Plant DNA Methyltransferase Genes: Multiplicity, Expression, Methylation Patterns
V. V. Ashapkin*, L. I. Kutueva, and B. F. Vanyushin
Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; fax: +7 (495) 939-3181; E-mail: basilashapkin@gmail.com; ashapkin@genebee.msu.ru* To whom correspondence should be addressed.
Received August 16, 2015; Revision received September 11, 2015
Expression and methylation patterns of genes encoding DNA methyltransferases and their functionally related proteins were studied in organs of Arabidopsis thaliana plants. Genes coding for the major maintenance-type DNA methyltransferases, MET1 and CMT3, and the major de novo-type DNA methyltransferase, DRM2, are actively expressed in all organs. Similar constitutively active expression was observed for genes encoding their functionally related proteins, a histone H3K9 methyltransferase KYP and a catalytically non-active protein DRM3. Expression of the MET1 and CMT3 genes is significantly lower in developing endosperm compared with embryo. Vice versa, expression of the MET2a, MET2b, MET3, and CMT2 genes in endosperm is much more active compared with embryo. A special maintenance DNA methylation system seems to operate in endosperm. The DNMT2 and N6AMT genes encoding putative methyltransferases are constitutively expressed at low levels. CMT1 and DRM1 genes are expressed rather weakly in all investigated organs. Most of the studied genes have methylation patterns conforming to the “body-methylated gene” prototype. A peculiar feature of the MET family genes is methylation at all three possible site types (CG, CHG, and CHH). The most weakly expressed among genes of their respective families, CMT1 and DRM1, are practically unmethylated. The MET3 and N6AMT genes have unusual methylation patterns, promoter region, and most of the gene body devoid of any methylation, and the 3′-end proximal part of the gene body is highly methylated.
KEY WORDS: DNA methylation, DNA methyltransferase, gene expression, quantitative PCR, bisulfite sequencing, Arabidopsis thalianaDOI: 10.1134/S0006297916020085
Abbreviations: m5C, 5-methylcytosine; Nm6A, N6-methyladenine; Nm4C, N4-methylcytosine; Nm2G, N2-methylguanine; RdDM, RNA-directed DNA methylation; siRNA, small interfering RNA.
DNA methylation is the best studied and most stable of all epigenetic
modifications in plants [1, 2].
Detailed maps of the Arabidopsis genome methylation (methylomes)
showed that 24% of the symmetric CG sites, 6.7% of the symmetric CHG
sites, and 1.7% of the asymmetric CHH sites are methylated in this
plant (where H is any nucleotide except G) [3, 4]. Considering the genome frequency of the respective
target sites, ~55% of all methylated cytosine residues are in CG, ~23%
are in CHG, and ~22% are in CHH sites. Thus, contrary to the popular
view, CG sites are not the predominant DNA methylation targets in
plants. The CG sites usually have a binary mode of methylation, being
either not methylated at all or nearly completely methylated. The
methylation levels of CHG sites are widely variable (between 0 and
100%), whereas the methylation levels of asymmetric sites rarely exceed
10%. All three types of the methylated sites are present in repeat- and
transposon-rich pericentromeric regions, whereas mostly CG sites are
methylated in gene bodies. DNA methylation in plants is carried out by
a large set of specific DNA methyltransferases, some of which have no
analogs in animals [1, 5]. For
example, a minimum of 10 genes encoding putative cytosine DNA
methyltransferases were found in the sequenced Arabidopsis
thaliana genome, more than in any other eukaryotic genome studied
by that time [5]. By a preference of either
unmethylated or hemimethylated substrates, the cytosine DNA
methyltransferases are classified as de novo or maintenance
type, respectively, whereas by the primary structure characteristics
five main families are recognized, named by their prototype members,
Dnmt1, Dnmt2, Dnmt3, chromomethyltransferase (CMT), and Masc1 [6, 7]. There are
methyltransferases of the first three families, common to plants and
mammals, and the plant-specific CMTs in Arabidopsis and other
plants [8]. Maintenance methylation of CG sites in
all genome compartments is carried out by DNA methyltransferase MET1, a
homolog of Dnmt1 playing a similar role in animals. It methylates the
daughter DNA chains immediately following replication. Three proteins
of the VARIANT IN METHYLATION family, VIM1-VIM3, that recognize
hemimethylated CG sites in the newly replicated DNA with the aid of a
special SRA domain, are helpers in this methylation [9]. The plant-specific DNA methyltransferase CMT3
carries out, analogously to MET1, maintenance methylation of symmetric
CHG sites, and it participates in CHH site methylation. Unlike MET1,
CMT3 has no strongly pronounced preference for the hemimethylated sites
and is considerably less effective in converting them to fully
methylated ones. A chromodomain of CMT3 recognizes nucleosomes
containing the H3 histone molecules methylated at the ninth lysine
residue (H3K9me1, H3K9me2, and H3K9me3) [10],
whereas the H3K9 methyltransferases KYP (SUVH4), SUVH5, and SUVH6
contain an SRA domain recognizing the DNA loci methylated at sites of
any type with a clear-cut preference for m5CHG and
m5CHH [11]. Thus, the CMT3 maintenance
activity in vivo is a consequence not of its own substrate
preferences, but rather of mutual positive influences of CMT3 and H3K9
methyltransferases. DRM1 and DRM2 DNA methyltransferases, homologs of
the animal de novo type DNA methyltransferases Dnmt3, are
responsible for de novo methylation at all sites [12]. Target sites to be de novo methylated are
recognized with the aid of 24-nucleotide small interfering RNAs
(siRNAs), complementary to parts of the respective loci, in a complex
multi-stage process, RNA-directed DNA methylation (RdDM) [13]. Interestingly, the final stages of RdDM are also
dependent on the SRA domain-containing proteins that recognize
methylated DNA sites [14]. The function of the
only Dnmt2 family methyltransferase in plants and many other organisms
is unknown.
The four families of DNA methyltransferases described above were found in all plants investigated and probably originated well before the divergence of monocotyledonous and dicotyledonous plants [8]. However, the DNA methyltransferase sets in each family, except Dnmt2, are variable. An analysis of DNA methylation in the Arabidopsis combined null-mutant plants cmt3 met1 and drm1/2 met1 showed MET1 DNA methyltransferase to be responsible for all CG methylation, and CMT3 together with DRM1 and DRM2 to be responsible for all CHG and CHH methylation [15]. Probably this is why the functions of other DNA methyltransferases in each family remained essentially unstudied.
In the present work, we have investigated the expression of genes encoding all known DNA methyltransferases in organs of Arabidopsis plants, as well as the methylation patterns of these genes probably controlling their expression.
MATERIALS AND METHODS
Cultivation of the A. thaliana (ecotype Columbia) plants was described earlier [16]. The embryo and endosperm plus seed coat fractions were isolated from developing seeds (7-10 flowering days) by the method of Perry and Wang [17]. Other plant parts were isolated by a manual dissection on ice-bath cooled Petri dishes and transferred to a liquid nitrogen containing mortar. After thorough homogenization in liquid nitrogen, the biomass was used for DNA and RNA isolation. DNA samples were obtained from leaves of early flowering stage plants with a GenElute Plant Genomic DNA Miniprep Kit (Sigma-Aldrich, USA) by the supplier-recommended protocol. The DNA modification with sodium bisulfite, PCR amplification of the gene segments of both bisulfite converted DNA strands, purification of the amplified segments, and their sequencing and quantification of the methylation levels were performed by the methods described earlier [18]. Arithmetic means of three independent determinations in biological parallels were rounded to the nearest multiple of five.
Total RNA was isolated from whole seeds and their parts by the method of Suzuki and coauthors [19], whereas the RNA samples from other organs were obtained with an RNeasy Plant Mini Kit (Qiagen, Germany) by the supplier-recommended protocol. Gene expression was studied by a TaqMan quantitative PCR method. The first cDNA strand was synthesized with a RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific Inc., USA) using 100 ng RNA in a 20-µl reaction mix. The 1-µl aliquots of the cDNA obtained were directly used as templates in 25-µl PCR reactions. A qPCRmix-HS Kit (Evrogen, Russia) and a real-time PCR detection system CFX96 (BioRad Laboratories, Inc., USA) were used. A web-service PrimerQuest (http://eu.idtdna.com/PrimerQuest/) was used for oligonucleotide primers and hybridization probe design. The hybridization probes of the genes studied were labeled with the fluorescent dye–quencher pair Fam–RTQ1, whereas the hybridization probe of the reference GAPDH mRNA – with a fluorescent dye–quencher pair R6G–BHQ1 (Syntol, Russia). The respective signals were detected in the same plate wells using Fam and Hex channels, respectively. Statistical analysis was performed in Microsoft Office Excel 2003. Three independently obtained RNA samples (biological parallels) and three or more PCR assays (technical parallels) were used for each determination.
RESULTS
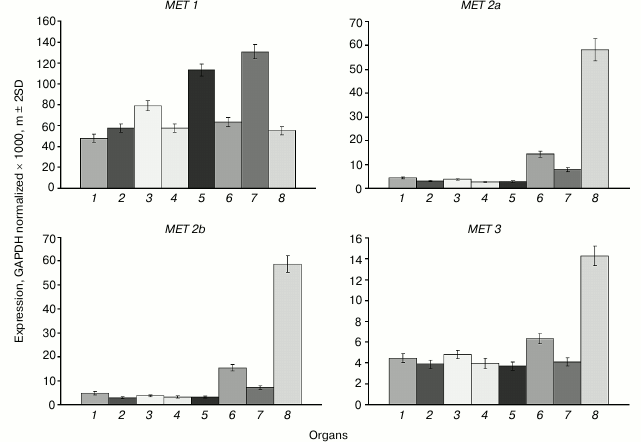
The main MET family gene MET1 is actively expressed in all organs investigated (Fig. 1). The variations of its expression levels between organs are rather small. The highest expression levels are observed in shoot apices and developing embryo, probably because of their high proliferative activity. The expression levels of the MET2a, MET2b, and MET3 genes in cells of vegetative organs are tens of times lower compared with MET1, whereas the expression levels of MET2a and MET2b in endosperm are similar to the expression levels of MET1 in vegetative organs. The expression level of MET3 in endosperm is four-fold lower compared with MET2a and MET2b, but still it is several-fold higher compared with its expression levels in vegetative organs.
Fig. 1. Expression levels of MET family DNA methyltransferase genes in Arabidopsis organs. Gene names are shown above charts. Organs are: 1) leaf; 2) root; 3) flower; 4) shoot; 5) shoot apices; 6) whole seed; 7) embryo; 8) endosperm + seed coat.
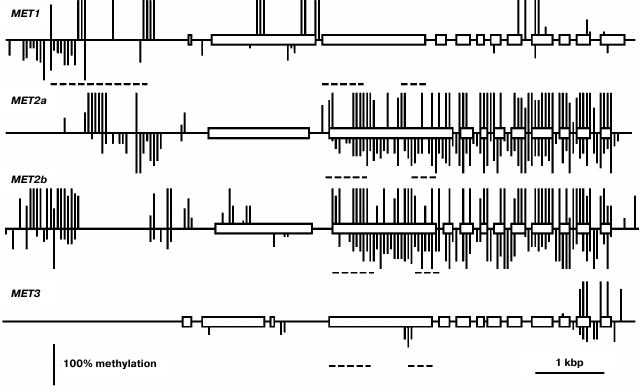
Using a Southern blot hybridization method, we found earlier that the MET family genes are nearly uniformly methylated in different Arabidopsis organs [20]. Thus, in the present work we have confined the study of their methylation patterns to the leaf. This time we used a bisulfite sequencing method that made it possible to assess the methylation levels of all cytosine residues in gene-coding sequences and their nearest neighborhood. The MET1 gene contains several fully methylated CG sites in the 5′-proximal part of the coding sequence (exon 2-intron 2) and sporadic variably methylated CG sites in the 3′-proximal part (Fig. 2). Several weakly methylated CHG and CHH sites were also detected in these regions. The proximal promoter region (from the transcription initiation site up to nucleotide −572) of the MET1 gene does not contain methylated residues. A cluster of four fully methylated CGs (from −572 to −651) and a partially methylated CAA site (−722) were found immediately before this region. There is a region containing several methylated CG sites (from −1083 to −2005) and multiple partially methylated CHG and CHH sites (from −1589 to −2583) further upstream. The methylation patterns of the MET2a and MET2b genes are quite similar to each other and very distinct from the MET1 gene methylation pattern described above. The coding parts of these genes are highly methylated at sites of all three types. The only exceptions are their first exons, that of the MET2a gene being completely unmethylated and that of the MET2b gene containing few partially methylated sites. The proximal promoter regions of both genes (~400 bp preceding the transcription initiation site) are unmethylated, whereas the sequences further upstream contain considerable numbers of methylated sites of all three types. The methylation pattern of MET3 is rather unusual. It is fully devoid of the methylated cytosine residues in the 5′-flank region and contains few methylated sites of all three types in the gene body, mainly in two last exons.
Fig. 2. Methylation patterns of the MET family genes in leaf cells. Exons are shown by rectangles, introns – by direct lines between. The methylated CG sites are shown by vertical lines above the gene sequence, the methylated CHG and CHH sites – by vertical lines below. The length of each line is proportional to the methylation level of respective site. Dashed lines below respective gene sequences show positions of repeat sequences. Positions of unmethylated CG, CHG, and CHH sites could not be appropriately shown on pictures on the scale used.
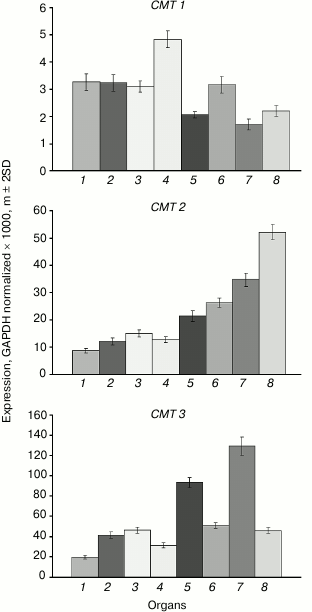
The expression levels of the CMT1 gene are very low in all investigated organs (Fig. 3). The CMT2 gene is moderately expressed at about equal levels in vegetative organs, at somewhat higher level – in the shoot apices, and at significantly higher levels – in developing seeds, especially in endosperm. The CMT3 gene is most actively expressed in rapidly growing organs (shoot apices, embryo). Its expression profile is nearly identical to that of the MET1 gene.
Fig. 3. Expression levels of the CMT family DNA methyltransferase genes in Arabidopsis organs. Organs are designated as in Fig. 1.
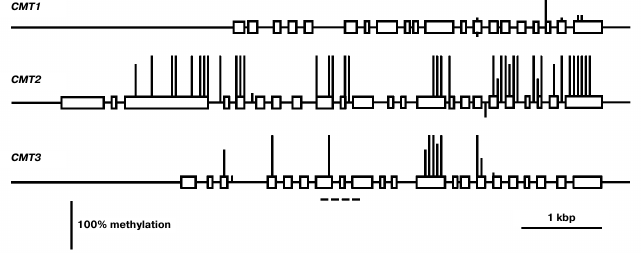
The least expressed gene of this family, CMT1, is practically unmethylated, except for a few weakly methylated CG sites in the 3′-proximal part of the coding region (Fig. 4). Evidently, its low expression in different organs is not caused by methylation but rather by some other repressive mechanisms. The CMT2 methylation pattern is typical of the enzyme-encoding genes that are constitutively expressed [2]. The promoter region and two first exons of this gene are completely unmethylated, whereas the most part of the coding sequence is methylated exclusively at CG sites. The CMT3 gene contains few methylated CG sites in its coding part and is completely unmethylated in the promoter region.
Fig. 4. Methylation patterns of CMT family genes in leaf cells. Designations are the same as in Fig. 2.
The DRM1 gene is uniformly lowly expressed in all organs (Fig. 5). The DRM2 gene expression levels are much higher, especially in developing plant parts (shoot apices, embryo, endosperm). The expression profile of the DRM3 gene is generally similar to that of DRM2, except for smaller inter-organ variations.
Fig. 5. Expression levels of DRM family DNA methyltransferase genes in Arabidopsis organs. Organs are designated as in Fig. 1.
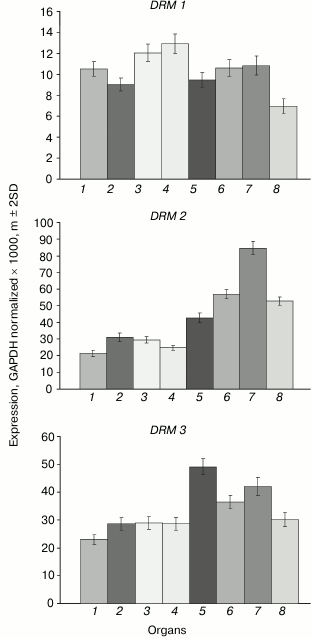
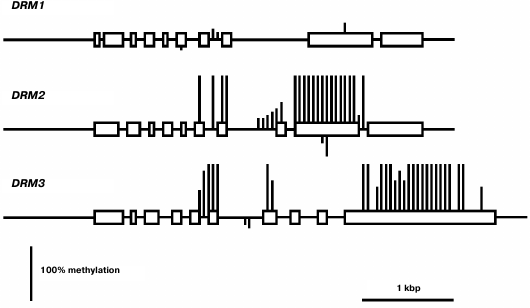
The least-expressed gene of the family, DRM1, is practically unmethylated except for a few weakly methylated sites in the middle part of gene body (Fig. 6). Apparently, in this case also low expression levels are not caused by gene methylation, but rather by some other repressive mechanism. The methylation pattern of the DRM2 gene is characteristic of genes that are constitutively expressed at high levels. Its promoter region and end-proximal parts of the coding sequence are unmethylated, whereas the middle part of the gene body is highly methylated exclusively at CG sites. A similar methylation pattern was found for the DRM3 gene, the only difference being that methylation of the gene body spreads somewhat farther toward the 3′-end-proximal part.
Fig. 6. Methylation patterns of DRM family genes in leaf cells. Designations are the same as in Fig. 2.
The DNMT2 gene is constitutively expressed at moderate levels in all organs, and somewhat higher in shoot apices and embryo compared with other plant parts (Fig. 7). A similar expression profile was found for the N6AMT gene encoding a putative adenine DNA methyltransferase, though in general the expression levels of this gene are 2-3 times higher compared with DNMT2. The KYP gene encoding the main H3K9 methyltransferase is rather actively expressed in all organs, most actively in rapidly growing ones (shoot apices, developing embryo, and endosperm) similarly to its major “partner” DNA methyltransferase CMT3 gene. The IBM1 gene encoding a H3K9 demethylase is expressed at moderate levels in vegetative organs and at lower levels in embryo and endosperm.
Fig. 7. Expression levels of genes encoding putative DNA methyltransferases DNMT2 and N6AMT, a histone H3K9 methyltransferase KYP, and H3K9 demethylase IBM1 in Arabidopsis organs. Organs are the same as in Fig. 1.
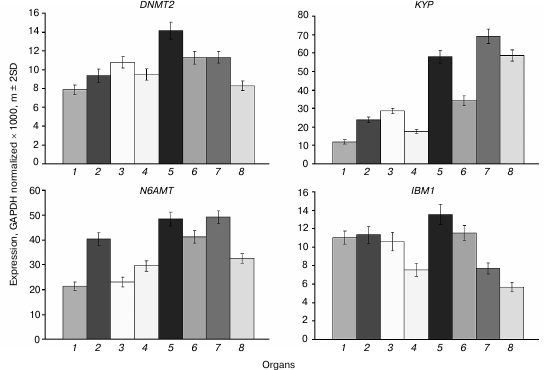
The DNMT2 gene methylation pattern is typical of a constitutively expressed at moderate levels gene (Fig. 8). The proximal promoter region (~650 bp upstream of transcription initiation site) and end-proximal exons of the gene are devoid of methylated residues, whereas the rest of the gene body is moderately methylated exclusively at CG sites. An extensively hypermethylated region is present in the distal part of the 5′-flank sequence (from −650 to −970), partially overlapping a dispersed repeat element (from −800 to −1600). The N6AMT gene methylation pattern is as unusual as that of the MET3 gene. It does not contain methylated cytosine residues in the 5′-flank and coding regions except for the last exon, which is highly methylated at CG sites. The KYP gene methylation pattern is typical of a moderately expressed gene. The promoter region of this gene and the 5′-end proximal part of the coding sequence (exons 1-3) are not methylated, whereas the rest of its coding sequence is highly methylated at CG sites. The IBM1 gene methylation pattern is also similar to this prototype, except for a local region in its longest intron highly methylated at sites of all three types.
Fig. 8. Methylation patterns of DNMT2, N6AMT, KYP, and IBM1 genes in leaf cells. Designations are the same as in Fig. 2.

DISCUSSION
The biological role of MET1 as the major maintenance DNA methyltransferase in plants is well established and supported by results of investigation of its gene expression levels. These levels are rather high and uniform in vegetative organs and are maximal in the rapidly growing parts of the plant (shoot apices, developing embryo), probably caused by high cell proliferation activity. Developing endosperm seems to deviate from this conformity, the MET1 expression level in endosperm being minimal. Probably it is still lower since, for technical reasons, we used endosperm fractions containing seed coats. The methylation pattern of the MET1 gene is atypical. It corresponds to actively expressed genes in being devoid of methylation in the proximal promoter region, but unlike most genes, the MET1 coding sequence contains a small number of methylated CG sites not in the middle part, but rather towards the ends. Another atypical feature is the presence of several partially methylated CHG and CHH sites, besides the methylated CG sites. A region locally hypermethylated at sites of all three types was found in the MET1 gene distal 5′-flank sequence (from −572 to −2583). As we noted earlier, this region overlaps with a long dispersed repeat element (from −672 to −2018) that was found in intergenic sequences in various Arabidopsis genome loci, were it was invariably highly methylated [20]. This region is targeted by several siRNAs that probably explains its methylation at sites of all types. Perhaps it serves as a border mechanism preventing aberrant read-through transcription from upstream sequences to the MET1 gene promoter.
The expression profiles of the MET2a, MET2b, and MET3 genes obviously indicate their specific roles in endosperm development. A first indication that DNA methyltransferase MET3 could possibly have a specific function was the finding of a MET3 (MEE57) gene mutation among the 130 mutations disrupting female gametophyte development [21]. Similarly to many other mutations, it caused an embryo development arrest at the single-cell zygotic stage and disrupted the endosperm development. Unfortunately, the exact cause of developmental arrest and therefore the specific role of DNA methyltransferase MET3 remained unknown.
Selective activation of maternal alleles of imprinted genes in endosperm involves a stepwise DNA demethylation by, first, a selective repression of the MET1 gene before the last syncytial division in the female gametophyte and later in the central cell, and, second, by subsequent activation of DNA demethylase DEMETER (DME) at early stages of endosperm development [2]. However, there are some imprinted genes that have an actively expressed paternal allele, for example PHERES1 (PHE1). This gene is among the rare examples when DNA methylation does not repress transcription but, on the contrary, is a prerequisite for active transcription [22]. In ~2.5 kb downstream of the PHE1 gene stop codon, there is a regulatory region containing a short triply repeated sequence that is methylated in the active paternal allele and is unmethylated in the repressed maternal one. It is also unmethylated in vegetative organs where the gene is not transcribed. Evidently, a de novo methylation of this sequence occurs during male gamete development, and the methylated state of the paternal allele is maintained in endosperm. The methylated state was also found to be a prerequisite for active expression in endosperm of the paternal alleles of some other imprinted genes [23]. MET1 gene transcription was shown, by deep transcriptome sequencing, to be tens of times higher in vegetative organs and embryo compared with other MET family genes. The MET1 gene expression level is considerably lower, whereas that of other MET family genes higher by an order of magnitude, in endosperm compared with other organs. These data are in good agreement with our results obtained with a more precise method. Of the five VIM family genes, only VIM1, -2, and -3 are expressed in vegetative organs, whereas VIM5 is the most active family member in endosperm. All these facts suggest the existence of a special maintenance DNA methylation system in endosperm. A met1 null-mutation decreased transcription levels of all MET family genes in endosperm, and vice versa, a dme null-mutation increased these levels. Thus, the high methylation levels of the MET family genes found not only do not prevent their expression, but also rather promote it.
As already noted, the expression profiles of the CMT3 and MET1 genes are nearly identical. This is not surprising, taking into account similar functions of the encoded DNA methyltransferases. The CMT3 gene methylation pattern perfectly conforms to the “body-methylated gene” prototype: unmethylated promoter, methylated at CG sites middle part of the gene body, weakly methylated end-proximal parts [2].
We found CMT1 gene to be practically silent. This is in a good accord with the results of its expression studied by other methods [24]. Besides, this gene was found to be defective in many ecotypes of Arabidopsis and thus cannot encode a catalytically active DNA methyltransferase. Strictly speaking, this does not prove this gene to be entirety useless, but its functional significance, if there is any, remains unknown.
For quite a long time, the CMT2 gene was not considered to have any specific significance. Expression of CMT2 as a green fluorescent protein fusion construct (pCMT2-GFP) was not detected in any vegetative organ or developing seed parts [24]. However, by deep transcriptome sequencing, the CMT2 gene was shown to be weakly expressed in egg cell, central cell, and vegetative organs. Unfortunately, its expression in developing endosperm was not studied. Generally, these data conform with our results. Recently, DNA methyltransferase CMT2 has been shown to be responsible, together with DRM2 methyltransferase, for all CHH methylation in the Arabidopsis genome. Moreover, these methyltransferases have distinct target preferences, CMT2 methylating mostly long transposable elements in the heterochromatic pericentromeric regions, whereas DRM2 – short transposons in euchromatic chromosome arms [25]. The CMT2 methyltransferases form a separate branch on the chromomethylase phylogenetic tree. Apparently, these methyltransferases evolved to be a special functional group prior to dicotyledonous and monocotyledonous plants divergence. Unlike CMT3, which preferentially methylates symmetric CHG sites, CMT2 has a clear-cut preference for asymmetric CHH sites [26]. Both enzymes are dependent on H3K9 methylation, but CMT2 preferentially binds to H3K9me2 and H3K9me3 and much weaker to H3K9me1, whereas CMT3 binds all three H3K9me forms equally well. Thus, the distribution of these histone marks significantly affects CMT2 and CMT3 targeting to various genome loci. The significance of the enhanced CMT2 gene expression in endosperm found in this work is unknown. Similarly to the MET2a and MET2b genes, CMT2 has an unmethylated proximal promoter region and a substantially methylated coding sequence. A significant difference between these genes is that the first ones are highly methylated at sites of all three types, whereas the CMT2 gene is methylated at CG sites only. Multiple siRNA target sites were found in the MET2a and MET2b gene sequences, whereas the CMT2 gene is devoid of such sites. Evidently, MET2a and MET2b are potential targets of RNA-dependent DNA methylation, but CMT2 is not.
Weak expression of DRM1 gene in some respect contradicts the general practice to consider DRM1 and DRM2 to be equivalent de novo type DNA methyltransferases. However, this practice is a casually arisen tradition rather than a view based on real experimental data. Distinct expression profiles of the DRM1 and DRM2 genes are indicative of different functions of their encoded proteins. Unlike DRM1, the DRM2 gene is actively expressed in all organs, especially in developing ones. DRM1 gene expression was detected earlier in the egg cell of the female gametophyte, but not in its other parts or in somatic tissues [24]. Methylation analysis of several genes in null-mutant drm1, drm2, or combined drm1 drm2 plants showed that both DRM1 and DRM2 significantly contribute to the RdDM pathway mediated CHH methylation at early (up to the heart stage) steps of embryo development. At later steps of embryo development and in vegetative organs of adult plants, no expression of DRM1 gene is observed, and DRM2 seems to be responsible for all methylation of this type. Weak expression of DRM1 in various organs found in our study obviously should be regarded as transcriptional noise detected owing to high sensitivity of PCR assays.
The DRM2 gene expression profile is somewhat different from those of two other “major” DNA methyltransferase genes, MET1 and CMT3. Of all organs studied, the highest DRM2 expression is observed in embryo, whereas the shoot apices are not much different from other vegetative organs. Apparently, de novo DNA methylation is most active in developing embryo. Nevertheless, the DRM2 gene is actively expressed in all organs, which is in good accordance with its methylation pattern by the body-methylated gene prototype. The DRM3 gene has a similar methylation pattern and is actively expressed in all organs, though somewhat more weakly compared with DRM2. Judging by the absence of some highly conserved amino acid residues, the DRM3 encoded protein could not be a catalytically active DNA methyltransferase [27]. It plays an auxiliary role in DNA methylation by DNA methyltransferase DRM2 and functionally is a direct analog of mammalian catalytically inactive protein Dnmt3L required for normal activity of DNA methyltransferase Dnmt3a. Phylogenetic analysis showed that DRM3 proteins evolved as specific members of the DRM family before the divergence of dicotyledonous and monocotyledonous plants, but DRM3 and Dnmt3L originated and evolved independently. DRM3 is important for methylation of a significant share but not all RdDM targets [28, 29]. This is in good accord with the DRM3 expression profile observed in our study.
The KYP gene expression profile is quite similar to that of CMT3. This similarity possibly reflects a close functional link between the encoded proteins. The KYP gene methylation pattern conforms to the “body-methylated gene” prototype. The IBM1 gene contains a region locally hypermethylated at sites of all three types. The middle part of this region is complementary to a 24-nucleotide siRNA, which probably accounts for its hypermethylation.
The DNMT2 gene is weakly expressed in different organs, the variance in its expression level being rather small. It has a methylation pattern conforming to the “body-methylated gene” prototype. There is a locally hypermethylated sequence in the distal 5′-flank region (from −654 to −964) of DNMT2 that overlaps with a dispersed repeat sequence element. This methylated region is complementary to several siRNAs, whereas the unmethylated downstream region contains recognition sites of some transcription factors. A locally hypermethylated region is quite possibly a border element separating the DNMT2 gene promoter region from that of an inversely oriented neighboring gene. The function of DNA methyltransferase DNMT2 in plants, like in other organisms, is not quite clear. On one hand, it contains all the conservative motifs characteristic of cytosine DNA methyltransferases. On the other hand, its DNA methylating activity is rather low in in vitro assays, and null-mutations of the only Dnmt2 gene in mice and Arabidopsis have no appreciable phenotypic manifestations [7]. Incubation of purified cloned human DNMT2 in the presence of labeled [3H]S-adenosyl-L-methionine did not lead to radioactive label incorporation into DNA, but it effectively labeled aspartic acid tRNA [30]. DNMT2 has a preferentially nuclear location in Arabidopsis and physically interacts with the HD2 family histone deacetylases [31]. It could be suggested that one of the DNMT2 functions in plants is connected with histone deacetylation.
A still more mysterious gene is N6AMT (AT3G26410). It was described for the first time in our laboratory when searching for putative adenine DNA methyltransferase genes in the genome of various eukaryotic organisms [32]. Genes encoding proteins containing all conservative motifs of adenine DNA methyltransferases have been found in genomes of very different eukaryotes, from yeast and nematode to higher plants and mammals. We discussed whether Nm6A could be present in DNA of plants and other eukaryotic organisms in detail earlier [2, 5]. We detected Nm6A residues in the Arabidopsis DRM2 gene [16], but not in other genes [20]. Apparently, Nm6A residues are present in plant and perhaps other eukaryotes genomes, but their quantities are very small. Unfortunately, the ability of the purified N6AMT protein to methylate adenine residues in DNA was never studied, and the only known consequences of the N6AMT gene null-mutations in Arabidopsis are reduction of Nm2G residue content in the 10th position of tRNA molecules and early flowering [33]. This finding together with an existing homology between N6AMT and the yeast tRNA methyltransferase Trm11p, which methylates the 10th guanine residue in tRNA molecules at the second position of the purine ring exocyclic amine, were grounds to give the AT3G26410 gene the name AtTRM11. Interestingly, the putative catalytic active center of Trm11p (motif IV) is more similar to those of DNA methyltransferases that methylate DNA at exocyclic amines (Nm2G, Nm6A, Nm4C) than to the active centers of other tRNA methyltransferases. This is reminiscent of the DNMT2 that have a typical cytosine DNA methyltransferase structure, but is “suspected” to be a tRNA methyltransferase. Quite possibly, both these enzymes have double functions, methylating both DNA and RNA. Their DNA methyltransferase functions are questioned on the grounds of the absence of respective activity in in vitro methylation assays. Another reason is unsuccessful attempts to detect any changes in DNA methylation in vivo when the respective genes are mutated. It must be stressed that similar grounds have existed for a long time for the DNA methyltransferase CMT2. Nevertheless, today its role as an important DNA methyltransferase is firmly established.
Recently it was shown that Nm6A residues are present in the nematode C. elegans genome at 0.01 to 0.4% of all adenine residues [34]. Genes were found that encode an Nm6A-specific DNA methyltransferase and Nm6A-specific DNA demethylase. Inactivation of these genes led to expected changes in the Nm6A content in DNA and to distinctive phenotypic consequences as well. Moreover, mutual positive links between Nm6A DNA methylation and H3K4me2 histone methylation were found, very reminiscent of the CMT3-KYP links in plants. The putative adenine DNA methyltransferase in C. elegans is encoded in gene C18A3.1. It is an evolutionarily conserved protein homologous to bacterial Nm6A-specific DNA methyltransferases with a circularly permuted order of the catalytic domain motifs (prototype – MunI). We did not identified this protein as a putative adenine DNA methyltransferase earlier because it contains catalytic motif DPPW instead of the more typical one, DPPY. C18A3.1 gene orthologs are found in genomes of many eukaryotes, including plants. Thus, for example, in the Arabidopsis genome it is the AT1G19340 gene. Unfortunately, the properties of the encoded protein have not been studied in plants.
This work was supported by the Russian Science Foundation (project No. 14-50-00029).
REFERENCES
1.Feng, S., and Jacobsen, S. E. (2011) Epigenetic
modifications in plants: an evolutionary perspective, Curr.
Opin. Plant Biol., 14, 179-186.
2.Vanyushin, B. F., and Ashapkin, V. V. (2011) DNA
methylation in higher plants: past, present and future, Biochim.
Biophys. Acta, 1809, 360-368.
3.Cokus, S. J., Feng, S., Zhang, X., Chen, Z.,
Merriman, B., Haudenschild, C. D., Pradhan, S., Nelson, S. F.,
Pellegrini, M., and Jacobsen, S. E. (2008) Shotgun bisulfite sequencing
of the Arabidopsis genome reveals DNA methylation patterning,
Nature, 452, 215-219.
4.Lister, R., O’Malley, R. C., Tonti-Filippini,
J., Gregory, B. D., Berry, C. C., Millar, A. H., and Ecker, J. R.
(2008) Highly integrated single-base resolution maps of the epigenome
in Arabidopsis, Cell, 133, 523-536.
5.Vanyushin, B. F., and Ashapkin, V. V. (2009) DNA
Methylation in Plants, Nova Science Publishers Inc., N. Y.
6.Buryanov, Ya. I., and Shevchuk, T. V. (2005) DNA
methyltransferases and structural-functional specificity of eukaryotic
DNA modification, Biochemistry (Moscow), 70, 730-742.
7.Goll, M. G., and Bestor, T. H. (2005) Eukaryotic
cytosine methyltransferases, Annu. Rev. Biochem., 74,
481-514.
8.Pavlopoulou, A., and Kossida, S. (2007) Plant
cytosine-5 DNA methyltransferases: structure, function, and molecular
evolution, Genomics, 90, 530-541.
9.Woo, H. R., Dittmer, T. A., and Richards, E. J.
(2008) Three SRA-domain methylcytosine-binding proteins cooperate to
maintain global CpG methylation and epigenetic silencing in
Arabidopsis, PLoS Genet., 4, e1000156.
10.Du, J., Zhong, X., Bernatavichute, Y. V., Stroud,
H., Feng, S., Caro, E., Vashisht, A. A., Terragni, J., Chin, H. G., Tu,
A., Hetzel, J., Wohlschlegel, J. A., Pradhan, S., Patel, D. J., and
Jacobsen, S. E. (2012) Dual binding of chromomethylase domains to
H3K9me2-containing nucleosomes directs DNA methylation in plants,
Cell, 151, 167-180.
11.Johnson, L. M., Bostick, M., Zhang, X., Kraft,
E., Henderson, I., Callis, J., and Jacobsen, S. E. (2007) The SRA
methylcytosine-binding domain links DNA and histone methylation,
Curr. Biol., 17, 379-384.
12.Cao, X., and Jacobsen, S. E. (2002) Role of the
Arabidopsis DRM methyltransferases in de novo DNA
methylation and gene silencing, Curr. Biol., 12,
1138-1144.
13.Cao, X., Aufsatz, W., Zilberman, D., Mette, M.
F., Huang, M. S., Matzke, M., and Jacobsen, S. E. (2003) Role of the
DRM and CMT3 methyltransferases in RNA-directed DNA methylation,
Curr. Biol., 13, 2212-2217.
14.Johnson, L. M., Law, J. A., Khattar, A.,
Henderson, I. R., and Jacobsen, S. E. (2008) SRA-domain proteins
required for DRM2-mediated de novo DNA methylation, PLoS
Genet., 4, e1000280.
15.Zhang, X., and Jacobsen, S. E. (2006) Genetic
analyses of DNA methyltransferases in Arabidopsis thaliana,
Cold Spring Harb. Symp. Quant. Biol., 71, 439-447.
16.Ashapkin, V. V., Kutueva, L. I., and Vanyushin,
B. F. (2002) The gene for domains rearranged methyltransferase (DRM2)
in Arabidopsis thaliana plants is methylated at both cytosine
and adenine residues, FEBS Lett., 532, 367-372.
17.Perry, S. E., and Wang, H. (2003) Rapid isolation
of Arabidopsis thaliana developing embryos,
BioTechniques, 35, 278-282.
18.Ashapkin, V. V., Linkova, N. S., Khavinson, V.
Kh., and Vanyushin, B. F. (2015) Epigenetic mechanisms of peptidergic
regulation of gene expression during aging of human cells,
Biochemistry (Moscow), 80, 310-322.
19.Suzuki, Y., Kawazu, T., and Koyama, H. (2004) RNA
isolation from siliques, dry seeds, and other tissues of Arabidopsis
thaliana, BioTechniques, 37, 542-544.
20.Ashapkin, V. V., Kutueva, L. I., and Vanyushin,
B. F. (2011) Is the cytosine DNA methyltransferase gene MET1
regulated by DNA methylation in Arabidopsis thaliana plants?
Russ. J. Genet., 47, 279-288.
21.Pagnussat, G. C., Yu, H.-J., Ngo, Q. A., Rajani,
S., Mayalagu, S., Johnson, C. S., Capron, A., Xie, L.-F., Ye, D., and
Sundaresan, V. (2005) Genetic and molecular identification of
genes required for female gametophyte development and function in
Arabidopsis, Development, 132, 603-614.
22.Makarevich, G., Villar, C. B. R., Erilova, A.,
and Koehler, C. (2008) Mechanism of PHERES1 imprinting in
Arabidopsis, J. Cell Sci., 121, 906-912.
23.Hsieh, T.-F., Shin, J., Uzawa, R., Silva, P.,
Cohen, S., Bauer, M. J., Hashimoto, M., Kirkbride, R. C., Harada, J.
J., Zilberman, D., and Fischer, R. L. (2011) Regulation of imprinted
gene expression in Arabidopsis endosperm, Proc. Natl. Acad.
Sci. USA, 108, 1755-1762.
24.Jullien, P. E., Susaki, D., Yelagandula, R.,
Higashiyama, T., and Berger, F. (2012) DNA methylation dynamics during
sexual reproduction in Arabidopsis thaliana, Curr. Biol.,
22, 1825-1830.
25.Zemach, A., Kim, M. Y., Hsieh, P.-H.,
Coleman-Derr, D., Eshed-Williams, L., Thao, K., Harmer, S. L., and
Zilberman, D. (2013) The Arabidopsis nucleosome remodeler DDM1
allows DNA methyltransferases to access H1-containing heterochromatin,
Cell, 153, 193-205.
26.Stroud, H., Do, T., Du, J., Zhong, X., Feng, S.,
Johnson, L., Patel, D. J., and Jacobsen, S. E. (2014) Non-CG
methylation patterns shape the epigenetic landscape in
Arabidopsis, Nat. Struct. Mol. Biol., 21,
64-72.
27.Henderson, I. R., Deleris, A., Wong, W., Zhong,
X., Chin, H. G., Horwitz, G. A., Kelly, K. A., Pradhan, S., and
Jacobsen, S. E. (2010) The de novo cytosine methyltransferase
DRM2 requires intact UBA domains and a catalytically mutated paralog
DRM3 during RNA-directed DNA methylation in Arabidopsis
thaliana, PLoS Genet., 6, e1001182.
28.Costa-Nunes, P., Kim, J. Y., Hong, E., and
Pontes, O. (2014) The cytological and molecular role of domains
rearranged methyltransferase3 in RNA-dependent DNA methylation of
Arabidopsis thaliana, BMC Res. Notes, 7, 721.
29.Zhong, X., Hale, C. J., Nguyen, M., Ausin, I.,
Groth, M., Hetzel, J., Vashisht, A. A., Henderson, I. R., Wohlschlegel,
J. A., and Jacobsen, S. E. (2015) Domains rearranged methyltransferase3
controls DNA methylation and regulates RNA polymerase V transcript
abundance in Arabidopsis, Proc. Natl. Acad. Sci. USA,
112, 911-916.
30.Goll, M. G., Kirpekar, F., Maggert, K. A., Yoder,
J. A., Hsieh, C. L., Zhang, X., Golic, K. G., Jacobsen, S. E., and
Bestor, T. H. (2006) Methylation of tRNAAsp by the DNA
methyltransferase homolog Dnmt2, Science, 311,
395-398.
31.Song, Y., Wu, K., Dhaubhadel, S., An, L., and
Tian, L. (2010) Arabidopsis DNA methyltransferase AtDNMT2
associates with histone deacetylase AtHD2s activity, Biochem.
Biophys. Res. Commun., 396, 187-192.
32.Shorning, B. Yu., and Vanyushin, B. F. (2001)
Putative DNA-(amino)methyltransferases in eukaryotes, Biochemistry
(Moscow), 66, 753-762.
33.Chen, P., Jager, G., and Zheng, B. (2010)
Transfer RNA modifications and genes for modifying enzymes in
Arabidopsis thaliana, BMC Plant Biol., 10,
201.
34.Greer, E. L., Blanco, M. A., Gu, L., Sendinc, E.,
Liu, J., Aristizabal-Corrales, D., Hsu, C.-H., Aravind, L., He, C., and
Shi, Y. (2015) DNA methylation on N6-adenine in C. elegans,
Cell, 161, 868-878.