometer from Varian (Australia).
Fluorescence polarization was measured with a VICTOR™ X5 Multilabel Plate Reader (Perkin Elmer, USA). The excitation wavelength was 485 nm, and the emission wavelength was 530 nm.
Analytical reverse phase HPLC was performed on a Milichrom A-02 chromatograph (Econova, Russia) with ProntoSIL-120-5-C18 AQ (2.0 × 75 mm, 5 µm) column in gradient of acetonitrile (MeCN) in 0.1% trifluoroacetic acid (TFA) with elution rate 100 µl/min, and detection at 214 and 260 nm.
Analytical and preparative reverse phase HPLC was performed on a Knauer semipreparative chromatograph (Germany) with Beckman Coulter Ultrasphere ODS (10 × 250 mm, 5 µm) column in gradient of MeCN with elution rate 5 ml/min, and detection at 214, 260, and 500 nm.
Optical rotation of substances was measured on a VNIEKIProdmash EPO 1A instrument (Russia) at 598 nm.
MALDI-TOF mass-spectra were recorded on an UltrafleXtreme MALDI-time-of-flight-mass-spectrometer (Bruker Daltonics, Germany) equipped with UV-laser (Nd), in the positive ion mode with reflectron.
LC-MS (liquid chromatography with mass-spectrometry) was carried out with a UPLC/MS/MS system containing Acquity UPLC System chromatograph (Waters, USA), Acquity BEH C18 (2.1 × 50 mm, 1.7 µm) column (0.5 ml/min, 20 mM formic acid, gradient 5-100% MeCN for 3 min), and quadrupole mass-spectrometer TQD (Waters) (ESI MS1 in the positive ion mode).
1H NMR spectra were registered on a Bruker Avance 400 NMR spectrometer (Bruker, USA) with 400 MHz operating frequency for 1H nuclei.
Synthesis of peptide derivatives of chloramphenicol amine. (1R,2R)-2-amino-1-(4-nitrophenyl)propane-1,3-diol (chloramphenicol amine) (V) [30]. A solution of 1.0 g (3.1 mmol) of chloramphenicol in 15 ml 1 M aqueous HCl was heated on a boiling water bath for 2 h, and after cooling to room temperature the mixture was extracted with ether (4 × 20 ml). The aqueous layer was evaporated to dryness in vacuo. The target product was isolated from the residue by column chromatography on silica gel eluting with CHCl3–19% NH4OH–MeOH, 65 : 5 : 25 (v/v). Yield: 0.61 g (93%); TLC: Rf (CHCl3–19% NH4OH–MeOH, 65 : 5 : 25) 0.54, Rf (CHCl3–H2O–MeOH, 65 : 4 : 25) 0.19; m.p. 162-164°C (160-162°C [30]); [α]20D –18.2 (c = 3; MeOH) (–22.8 (c = 3; MeOH) [30]); 1H-NMR: (DMSO-d6, 294K, 400 MHz), δ (ppm): 1.74 (bs, 2H, -NH2), 2.72 (m, 1H, -CHNH2), 3.18 (dd, Jdd = 10.4 Hz, 6.0 Hz, 1H, -CH2aOH), 3.35 (dd, Jdd = 10.4 Hz, 5.7 Hz, 1H, -CH2bOH), 4.68 (d, Jd = 4.2 Hz, 1H, -CHOH), 7.59 (d, Jd = 8.6 Hz, 2H, m-H-NO2-Phe), 8.18 (d, Jd = 8.6 Hz, 2H, o-H-NO2-Phe).
AcMetArg(Pbf)Leu-CAM (VI). To a cold solution of 20 mg (28 µmol) of AcMetArg(Pbf)LeuOH and 3.2 mg (28 µmol) of N-hydroxysuccinimide in 300 µl of anhydrous CH2Cl2, 7 mg (34 µmol) of DCC was added at 0°C. The mixture was stirred for 2 h at 0°C, then overnight at 4°C. The formed precipitate was filtered off, and solvent was removed in vacuo. The residue was dissolved in 250 µl of DMF and added to a solution of 6 mg (28 µmol) of V and 4.9 µl (28 µmol) of DIPEA in 40 µl of DMF. The resulting mixture was stirred for 7 h at room temperature, then overnight at 4°C. The reaction mixture was diluted with 3 ml of CHCl3, washed with water (3 × 2 ml). Organic layer was dried with Na2SO4, and the volatiles were evaporated in vacuo. The target product was isolated from the residue by purification on a silica gel column eluting with CHCl3–MeOH, 9 : 1. Yield: 20 mg (78%); TLC: Rf (CHCl3–MeOH, 9 : 1) 0.24, Rf (CHCl3–EtOAc–MeOH, 6 : 3 : 0.5) 0.11, Rf (CHCl3–19% NH4OH–MeOH, 65 : 5 : 25) 0.86, Rf (CHCl3–MeOH, 4 : 1) 0.91; UV (H2O): λmax – 224 nm, 258 nm; LC-MS, m/z calculated for C41H63N8O11S2+ – 907.4; found – 908.0; tR = 1.96 min.
TFA·AcMetArgLeu-CAM (I). A mixture of 2.48 ml of TFA, 150 µl of phenol, 150 µl of water, 150 µl m-cresol, and 75 mg of dithiothreitol was deaerated by bubbling with nitrogen for a few minutes. The resulted mixture was added to 20 mg (22 µmol) of VI and stirred for 4 h at room temperature under nitrogen. The volatiles were evaporated in vacuo, and the residue was dissolved in a minimal amount of water and treated with ether. The resulting precipitate was filtered. The target product was isolated by HPLC. Yield: 14.5 mg (86%); TCX: Rf (CHCl3–H2O–MeOH, 65 : 4 : 25) 0.16, Rf (CHCl3–19% NH4OH–MeOH, 65 : 5 : 25) 0.29; HPLC: tR = 16 min (Beckman Coulter Ultrasphere ODS (10 × 250 mm, 5 µm), 5 ml/min, 0-60% MeCN at 20 min); UV (H2O): λmax – 279 nm; LC-MS, m/z calculated for C28H47N8O8S+ – 655.3; found – 655.7; tR = 1.10 min.
TFA·AcIleArgAla-CAM (II) was obtained as I, from 50 mg (77 µmol) of AcIleArg(Pbf)AlaOH, 9 mg (77 µmol) of N-hydroxysuccinimide, 19 mg (92 µmol) of DCC, 13 µl (77 µmol) of DIPEA, and 16 mg (77 µmol) of V. Yield: 31 mg (57%); TLC: Rf (BuOH–H2O–AcOH, 4 : 1 : 1) 0.19, Rf (CHCl3–H2O–MeOH, 65 : 4 : 25) 0.11; UV (H2O): λmax – 280 nm; LC-MS, m/z calculated for C26H43N8O8+ – 595.3; found – 595.7; tR = 0.89 min.
AcIleTrpPro-CAM (III) was obtained as I, from 50 mg (90 µmol) of AcIleTrp(Boc)ProOH, 10 mg (90 µmol) of N-hydroxysuccinimide, 22 mg (108 µmol) of DCC, 15.6 µl (90 µmol) of DIPEA, and 19 mg (90 µmol) of V. Yield: 51 mg (87%); TLC: Rf (CHCl3–MeOH, 9 : 1) 0.33; UV (H2O): λmax – 224 and 280 nm; LC-MS, m/z calculated for C33H43N6O8+ – 651.3; found – 651.8; tR = 1.53 min.
Syntheses of AcMRL-dACaegCaeg (IV) and MRL-OMe peptide (VII) are described in the Supplement (see Figs. S1 and S2).
BODIPY-MetArgLeu-OMe (VIII). To a solution of 0.5 mg (0.76 µmol) MetArgLeu-OMe × 2TFA in 100 µl of 0.1 N aqueous NaHCO3, 0.58 mg (1.39 µmol) BODIPY-OSu in 80 µl MeCN was added, and the resulting mixture was stirred for 3 h at room temperature. The target compound was isolated by HPLC (Beckman Coulter Ultrasphere ODS; 10 × 250 mm, 5 µm; 5 ml/min, 0-100% MeCN at 20 min). Yield: 0.32 mg (57%): TLC: Rf (CHCl3–MeOH, 9 : 1) 0.05, Rf (CHCl3–MeOH, 3 : 1) 0.37; HPLC: tR = 16.4 min; UV (H2O): λmax – 227, 365, and 505 nm; LC-MS, m/z calculated for C34H54BF2N8O5S+ – 735.4, found – 735.8.
Molecular docking was performed in AutoDock Vina [31]. The crystal structure of the E. coli ribosome with chloramphenicol (PDB: 3OFC) [32], from which the water molecules, inorganic ions, and low molecular weight ligands were removed, was used as a receptor. The molecular docking region was determined as a cube with the center in the chloramphenicol-binding site and with 34 Å sides. The initial conformations of ligands were pre-optimized by a semiempirical method in MOPAC2012 with PM7 Hamiltonian [33]. The “exhaustiveness” parameter, characterizing the docking accuracy, was equal to 100. The data were graphically displayed using PyMOL 1.5.
Investigation of ligand binding to bacterial E. coli ribosomes [34]. 70S ribosomes of E. coli strain MRE-600 were kindly provided by A. L. Konevega (Laboratory of Protein Biosynthesis, Molecular and Radiation Biophysics Division, Petersburg Nuclear Physics Institute (PNPI)). Ribosome concentration was determined by optical density at 260 nm (A260 = 1 a.u. at C(Rs) = 24 nM). BIND buffer was used with the following composition: 20 mM HEPES, pH 7.5, 50 mM NH4Cl, 10 mM MgCl2, 0.05% Tween 20. Escherichia coli 70S ribosomes were thawed at 0°C, followed by incubation for 15 min at 37°C.
The dissociation constants of the investigated compounds were determined by replacement of fluorescently labeled erythromycin (BODIPY-Ery) from the complex with ribosomes. BODIPY-Ery (4 nM) was incubated with ribosomes (24 nM) for 30 min at 25°C in 384-well plate. The solution of rival ligand in concentrations from 1 µM to 1 mM was added to the formed ribosomal complex. The mixture was incubated for 2 h until equilibrium was reached, and the values of the polarization of fluorescence were measured and then converted to anisotropy (mA). The calculations of dissociation constants of the complexes of peptide derivatives with ribosomes were performed basing on two replicate measurements. The confidence interval (95%) was calculated for the presented dissociation constant values.
With the goal to obtain BODIPY-MetArgLeu-OMe binding curves, solutions of ribosomes in different concentrations (from 1 to 7000 nM) in BIND buffer were incubated with the fluorescently labeled peptide (4 nM) solution at 25°C for 1 h to reach equilibrium, and then the fluorescence polarization values were measured.
The data for binding and fluorescent substrate replacement experiments were processed using the GraphPad Prizm 6 computer program.
RESULTS
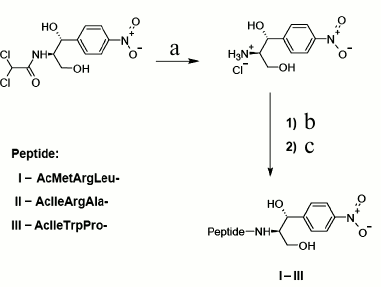
Synthesis. Novel chloramphenicol amine analogs were synthesized: N-acetyl-L-methionyl-L-arginyl-L-leucyl-[(1R,2R)-2-hydroxy-2-(4-nitrophenyl)-1-hydroxymethyl-ethyl]-amide (AcMRL-CAM, I), N-acetyl-L-isoleucyl-L-arginyl-L-alanyl-[(1R,2R)-2-hydroxy-2-(4-nitrophenyl)-1-hydroxymethyl-ethyl]-amide (AcIRA-CAM, II), N-acetyl-L-isoleucyl-L-tryptophyl-L-prolyl-[(1R,2R)-2-hydroxy-2-(4-nitrophenyl)-1-hydroxymethyl-ethyl]-amide (AcIWP-CAM, III). The synthetic scheme comprised chloramphenicol hydrolysis to chloramphenicol amine [30], acylation of the latter by N-acetyl-peptides with the side groups protected, and final deprotection (Fig. 3).
Fig. 3. Scheme of synthesis of peptide chloramphenicol analogs. AcMRL-CAM (I): a) 1 M HCl, 100°C; b) AcMetArg(Pbf)Leu-OSu, DMF, DIPEA; c) TFA, PhOH, H2O, m-cresol, DTT, 33 : 2 : 2 : 2 : 1 (v/v). AcIRA-CAM (II): a) 1 M HCl, 100°C; b) AcIleArg(Pbf)Ala-OSu, DMF, DIPEA; c) TFA, PhOH, H2O, m-cresol, DTT, 33 : 2 : 2 : 2:1 (v/v). AcIWP-CAM (III): a) 1 M HCl, 100°C; b) AcIleTrp(Boc)Pro-OSu, DMF, DIPEA; c) TFA, PhOH, H2O, m-cresol, DTT, 33 : 2 : 2 : 2 : 1 (v/v).
The conjugate of PNA with “stop peptide” AcMetArgLeu-dACaegCaeg (IV) was also obtained synthetically. Its structure is shown in Fig. 2a, and its synthesis is depicted in Fig. S1 in the Supplement.
The fluorescent derivative BODIPY-MetArgLeu-OMe (VIII) was synthesized by a reaction between succinimide ester BODIPY-OSu and peptide MRL-OMe (VII), obtained according to the scheme shown in Fig. S2 in the Supplement.
Study of binding of chloramphenicol analogs and PNA to E. coli ribosomes. The interaction between peptide derivatives and ribosomes was studied via replacement of BODIPY-Ery from its complexes with E. coli ribosomes by these compounds. There was a decrease in fluorescence polarization, indicating competition of the tested compounds with erythromycin for the corresponding binding site in RT (Fig. 4).
Fig. 4. Curves of replacement of BODIPY-Ery from its complex with 70S E. coli ribosomes by the investigated compounds. a: 1) chloramphenicol; 2) AcIRA-CAM (II); 3) AcMRL-CAM (I); 4) AcIWP-CAM (III); b: 5) AcMRL-dACaegCaeg (IV); 6) MRL-OMe (VII); 7) chloramphenicol amine (V). BODIPY-Ery concentration was 4 nM, ribosomes – 24 nM. For each point the mean value ± s.d. is given.
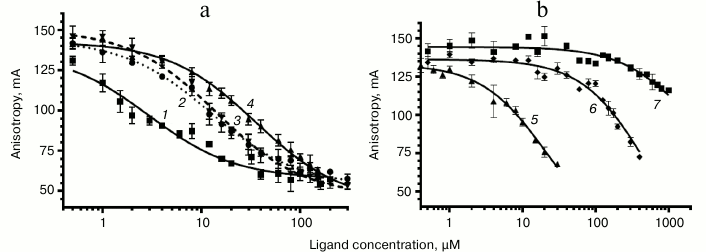
To quantify the affinity of the peptide derivatives to the ribosomes, the data on BODIPY-Ery replacement was approximated using a function corresponding to the solution of the cubic equation that describes the competitive binding of two ligands in one site [35]. We choose this model based on the fact that the ribosomal binding sites of chloramphenicol peptide derivatives and erythromycin overlap. The binding areas of chloramphenicol and erythromycin differ spatially; however, it appears that both antibiotics cannot simultaneously be complexed with the ribosome. This indicates the allosteric nature of chloramphenicol displacement of fluorescently labeled erythromycin, for which the same mathematical description can be applied. The dissociation constant value of the BODIPY-Ery ribosomal complex used for this calculation was measured previously and was 16.4 nM [36]. The KD obtained for the test compounds are presented in the table.
Peptide derivatives and dissociation constants of their complexes with
ribosomes (± 95% confidence interval)
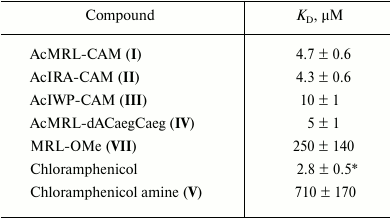
* The corresponding KD value determined by
[14C]chloramphenicol replacement is 1.7 µM [19].
As seen from the table, the dissociation constants of chloramphenicol peptide analogs lie in the range of 4.3-10 µM. The data suggests that the peptide fragment increases the affinity of the compounds to the ribosome around two hundred times more to the level characteristic for chloramphenicol amine. At the same time, the MRL-OMe (VII) peptide itself does not exhibit any affinity to RT, underscoring the importance of the anchor role of the chloramphenicol amine. This is confirmed by the fact that the fluorescently labeled peptide BODIPY-MetArgLeu-OMe (VIII) also very poorly interacts with E. coli ribosomes (see Fig. S3 in Supplement). Binding of compound IV with the RT is flushed with chloramphenicol analogs I and II, which is apparently due to similarity in structures of the peptide moieties.
Molecular docking. Molecular docking in 70S ribosomes was performed for peptide derivatives of chloramphenicol amine. The X-ray data of the ribosomal complex with chloramphenicol bound in the PTC A-site (PDB: 3OFC) were used for the investigation. From the molecular docking, 20 structures for every compound were obtained and ranged by the energy of binding with the ribosome. The structures where peptide fragments were oriented towards the RT and the nitrophenyl group was located in the chloramphenicol-binding site were chosen for further analysis.
According to the molecular docking results, the peptide fragment of the AcMRL-CAM molecule (I) is surrounded by residues C2610-2612, G2505, U2506, and G2581 (Fig. 5a). Thus, the hydrogen atom at the methionine α-nitrogen atom forms a hydrogen bond with atom O1P G2505 (not shown), while the carbonyl oxygen atom of the amino acid can form hydrogen bonds with bases C2611 and C2612. The arginine guanidine group is close to the C2611 phosphate and forms hydrogen bonds with the O2 atom of C2610 and O6 atom of G2581.
Fig. 5. Structures of E. coli ribosome complexes with peptide derivatives of chloramphenicol amine obtained by molecular docking. a) AcMRL-CAM (I); b) AcIRA-CAM (II); c) AcIWP-CAM (III). The dotted lines show possible hydrogen bonds.
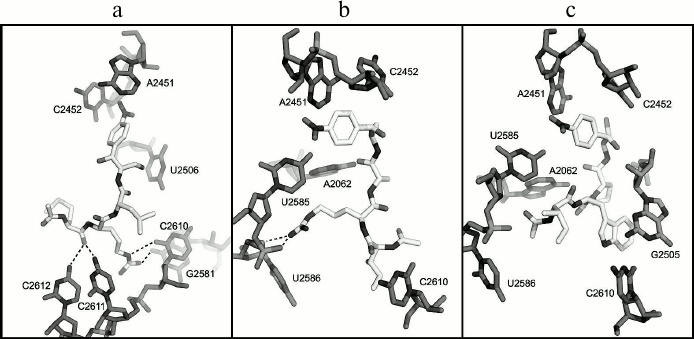
In the case of the AcIRA-CAM (II) derivative, the arginine side chain is surrounded by nucleotides A2062, U2585, U2586, and its guanidine group can form hydrogen bonds with O4′, O5′, and O2P atoms of U2586 (Fig. 5b). The α-N hydrogen atom of isoleucine forms a hydrogen bond with the U2506 O4 atom (not shown), and the amino acid side chain is in close proximity to the C2610 base.
It was revealed for compound AcIWP-CAM (III) that its tryptophan indole ring comes into stacking with the G2505 base (Fig. 5c).
DISCUSSION
It is known that the removal of the dichloroacetyl moiety from the chloramphenicol molecule results in a significant decrease in biological activity [30] that is apparently due to a sharp decrease in the ability of the chloramphenicol amine to bind to the ribosome (table). These results confirm our hypothesis that a short peptide chain administration instead of dichloroacetyl fragment into the chloramphenicol analog molecular structure can compensate interactions critical for antibiotic binding to the ribosome (table) [37, 38]. It has been earlier elucidated that pentapeptide chloramphenicol analogs, different in amino acid sequence from the known “stop peptides”, are able to bind to the E. coli ribosome in a way that the peptide fragment of the molecule is located in the RT [19]. The values of the dissociation constants of complexes of these compounds with ribosomes were in the range of 2.5-36 µM.
Greater or lesser affinity of the investigated derivatives to RT elements is likely explained by the nature of the amino acid side chains. As seen from Fig. 1b, the first amino acid residue of the peptide attached to chloramphenicol amine corresponds to the position of the third amino acid from C-terminus of the peptidyl-tRNA, which is bound to the ribosome. Thus, the MRL, IRA, and IWP peptides linked to chloramphenicol amine amino group will correspond to peptides MRLXX, IRAXX, and IWPXX attached to the tRNA. With this in mind, and from comparison of the dissociation constants of compounds II and III, it follows that the presence of the Arg residue at the second position from the C-terminus (at the fourth from the 3′-tRNA) is more preferable for strong binding than Trp. This is confirmed by the fact that the Arg7 residue of regulatory peptide ErmBL, being at the fourth position from the 3′-tRNA end, is crucial for translation arrest [20]. Judging by cryoelectron microscopy data, it apparently interacts with U2586 nucleotide, and it is also observed for the arginine residue of peptide chloramphenicol analog II based on molecular docking (Fig. 5b). Moreover, the arginine side chain in the complex of II with the E. coli ribosome is located approximately in the same environment as Arg163 of SecM peptide [26]. This suggests that residues Arg7 in ErmBL and Arg163 in SecM form with rRNA nucleotide moieties an ensemble of hydrogen bonds similar to that formed by the arginine residue in compound II.
It should also be noted that according to the molecular docking results, in most structures the peptide fragment of the synthesized compounds locates analogously to the nascent polypeptide chain in ribosomal complexes of the regulatory “stop peptides” TnaC (PDB: 4V5H) [39] and MifM (PDB: 3J9W) [40], whose structure was obtained by cryoelectron microscopy.
The affinity of PNA conjugate with MRL “stop peptide” (IV) to the ribosome is flush with the corresponding analog of chloramphenicol (I), which likely mimics a similar orientation of peptide residues of investigated compounds in the RT.
Thus, using tripeptide analogs of chloramphenicol with the “stop peptides” it was demonstrated that short peptide fragments are able to provide the interaction of these compounds with E. coli ribosomes at a level close to the original antibiotic. Substances of this type are quite promising for the study of the interaction between regulatory peptides and the ribosomal tunnel.
The authors are grateful to M. N. Zhmak for tripeptide synthesis, to A. L. Konevega for ribosomes given for investigation, to M. V. Serebryakova for mass-spectrometry analyses, and to Yu. K. Grishin for NMR spectra.
This research was supported by the Russian Science Foundation (project No. 14-50-00029, molecular modeling) and by the Russian Foundation for Basic Research (projects Nos. 13-04-00986-a, synthesis of chloramphenicol analogs; 13-04-40211-H, study of analogs binding to bacterial ribosomes). The work was carried out using equipment purchased with funds of Lomonosov Moscow State University Program of Development.
REFERENCES
1.Ban, N., Nissen, P., Hansen, J., Moore, P. B., and
Steitz, T. A. (2000) The complete atomic structure of the large
ribosomal subunit at 2.4 Å resolution, Science,
289, 905-920.
2.Nissen, P., Hansen, J., Ban, N., Moore, P. B., and
Steitz, T. A. (2000) The structural basis of ribosome activity in
peptide bond synthesis, Science, 289, 920-930.
3.Harms, J., Schluenzen, F., Zarivach, R., Bashan,
A., Gat, S., Agmon, I., Bartels, H., Franceschi, F., and Yonath, A.
(2001) High-resolution structure of the large ribosomal subunit from a
mesophilic eubacterium, Cell, 107, 679-688.
4.Bogdanov, A. A., Sumbatyan, N. V., Shishkina, A.
V., Karpenko, V. V., and Korshunova, G. A. (2010) Ribosomal tunnel and
translation regulation, Biochemistry (Moscow), 75,
1501-1516.
5.Kolb, V. A. (2010) Properties of intraribosomal
part of nascent polypeptide, Biochemistry (Moscow), 75,
1517-1527.
6.Wilson, D. N., and Beckman, R. (2011) The ribosomal
tunnel as a functional environment for nascent polypeptide folding and
translational stalling, Curr. Opin. Struct. Biol., 21,
274-282.
7.Subramanian, S. L., Ramu, H., and Mankin, A. S.
(2012) in Antibiotic Discovery and Development (Dougherty, T.
J., and Pucci, M. J., eds.) Springer, pp. 455-484.
8.LaMarre, J., Mendes, R. E., Szal, T., Schwarz, S.,
Jones, R. N., and Mankin, A. S. (2013) The genetic environment of the
cfr gene and the presence of other mechanisms account for the
very high linezolid resistance of Staphylococcus epidermidis
isolate 426-3147L, Antimicrob. Agents Chemother., 57,
1173-1179.
9.Mankin, A. S. (2006) Nascent peptide in the
“birth canal” of the ribosome, Trends Biochem. Sci.,
31, 11-13.
10.Cruz-Vera, L. R., Sachs, M. S., Sguires, C. L.,
and Yanofsky, C. (2011) Nascent polypeptide sequences that influence
ribosome function, Curr. Opin. Microbiol., 14,
160-166.
11.Ito, K., and Chiba, S. (2013) Arrest peptides:
cis-acting modulators of translation, Annu. Rev.
Biochem., 82, 171-202.
12.Arenz, S., Meydan, S., Starosta, A. L.,
Berninghausen, O., Beckmann, R., Vazquez-Laslop, N., and Wilson, D. N.
(2014) Drug sensing by the ribosome induces translational arrest via
active site perturbation, Mol. Cell, 56, 446-452.
13.Roy, R. N., Lomakin, I. B., Gagnon, M. G., and
Steitz, T. A. (2015) The mechanism of inhibition of protein synthesis
by the proline-rich peptide oncocin, Nat. Struct. Mol. Biol.,
22, 466-469.
14.Seefeldt, A. C., Nguyen, F., Antunes, S.,
Perebaskine, N., Graf, M., Arenz, S., Inampudi, K. K., Douat, C.,
Guichard, G., Wilson, D. N., and Innis, C. A. (2015) The proline-rich
antimicrobial peptide Onc112 inhibits translation by blocking and
destabilizing the initiation complex, Nat. Struct. Mol. Biol.,
22, 470-475.
15.Hansen, J. L., Moore, P. B., and Steitz, T. A.
(2003) Structures of five antibiotics bound at the peptidyl transferase
center of the large ribosomal subunit, J. Mol. Biol.,
330, 1061-1075.
16.Schlunzen, F., Zarivach, R., Harms, J., Bashan,
A., Tocilj, A., Albrecht, R., Yonath, A., and Franceschi, F. (2001)
Structural basis for the interaction of antibiotics with the peptidyl
transferase center in eubacteria, Nature, 413,
814-821.
17.Lu, J., Hua, Z., Kobertz, W. R., and Detsch, C.
(2013) Nascent peptide side-chains induce rearrangements in distinct
locations of the ribosomal tunnel, J. Mol. Biol., 411,
499-510.
18.Woolstenhulme, C. J., Parajuli, S., Healey, D.
W., Valverde, D. P., Petersen, E. N., Starosta, A. L., Guydosh, N. R.,
Johnson, W. E., Wilson, D. N., and Buskirk, A. R. (2013) Nascent
peptides that block protein synthesis in bacteria, Proc. Natl. Acad.
Sci. USA, 110, 878-887.
19.Mamos, P., Krokidis, M. G., Papadas, A.,
Karahalios, P., Starosta, A. L., Wilson, D. N., Kalpaxis, D. L., and
Dinos, G. P. (2013) On the use of the antibiotic chloramphenicol to
target polypeptide chain mimics to the ribosomal exit tunnel,
Biochimie, 95, 1765-1772.
20.Arenz, S., Ramu, H., Gupta, P., Berninghausen,
O., Beckmann, R., Vazquez-Laslop, N., Mankin, A. S., and Wilson, D. N.
(2014) Molecular basis for erythromycin-dependent ribosome stalling
during translation of the ErmBL leader peptide, Nat. Commun.,
5, 3501.
21.Fischer, N., Neumann, P., Konevega, A. L., Bock,
L. V., Ficner, R., Rodnina, M. V., and Stark, H. (2015) Structure of
the E. coli ribosome–EF-Tu complex at <3 Å
resolution by Cs-corrected cryo-EM, Nature, 520,
567-570.
22.Sumbatyan, N. V., Korshunova, G. A., and
Bogdanov, A. A. (2003) Peptide derivatives of antibiotics tylosin and
desmycosin, protein synthesis inhibitors, Biochemistry (Moscow),
68, 1156-1158.
23.Starosta, A. L., Karpenko, V. V., Shishkina, A.
V., Mikolajka, A., Sumbatyan, N. V., Schluenzen, F., Korshunova, G. A.,
Bogdanov, A. A., and Wilson, D. N. (2010) Interplay between the
ribosomal tunnel, nascent chain, and macrolides influences drug
inhibition, Chem. Biol., 17, 504-514.
24.Shishkina, A., Makarov, G., Tereshchenkov, A.,
Korshunova, G., Sumbatyan, N., Golovin, A., Svetlov, M., and Bogdanov,
A. (2013) Conjugates of amino acids and peptides with
5-O-mycaminosyltylonolide and their interaction with the ribosomal exit
tunnel, Bioconj. Chem., 24, 1861-1869.
25.Vazquez-Laslop, N., Ramu, H., and Mankin, A.
(2011) in Ribosomes: Structure, Function and Dynamics (Rodnina,
M. V., Wintermeyer, W., and Green, R., eds.) Springer, Wien-N.Y., pp.
377-392.
26.Gumbart, J., Schreiner, E., Wilson, D., Beckmann,
R., and Schulten, K. (2012) Mechanism of SecM-mediated stalling in the
ribosome, Biophys. J., 103, 331-341.
27.Nielsen, P. E. (1998) Structural and biological
properties of peptide nucleic acid (PNA), Pure Appl. Chem.,
70, 105-110.
28.Lundin, K. E., Good, L., Stromberg, R., Graslund,
A., and Smith, C. I. E. (2006) in Advances in Genetics (Hall, J.
C., ed.), Vol. 56, Academic Press, Waltham, pp. 1-51.
29.Good, L., and Nielsen, P. E. (1998) Inhibition of
translation and bacterial growth by peptide nucleic acid targeted to
ribosomal RNA, Proc. Natl. Acad. Sci. USA, 3,
2073-2076.
30.Rebstock, M. C., Crooks, H. M., Controulis, J.,
and Bartz, Q. R. (1949) Chloramphenicol (Chloromycetin). IV. Chemical
Studies, J. Am. Chem. Soc., 71, 2458-2462.
31.Trott, O., and Olson, A. J. (2010) AutoDock Vina:
improving the speed and accuracy of docking with a new scoring
function, efficient optimization, and multithreading, J. Comput.
Chem., 31, 455-461.
32.Dunkle, J. A., Xiong, L., Mankin A. S., and Cate,
J. H. D. (2010) Structures of the Escherichia coli ribosome with
antibiotics bound near the peptidyl transferase center explain spectra
of drug action, Proc. Natl. Acad. Sci. USA, 107,
17152-17157.
33.Stewart, J. J. (2013) Optimization of parameters
for semiempirical methods VI: more modifications to the NDDO
approximations and re-optimization of parameters, J. Mol.
Model., 19, 1-32.
34.Yan, K., Hunt, E., Berge, J., May, E., Copeland,
R. A., and Gontarek, R. R. (2005) Fluorescence polarization method to
characterize macrolide–ribosome interactions, Antimicrob.
Agents Chemother., 49, 3367-3372.
35.Wang, Z. X. (1995) An exact mathematical
expression for describing competitive binding of two different ligands
to a protein molecule, FEBS Lett., 360, 111-114.
36.Shishkina, A. V., Tereshchenkov, A. G.,
Sumbatyan, N. V., Korshunova, G. A., and Bogdanov, A. A. (2013)
Characterization of tylosin-related macrolides–ribosome
interactions by fluorescence polarization method, FEBS J.,
280 (Suppl. 1), 356.
37.Tereshchenkov, A., Sergeeva, V., Shishkina, A.,
Sumbatyan, N., and Bogdanov, A. (2014) in EMBO Conference Series:
Chemical Biology 2014, Mera Druck GmbH, Sanghausen, pp.
263-263.
38.Tereshchenkov, A. G. (2013) in Kazan Summer
School on Chemoinformatics, Innovation Publishing House
“Butlerov Heritage”, Kazan, pp. 33-33.
39.Seidelt, B., Innis, C. A., Wilson, D. N.,
Gartmann, M., Armache, J.-P., Villa, E., Trabuco, L. G., Becker, T.,
Mielke, T., Schulten, K., Steitz, T. A., and Beckmann, R. (2009)
Structural insight into nascent polypeptide chain-mediated
translational stalling, Science, 326, 1412-1415.
40.Sohmen, D., Chiba, S., Shimokawa-Chiba, N.,
Innis, C. A., Berninghausen, O., Beckmann, R., Ito, K., and Wilson, D.
N. (2015) Structure of the Bacillus subtilis 70S ribosome
reveals the basis for species-specific stalling, Nat. Commun.,
6, 6941.
Supplementary Table and Figures (PDF)