REVIEW: Cellular Energetics as a Target for Tumor Cell Elimination
P. V. Maximchik1, A. V. Kulikov1, B. D. Zhivotovsky1,2, and V. G. Gogvadze1,2*
1Faculty of Basic Medicine, Lomonosov Moscow State University, 119991 Moscow, Russia; fax: +7 (499) 726-5547; E-mail: Vladimir.Gogvadze@ki.se2Institute of Environmental Medicine, Karolinska Institutet, Box 210, 17177 Stockholm, Sweden
* To whom correspondence should be addressed.
Received June 12, 2015; Revision received July 9, 2015
Investigation of cancer cell metabolism has revealed variability of the metabolic profiles among different types of tumors. According to the most classical model of cancer bioenergetics, malignant cells primarily use glycolysis as the major metabolic pathway and produce large quantities of lactate with suppressed oxidative phosphorylation even in the presence of ample oxygen. This is referred to as aerobic glycolysis, or the Warburg effect. However, a growing number of recent studies provide evidence that not all cancer cells depend on glycolysis, and, moreover, oxidative phosphorylation is essential for tumorigenesis. Thus, it is necessary to consider distinctive patterns of cancer metabolism in each specific case. Chemoresistance of cancer cells is associated with decreased sensitivity to different types of antitumor agents. Stimulation of apoptosis is a major strategy for elimination of cancer cells, and therefore activation of mitochondrial functions with direct impact on mitochondria to destabilize them appears to be an important approach to the induction of cell death. Consequently, the design of combination therapies using acclaimed cytotoxic agents directed to induction of apoptosis and metabolic agents affecting cancer cell bioenergetics are prospective strategies for antineoplastic therapy.
KEY WORDS: tumor cells, bioenergetics, mitochondria, Warburg effect, glycolysisDOI: 10.1134/S0006297916020012
Abbreviations: ANT, adenine nucleotide translocase; α-TOS, α-tocopheryl succinate; CypD, cyclophilin D; DCA, dichloroacetate; 2-DG, 2-deoxyglucose; HIF, hypoxia inducible factor; HK, hexokinase; MOM(P), mitochondrial outer membrane (permeabilization); MPT(P), mitochondrial permeability transition (pore); OXPHOS, oxidative phosphorylation; ROS, reactive oxygen species; VDAC, voltage-dependent anion channel.
Cancer cells have a range of characteristic features distinguishing them
from normal cells of healthy tissues. Among these features are
suppression of cell death mechanisms, elimination of growth-inhibitory
signals, hyperactivation of proliferation signaling cascades, induction
of angiogenesis, limitless replicative potential, and activation of
invasive and metastasis processes. In addition, alterations in
bioenergetics that lead to increased glucose consumption, upregulation
of glycolytic cascade, and augmentation of lactate production with
reduced or diminished oxidative phosphorylation (OXPHOS) in the
background even in the presence of ample oxygen should be mentioned [1-3]. Otto Warburg hypothesized
that defects in mitochondria of fast-growing cancer cells account for
their bioenergetic phenotype. However, according to current knowledge,
mitochondria in the majority of cancer cell lines are functionally
active, and suppression of metabolic pathways involved in the
regulation of mitochondrial processes determine their decreased
contribution to cell bioenergetics. Furthermore, it was demonstrated
later that the degree of glycolytic contribution to the energy supply
largely depends on tumor etiology and, in some cancer cell lines,
oxidative phosphorylation may predominate as a major source of ATP [3-5]. Investigation of cancer cell
bioenergetics demonstrated variability of metabolic profile in
different cancer cell lines, as well as within the same cell type,
depending on the mechanisms involved and substrates utilized for energy
production in different conditions. In any case, no matter whether the
bioenergetics is based on glycolysis or OXPHOS, cancer cells consume
significantly more glucose than normal cells. This observation
underlies the approach for cancer cell visualization using positron
emission tomography and the glucose analog
2-deoxy-2-(18F)fluoro-D-glucose.
Considering the leading role of glycolysis in cellular energetics in the majority of cancer types, inhibition of glycolytic pathways for tumor cell elimination has attracted much attention from researchers. Thus, Otto Warburg believed that moderate doses of ionizing therapy could suppress the mitochondrial activity of cancer cells below the permissible threshold necessary for normal cell function without affecting normal cells. It should be noted that ATP production is not the sole function of mitochondria. Mitochondria are involved in the regulation of Ca2+ homeostasis, production of reactive oxygen species (ROS), and are crucial for realization of programmed cell death. In particular, mitochondrial outer membrane permeabilization (MOMP) and release of proteins from the intermembrane space are a point of no return in one of the types of programmed cell death – apoptosis. That makes mitochondria a prospective target in chemotherapy.
CONTRIBUTION OF GLYCOLYSIS AND OXIDATIVE PHOSPHORYLATION TO CELL
ENERGETICS IN DIFFERENT CANCER CELL TYPES
Rapidly proliferating cancer cells easily become hypoxic as the vascular system fails to provide these cells with an adequate amount of oxygen. Therefore, in the majority of cells the glycolytic pathway of ATP production is stimulated (Fig. 1). However, as mentioned above, several types of tumors keep relying on OXPHOS. It was demonstrated that contribution of OXPHOS to ATP production in HeLa cells was about 80%, while in hypoxic conditions it decreased to 30% [6]. Pyruvate, glutamine, glycine, alanine, proline, and glutamate are mainly used as substrates in OXPHOS, while in addition to fatty acids, ketone bodies, short-chained carboxylic acids, propionate, acetate, and butyrate can serve as alternative sources [7, 8].
Fig. 1. Suppression of glycolysis as a strategy for stimulation of tumor cell death. LDH, lactate dehydrogenase; PDH, pyruvate dehydrogenase; RC, respiratory chain; 2-DG, 2-deoxyglucose; OXPHOS, oxidative phosphorylation.
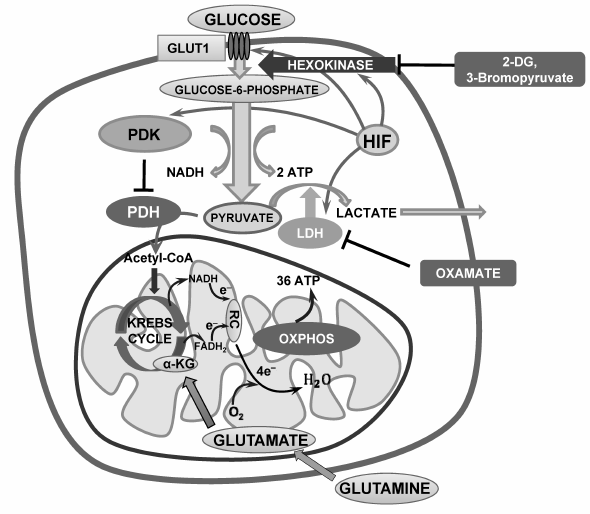
Glutamine is required for intensive cancer cell proliferation. It is converted into glutamate by glutaminase upon entry into the cell. Then, the enzyme glutathione-cysteine-ligase converts glutamate to glutathione, a crucial component of the antioxidant system, which is necessary for redox control of all subcellular compartments [9]. Glutamate can be converted to α-ketoglutarate, which is one of the Krebs cycle metabolites. This process of anaplerosis provides a source of carbon, which is essential for the Krebs cycle as a biosynthetic center and contributes to the synthesis of amino and fatty acids [10]. Neoplastic aberration stimulates glutaminolysis. The oncogene Myc plays a crucial role in the regulation of this process and is able to enhance the supply of glutamine by stimulating expression of glutamine transporters SLC5A1 and SLC7A1 (the latter also known as CAT1) [11]. Moreover, Myc indirectly stimulates expression of glutaminase-1, an enzyme of the first step of glutaminolysis, by suppressing glutaminase-1 inhibitors microRNA-23A and microRNA-23B [11, 12]. In addition, stimulation of glutaminase by Myc in the human B-cell line was demonstrated, although the level of mRNA was not altered significantly; this indicates that Myc regulates glutaminase at the posttranscriptional level [13]. Therefore, Myc can be involved in maintenance of the cell antioxidant capacity through stimulation of NADPH production in the pentose phosphate cycle because of upregulation of pyruvate kinase PKM2 and increase of glutathione synthesis by glutaminolysis. Stimulation of glutathione metabolism is crucial for biosynthesis of nucleotides and essential amino acids and for maintaining the normal redox potential of the cell.
Experimental studies demonstrated that small tumors were characterized by a low conversion of glucose into lactate, whereas the conversion of glutamine to lactate was high. In medium sized tumors, the conversion of glucose to lactate as well as oxygen utilization was increased, whereas glutamine and serine consumption were reduced. Large tumors were characterized by low oxygen and glucose supply, but a high glucose and oxygen utilization rate [14]. Apparently, in the inner layers of solid tumors, significant substrate and oxygen limitation occurs; however, experiments designed to model these conditions in vitro revealed that nutrient and oxygen limitation does not affect OXPHOS and cellular ATP levels. Furthermore, the growth of HeLa cells (cervical cancer), HepG2 (liver cancer) cells, and HTB126 cells (breast cancer) in aglycemia and/or hypoxia even triggered a compensatory increase in OXPHOS [6].
It was mentioned above that such substrates as glucose and glutamine are primary utilized by fast-growing cancer cells. Nonetheless, it remains unclear which of them (or any other oxidizable substrate) is predominantly utilized by the cell to sustain intense proliferation. Sometimes, glycolytic tumors demonstrate high rates of glutamine oxidation [15]. Several tumor cell types, such as HeLa, are able to adapt their metabolism in accordance with availability of external sources of carbon; a deficiency of glucose in the medium stimulates de novo mitochondrial DNA synthesis in HeLa cells, which leads to synthesis of respiratory chain complexes and citrate synthase [16]. It should be mentioned that HeLa cells utilize both glucose and glutamine for ATP production, and this indicates the dependence of cells on OXPHOS, as well as on glycolysis at the same time [5].
Evidence of the presence of active mitochondrial enzymes (NADP+-malic enzyme, glutaminase and glutamine transporter) in tumors suggests normal functioning of mitochondria in the cells. Moreover, malic enzyme activity is 10-20 times higher in several types of cancer than in normal tissues [17]. The role of malic enzyme in cancer cells remains to be elucidated; however, it is suggested that this enzyme is essential for preventing malate overload by converting malate to pyruvate for further usage in OXPHOS [18].
As discussed above, insufficient tumor vascularization leads to decreases in both oxygen level and nutrients in the microenvironment. Glucose deprivation and glycolysis inhibition by iodoacetic acid result in a metabolic switch toward glutamine utilization accompanied by rapid suppression of lactate production in HeLa cells [19]. This demonstrates how tumors can use alternative pathways of energy production, such as glutaminolysis, to adapt cells to glucose deprivation.
Investigation of cancer cell metabolism demonstrated that the amount of glucose consumed by tumors significantly exceeds cellular energetic demands. Contribution of glycolytic ATP entirely depends on the cell microenvironment and varies greatly (0.31-64%) in relation to the cell/tissue types and experimental conditions [20]. Most of the glucose is consumed for maintaining lipid storage, pentose phosphate cycle, and production of ribose, which is essential for the biosynthesis of nucleotides and sugars used by rapidly proliferating cells. Thus, the main role of stimulated glycolysis in rapidly proliferating cells could be maintenance of an adequate level of intermediates required for biosynthetic processes [21]. It must be emphasized that there is no reason to apply these mechanisms of metabolism regulation to all cancer cell lines, as in different tumors various combinations of regulatory pathways and diverse rates of enhancement of glycolysis or other alternative energetics pathways may be operating.
It should be mentioned that alterations of cell bioenergetics occur during tumor growth. Metabolomic studies in vivo suggest the occurrence of continuous metabolic remodeling in cancer cells according to tumor size and rate of growth [14]. Considering the mechanisms of ATP supply in particular, cancer cell lines are crucial for determining adequate and effective application of chemotherapy targeting tumor bioenergetics.
MECHANISMS OF BIOENERGETIC REPROGRAMMING
Stimulation of glycolysis and suppression of mitochondrial activity by hypoxia-inducible factor. Oxygen concentration in the cancer cell microenvironment is highly heterogeneous and varies greatly in different tissues from normoxic (2-4% oxygen concentration) to anoxic (≤0.1%) conditions. Average oxygen concentration in the cell microenvironment from the nearest capillary is nearly 2%, and hypoxia occurs below this level. Cells that are located 200 µm from the nearest capillary are already hypoxic. However, oxygen concentration reaches 8-57 µM in the central regions of gliomas, carcinomas, and multicellular spheroids (model of avascular solid tumors) and in the hypoxic regions of human tumors [22-24], which corresponds to the oxygen level in normal vascular tissues (mammary gland and femoral muscle) [25]. As previously mentioned, decreased oxygen content and vascularization in solid tumors, particularly at early stages of (frequently avascular) cancer, affect the nutrient content (glucose availability), leading to a rapid decline of glycolytic ATP production [26].
OXPHOS does not function properly if oxygen concentration reaches levels below 1 µM, as the Km of cytochrome-c-oxidase is 0.10-0.15 µM for pure protein and submitochondrial particles [27], 0.4-0.8 µM in human umbilical vein endothelial cells [28], and 0.39 µM in intact human dermal fibroblast [29]. In turn, oxygen saturation concentration for cytochrome-c-oxidase and OXPHOS is more than 4-8 µM (i.e. it is 10 times higher than Km). Therefore, tumor mitochondrial metabolism would not be affected by the hypoxia level found in tumors unless prolonged exposure to the hypoxic microenvironment somehow alters the expression of mitochondrial enzymes, perhaps through a p53-mediated mechanism [30]. Oxygen concentration in the tumor microenvironment does not always reach such critically low levels, unless a concentration gradient is created when the concentration of oxygen in nearby mitochondria falls below a threshold level of 1 µM [5]. Thus, cancer cells are able to adapt to diverse microenvironmental conditions by switching between several mechanisms of ATP production.
Oxygen deficiency leads to stabilization of hypoxia-inducible factor (HIF), which is composed of a constitutively expressed HIF1-β subunit and oxygen-sensitive HIF1-α or HIF2-α subunits. Under aerobic conditions, the HIF-α subunit is hydroxylated at the level of conserved proline and asparagine residues by prolyl hydroxylases. After proline hydroxylation, the HIF-α subunits are ubiquitinated by the VHL tumor-suppressor protein, pVHL, which targets them for degradation by the proteasome. Under hypoxia, where HIF-α hydroxylation is reduced, or in the absence of pVHL, where ubiquitination does not occur, the HIF-α subunit is stabilized and translocated to the nucleus, where it forms a dimer with β-subunit and promotes expression of target genes such as growth factors, angiopoietin, and vascular endothelial growth factor via binding to hypoxia-responsive elements. Expression of a wide range of other enzymes such as glucose transporters (GLUT-1), hexokinase (HK), phosphofructokinase, lactate dehydrogenase, and others are regulated by HIF. In addition to this, HIF activation leads to suppression of mitochondrial activity. Pyruvate dehydrogenase (PDH), which oxidizes the endproduct of glycolysis, pyruvate, is regulated by an enzyme called pyruvate dehydrogenase kinase (PDK), which suppresses PDH activity by its phosphorylation. HIF stimulates PDK activity, causing inhibition of pyruvate oxidation and stimulating its conversion into lactate (Fig. 1).
It should be noted that there are other ways of HIF-1α stabilization besides those mentioned above. Thus, HIF-1α can be stabilized under aerobic conditions by cytokines, growth factors, ROS, and nitroxides or metabolic intermediates such as pyruvate, succinate, fumarate, lactate, or oxaloacetate. Thus, in cancer cells, in which enzymes of the Krebs cycle such as succinate dehydrogenase and fumarate dehydrogenase are mutated, the levels of succinate and fumarate increase, and this leads to HIF stabilization even under normoxic conditions. This situation is referred to as pseudohypoxia [31]. It is notable that the metastatic cancer cell lines (breast cancer lines MDA, U87 glioblastoma, DU145 prostate cancer, and renal cell carcinomas RCC4 and CaSKi) have an increased level of HIF-1 expression, upregulation of glycolytic enzymes, and enhanced glycolysis regardless of oxygen concentration, whereas nonmetastatic cell lines (breast cancer MCF-7, HT-29 colon cancer, MiaPaCa pancreatic cancer, A549 lung cancer, and BX-PC3 prostate cancers) demonstrate HIF-1 stabilization and glycolytic protein upregulation only in hypoxic conditions [32].
As mentioned earlier, evidence points to the fact that mitochondria remain functionally active in cancer cells, and their low contribution to cell bioenergetics is associated with suppression of the metabolic pathways responsible for regulation of bioenergetic processes in the cell. Nonetheless, it should be noted that decreased mitochondrial activity might be caused by defects in the mitochondrion itself. A decrease in mtDNA level, suppression of mitochondrial genome transcription, and accumulation of mutations and deletions frequently occur in tumor cells [33]. Accumulation of defects in the mitochondrial genome may cause alterations in the mitochondrial respiratory chain, which triggers the glycolytic shift in cancer cell metabolism.
Another possible mechanism of OXPHOS suppression is overexpression of the ATPase inhibitory protein (IF1) [34]. Decrease in H+-ATP synthase (β-F1-ATPase) is a proteomic signature of declined OXPHOS and is characteristic of cancer cell bioenergetics, which can predict the prognosis of colon, lung, and breast cancers [34, 35].
Abnormalities in Krebs cycle functioning. Parlo and Coleman explained high glycolytic activity of cancer cells by mitochondrial dysfunctions at the level of the Krebs cycle, which results in decrease of reducing equivalents essential for respiratory chain functioning and, consequently, to the suppression of OXPHOS [36]. They found that citrate, generated from pyruvate, was removed from tumor mitochondria of Morris 3924A hepatomas (up to four times higher than in normal cells) because of suppression of citrate conversion to 2-oxuglutarate (i.e. malfunctions in aconitase and isocitrate dehydrogenase). This caused citrate accumulation in the mitochondrial matrix and its subsequent removal from mitochondria. In the cytosol, citrate is involved in the biosynthesis of cholesterol, triacylglycerides, and phospholipids. Citrate is also known as an inhibitor of phosphofructokinase, but in many cases, in tumors is expressed as the phosphofructokinase-1 isoform, which is insensitive to citrate, and glycolysis is not suppressed. However, results presented by other researchers [37, 38] contradicted the data from Parlo and Coleman. Thus, the intensity of decarboxylation of pyruvate, malate, citrate, acetoacetate, and acetate in AS-30D hepatoma cells was similar to that in normal mitochondria, indicating normal metabolism at least in this cell line. Furthermore, the activity of some other Krebs cycle enzymes in AS-30D hepatoma cells was 1-30 times higher than in normal hepatocytes [38]. Apparently, alterations in Krebs cycle activity depend on the cancer type, as well as stage of tumor development.
Reduced number of mitochondria in cells and defects in mitochondrial proteins. The reduced number of mitochondria may account for low mitochondrial impact [39]. For instance, studies have demonstrated that the activity of cancer mitochondria was equal to normal mitochondria from healthy tissues, but the OXPHOS intensity in cancer cells was diminished because of decreased numbers of mitochondria per cell (number of mitochondria is decreased by 20-50% compared to normal cells) [40]. This can be explained by either increased degradation (mitophagy) or attenuation in organelle proliferation. The precise mechanisms remain to be elucidated. In addition, such a decrease was not observed in all tumors. For instance, mitochondrial content in Morris hepatoma 16 and 7800 is similar to normal cells.
Although investigation of respiratory chain complexes functioning in cancer cells has become a matter of interest in recent years, studies reported so far have not yielded a consensus. Thus, marked deficiencies have been identified in some respiratory chain components (iron–sulfur centers, NADH cytochrome c reductase, succinate dehydrogenase, and cytochrome c oxidase) of mitochondria from several tumor types [5, 41]. However, an increase (from two- to five-fold) in the activity of NADH cytochrome c reductase has also been determined in the same tumors [5, 42]. No differences in the activity of OXPHOS enzymes between normal cells and Morris hepatomas 3924A, 9618A, and 7800 and Novikoff hepatoma have been found [5]. At the same time, the synthetic and hydrolytic activities of ATP synthase in the mitochondria of human hepatocellular carcinoma were reduced by 50-70% as compared to normal human hepatocytes. Presumably, depending on the cancer cell type, alterations in the respiratory chain complexes activity can increase or decrease.
Crabtree effect (inhibition of oxidative phosphorylation by glycolysis). The suppression of OXPHOS by the addition of glucose, described by Crabtree in 1928, is a possible mechanism of the inhibition of mitochondrial activity. Glucose and other hexoses cause partial inhibition of OXPHOS in fast-growing tumors [43-45] and normal proliferating cells [46, 47]. After glucose addition to AS-30D hepatoma cells, the glycolytic flux was stimulated and the concentrations of phosphorylated hexoses (glucose-6-phosphate, fructose-6-phosphate, fructose-1,6-bisphosphate) showed a substantial increase, but the ATP and Pi contents decreased and the cytosolic pH lowered from 7.2 to 6.8 [48]. The alterations of ATP, Pi, and hexoses levels correspond to the enhancement of glycolysis and decrease of mitochondrial ATP production. This may be explained by the competition for Pi between glycolysis and OXPHOS. Lowering of the pH caused by lactate production can alter activity of pH-sensitive mitochondrial proteins, such as 2-oxoglutarate dehydrogenase and cytochrome bc1 [49].
After analysis of the mitochondrial activity in diverse cancer cell types, the conclusion can be made that mitochondrial silencing is not caused by mitochondrial damage, but mostly by alteration of the mechanisms regulating mitochondrial energy producing pathways. Considering the leading role of the mitochondria in the regulation of various modes of programmed cell death, it is worthwhile to elucidate how suppression of mitochondrial activity can affect cell death pathways.
APOPTOSIS AS ANTAGONIST OF MALIGNANT TRANSFORMATION
In addition to the reprogramming of bioenergetics, suppression of cell death is another characteristic feature of the malignant transformation. Maintenance of tissue homeostasis is determined by three fundamental processes of cell biology: proliferation, differentiation, and programmed cell death. Apoptosis is one of the cell death modes and may account for spontaneous tumor regression, whereas defective apoptotic mechanisms are involved in the formation of tumors and their resistance to chemotherapy. There are several signaling cascades of apoptosis initiation. The extrinsic (receptor-dependent) pathway of apoptosis is mediated by death receptors, membrane receptor proteins, the cytoplasmic part of which is composed of 80 a.a. and represents the so-called death domain. These molecules belong to the TNFα receptor family. There are six members in this group: Fas-receptor (APO-1/CD95/DR2), TNF-R1 (p55/CD120a/DR1), DR3, DR4, DR5, and DR6. All the ligands are presented as trimers (Fas-ligands), and ligand-receptor binding leads to receptor trimerization, which triggers a signaling cascade. Then, death domains become able to interact with analogous adaptor domains FADD (Fas-associated death domain) and TRADD (TNF-receptor death domain). The interaction of FADD with procaspase-8 results in the activation of caspase-8, a member of the caspase family and one of the key enzymes in apoptosis. TRADD functions analogously, but through the FADD-mediated pathway. Consequently, these interactions are followed by formation of the DISC (death-inducing signaling complex).
In the intrinsic pathway, apoptosis is triggered by the release of cytochrome c and other proteins, in particular apoptosis-inducing factor (AIF), from the intermembrane space of mitochondria because of mitochondrial outer membrane permeabilization (MOMP). Released cytochrome c forms an apoptosome complex together with cytosolic protein Apaf-1, dATP (or ATP), and procaspase-9. In this complex procaspase-9 is processed and activated. Active caspase-9 cleaves and thereby activates another member of the caspase family – caspase-3, which cleaves target proteins responsible for a variety of biochemical and morphological characteristics of apoptosis. Defects in any of the pathways described above lead to ineffective detection and elimination of potentially dangerous cells that may be transformed into malignant tumors and are responsible for cancer cell chemoresistance.
There is certain intercrossing between intrinsic and extrinsic pathways of apoptosis. Thus, caspase-8, activated in the receptor-mediated pathway, can cleave cytosolic proapoptotic protein Bid, and its cleaved fragment tBid (truncated Bid) triggers Bax and/or Bak oligomerization and incorporation into outer mitochondrial membrane, leading to pore formation and the release of proteins from the intermembrane space. Proteins described previously refer to the Bcl-2 family proteins. They play a crucial regulatory role in apoptosis induction. Originally, the first evidence that genes and proteins that are involved in oncogenesis negatively regulate cell death mechanisms was demonstrated in chromosomal translocation-containing B-cell lymphoma with Bcl-2 protein overexpression [50]. The Bcl-2 protein family is comprised of more than 30 members, which have at least one of four conserved Bcl-2 homology domains: BH1-BH4. Three main groups can be defined: 1) proapoptotic proteins that act as activators (Bax, Bak, and the less investigated Bok) and form pores in the mitochondrial outer membrane (MOM) by oligomerization, leading to cytochrome c release and caspase activation; 2) antiapoptotic proteins (Bcl-2, Bcl-XL, Bcl-W, MCL1, A1/BFL1, and Bcl-B in humans) bind proapoptotic members in the BH3 domain, preventing their oligomerization and apoptosis; and 3) proteins with only one 26-a.a. BH3 domain (Bad, Bik, Hrk, Bid, Bim, Bmf, Noxa, and Puma). In particular, the BH3-only family proteins play a regulatory role by displacing antiapoptotic proteins from their complexes with proapoptotic members that result in pore formation. The binding selectivity and affinity of the antiapoptotic proteins to the proapoptotic is determined by hydrophobic and electrostatic interactions between the proapoptotic protein BH3 domain and the binding groove formed by BH1, BH2, and BH3 domains of the antiapoptotic protein [51-53]. All of the antiapoptotic proteins bind to Bax, whereas only Bcl-XL and MCL-1 are able to bind Bak. Noxa is bound solely by Mcl-1 and A1, Bad binds to Bcl-2, Bcl-XL, and Bcl-W but not to Mcl-1 or A1, while Bim, Puma, and probably tBid bind to all five antiapoptotic members. Evidently, the upregulation of Bim or Puma leads to cell death initiation, whereas the simultaneous expression of both Bad and Noxa is required for the induction of apoptosis [54].
Thus, the balance between proapoptotic and antiapoptotic proteins in the MOM is crucial for apoptosis induction, and the disproportion towards an increase of the antiapoptotic factors leads to the suppression of the mitochondrial pathway of apoptosis [55, 56]. Indeed, elevated level of antiapoptotic and decreased content of proapoptotic proteins has been demonstrated for some tumors. The shifting of this balance towards the increase of proapoptotic protein levels resulting in apoptosis induction represents one of the strategies of fighting cancer [57].
MOMP may be achieved also through the induction of the mitochondrial permeability transition (MPT) as a result of the opening of nonspecific pores in the inner mitochondrial membrane (MPTP). Accumulation of Ca2+ by mitochondria leads to MPT, which results in the entry of water and soluble components into the mitochondria and swelling of the organelle. Therefore, the outer mitochondrial membrane is disrupted and the release of proteins from the intermembrane space occurs. According to widely held knowledge, MPTP consists of Voltage Dependent Anion Channel (VDAC) in the outer mitochondrial membrane, adenine nucleotide transporter (ANT) in the inner membrane, and cyclophilin D, a soluble protein in the matrix.
It has been established that both mechanisms of MOMP can be induced by ROS, and the anticancer effect of a wide range of therapeutic agents is based on their ROS-inducing activity (e.g. ionizing radiation, etoposide, and arsenic derivatives). Thus, α-tocopheryl succinate (α-TOS), an analog of vitamin E, targets respiratory chain complex II, leading to ROS production, Bax activation, and its translocation from cytosol to mitochondria, which results in cytochrome c release and the activation of caspases [8]. The anticancer activity of arsenic trioxide is based on the oxidative modification of thiol groups in ANT and subsequent release of cytochrome c through MPT induction.
Perhaps these two pathways of the MOMP overlap. Thus, recombinant Bax facilitates the induction of pore in isolated rat liver mitochondria [58], and Bax and Bcl-XL can interact with VDAC leading to release of cytochrome c from isolated mitochondria [59, 60]. ANT was suggested to act as a Bcl-2 and Bax partner as well [61-63]. A detailed analysis of the interaction between Bcl-2 proteins and VDAC was performed by Rostovtseva and colleagues [64]. They convincingly demonstrated that only tBid can interact with VDAC, and that this interaction results in VDAC closure.
MITOCHONDRIAL MEMBRANE STABILIZATION IN CANCER CELLS
Stabilization of the outer mitochondrial membrane against permeabilization is one of the consequences of the glycolytic shift, thus contributing to the resistance of cancer cell mitochondria to MOMP [65]. The connection between glycolysis and MOMP remains to be investigated, and different explanations exist.
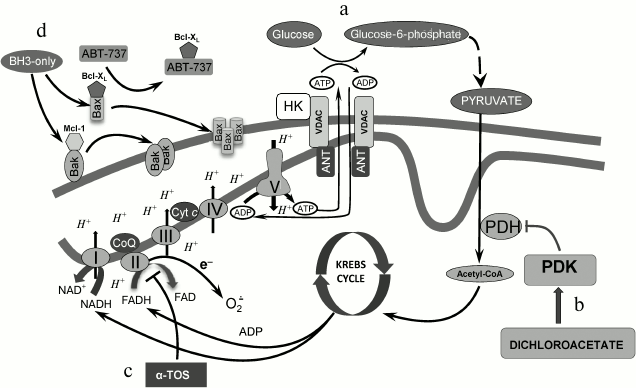
Hexokinase II and the MOM stabilization. As mentioned above, HIF stabilization leads to the upregulation of hexokinase (HK) expression, which is the first glycolytic enzyme. There are four isoforms (I-IV); two of them (I and II) directly bind to the mitochondrial membrane, in particular to VDAC, whereas HK III does not possess the hydrophobic N-terminal domain, which is essential for membrane binding. HK II is overexpressed in fast-growing tumors, with the exception of the brain tumors where HK I is expressed. This HK–VDAC interaction facilitates glucose phosphorylation, consuming ATP synthesized in mitochondria [66]. As demonstrated earlier [67], growth factor removal induced VDAC closure, which leads to cell death, whereas Bcl-XL, by binding to VDAC, sustains its open state and prevents cell death. It remains to be investigated how closure of VDAC stimulates cell death. One of the explanations suggests that VDAC has higher Ca2+ permeability in the closed state than in the open state [68], and enhanced Ca2+ accumulation might trigger MPT induction and MOMP.
HK II acts similarly to Bcl-XL by preventing VDAC closure through binding to the channel (Fig. 2a). In addition, HK II can occupy proapoptotic protein binding sites. As mentioned above, tBid is the only proapoptotic protein that can bind to VDAC and initiate its closure [64]. The importance of VDAC–HK II interaction has been demonstrated in various studies, when apoptosis was initiated upon dissociation of HK II from VDAC. Thus, Shulga et al. [69] used the 15-a.a. N-terminal peptide of HK II (N-HK II peptide), which binds to VDAC competitively with the full-size protein. Therapy of HCT116 colon cancer cells with N-HK II peptide caused HK to dissociate from its complex with VDAC. Furthermore, combined administration of cisplatin and N-HK II peptide led to synergistic enhancement of the cytotoxic effect of the drug. These data demonstrates that HK dissociation from the VDAC complex can sensitize cancer cells to chemotherapeutic agents and increase their anticancer activity [69]. In contrast, phosphorylation of HK II by Akt enhanced VDAC–HK II interaction [70]. As a result, HK II can compete with Bcl-XL for VDAC binding sites. This might lead to the release of this antiapoptotic protein, facilitating its interaction with proapoptotic protein Bax that prevents Bax–Bax oligomerization and Bax–Bak interaction. Detachment of HK II upon its phosphorylation by glycogen-3β synthase increased Bcl-XL and VDAC interaction and induced release of Bax, which, in turn, can interact with Bak/Bax, leading to pore formation and subsequent cytochrome c release [71].
Fig. 2. Mitochondria as a target for cell death stimulation. a) Stabilization of the open state of VDAC after hexokinase II binding, which optimizes glucose phosphorylation and prevents interaction of proapoptotic proteins with VDAC; b) activation of the mitochondrial pathway in energy production as a result of pyruvate dehydrogenase kinase inhibition by dichloroacetate; c) suppression of complex II of the mitochondrial respiratory chain and stimulation of oxidative stress as a result of electron leakage and superoxide radical formation; d) permeabilization of the outer mitochondrial membrane caused by dissociation of pro- and antiapoptotic protein complexes caused by BH3 protein mimetics (see details in text).
HK I binding to VDAC attenuates apoptosis, while downregulation of HK I by siRNA or inhibition by clotrimazole [72] stimulated TNF-mediated cell death. Therefore, the HK I–VDAC interaction accounts for mitochondrial membrane stabilization, thus suppressing mitochondrial pathways in cell death. This interaction can be enhanced by overexpression of oncogenic isoforms of hexokinase, or Akt activation [73]. Several VDAC isoforms with similar kinetic characteristics are known, which indicates that their interaction with HK and subsequent enhancement of glucose phosphorylation depend on the number of available binding sites [70, 74]. The most frequently expressed isoform associated with HK is VDAC1; it is also used by other mitochondria-anchoring proteins involved in the regulation of apoptosis.
Akt in mitochondrial membrane stabilization. Akt (or protein kinase B) is involved in mitochondrial stabilization. A decrease in mitochondrial respiration elevates NADH content, which results in inactivation of phosphatase and tensin homolog, PTEN, and activation of Akt. Akt activation leads to the suppression of cytochrome c release and apoptosis initiation. At the same time, Akt did not prevent apoptosis following cytochrome c injection into the cell [75], indicating that antiapoptotic activity of Akt is upstream of MOMP, and Akt contributes to mitochondrial stabilization. Besides, Akt inhibits p53-mediated expression of Bax, which also decreases the probability of MOMP [76]. Activated Akt phosphorylates proapoptotic protein Bad [77], thus preventing its interaction with MOM. Bad binds to antiapoptotic Bcl-2 family proteins, but in the phosphorylated state Bad can bind 14-3-3 proteins, which results in Bad relocalization to the cytosol and suppression of its proapoptotic activity. Furthermore, phosphorylation of Bad alters its affinity to Bcl-2 family proteins, which was demonstrated by the inability of constitutively phosphorylated Bad mutants to suppress antiapoptotic Bcl-2 proteins even in the absence of 14-3-3 protein [78].
Phosphoinositide 3-kinase (PI3-K) activation leads to Akt accumulation in mitochondria, not only on the MOM, but in the matrix and inner mitochondrial membrane as well. Besides, Akt can prevent Bax oligomerization and stimulate HK II translocation to the mitochondria, where it interacts with VDAC, which results in MOMP suppression [79-81].
Thus, cancer cell bioenergetic reprogramming is involved in the suppression of the mitochondrial pathways of apoptosis by mitochondrial membrane stabilization. Therefore, both stimulation of mitochondrial activity by inhibition of glycolysis and glutaminolysis, and direct impact on mitochondria towards its destabilization, offer promising strategies for cancer cell elimination.
TARGETING CANCER CELL BIOENERGETICS
Aerobic glycolysis. The predominant use of glycolysis for energy supply by cancer cells suggests that inhibition of glycolysis would lead to suppression of cell proliferation and to cell death. Thus, 2-deoxyglucose (2-DG), a non-metabolizable glucose analog, inhibits phosphorylation of glucose by hexokinase in a competitive manner, decreasing glucose-6-phosphate level and glycolytic capacity (Fig. 1). In addition, 2-DG enhances cytotoxic effects of doxorubicin and paclitaxel and acts synergistically with histone deacetylase inhibitors [55]. Thus, the investigation of combined therapy by 2-DG and the antidiabetic drug metformin suggests prospects for its success in clinical trials [82].
3-Bromopyruvate (3-BP), a synthetic analog of pyruvate, acts in a similar way; it covalently binds to HK II, thus inducing suppression of enzyme activity and its dissociation from VDAC, facilitating MOMP. Besides, 3-BP-induced cell death is associated with a rapid decrease in ATP content and suppression of cancer cell proliferation, which makes this agent a prospective drug for cancer cell elimination [70, 83]. 2-DG and 3-BP are already being tested in clinical trials [84-87]. It is notable that studies of cancer cell resistance to 3-BP revealed monocarboxylate transporter 1 (MCT1, SLC16A1 gene product), which is necessary for 3-BP entry into the cell, as a major factor associated with cancer cell sensitivity to 3-BP [88]. Upregulation of MCT1 expression may increase cancer cell sensitivity to treatment.
A decrease in ATP content due to inhibition of glycolysis in cancer cells with glycolytic metabolic profiles can initiate cell death in multiple ways [89]. One of the possible mechanisms is dephosphorylation of proapoptotic protein Bad followed by its activation, migration to mitochondria, MOMP, and cell death with minor toxicity for normal cells [90]. A decrease in ATP, caused by 2-DG or glucose-deprived medium, stimulated Fas-induced apoptosis as assessed by phosphatidylserine externalization on plasma membrane [91]. Notably, a significant decline in ATP content will cause necrotic cell death.
It has been mentioned previously that the metabolic shift towards glycolytic ATP production characterizes fast-growing cancer cells in contrast to slower growing cancer cells using OXPHOS as a major source of ATP [92, 93]. Thus, it appears that applying glycolytic inhibitors would not always be an effective therapy. Inhibition of glycolysis would be most effective in tumors with defective mitochondria; otherwise, stimulation of mitochondrial activity followed by inhibition of glycolysis will compensate for ATP deficiency. Thus, it has been demonstrated that gliomas that predominantly use glycolysis (glycolytic gliomas) are able to switch to OXPHOS with preferential pyruvate and glutamine utilization [94-96]. A similar switch between glycolysis and OXPHOS was found in cervical cancer cells, breast carcinoma, and pancreatic cancer [3]. In addition, in studies performed by Reitzer and colleagues, it was shown that glutamine, not glucose, is preferentially used by HeLa cells for energy supply. At the same time, OXPHOS is a predominant pathway of energy production in cervical cancer [97]. Thus, cell death may be induced by affecting glutamine metabolism. Glutamine deprivation caused apoptosis because of depletion of Krebs cycle intermediates, and not due to other consequences, such as alterations in protein, glutathione, and nucleotide biosynthesis or redox state regulation [98, 99]. It should be mentioned that the addition of two Krebs cycle intermediates, such as pyruvate or oxaloacetate, prevented cell death. Apoptosis induced by depletion of Krebs cycle intermediates is mediated by the following main functions of the cycle: (i) NADH and FADH supply for OXPHOS through reduction of NAD+ and FAD, and (ii) maintaining cooperation of different cell metabolic pathways. Apparently, alterations of NADH/NAD+ and FADH2/FAD ratios can affect regulation of transcription of genes [100] and MOMP [101]. In this way, glutamine may be involved in promotion of cell survival by providing biosynthetic precursors as well as supporting Krebs cycle functioning, which act as a regulator of the redox processes [102]. This hypothesis is supported by the fact that only a small percentage of consumed glutamine is utilized for biosynthetic processes by proliferating cells [103]. This evidence suggests that Krebs cycle substrates play crucial roles in cell bioenergetics and are involved in the regulation of cell death mechanisms [104].
Cancer cells are forced to stimulate activity of mitochondria following suppression of glycolysis and glutaminolysis. Thus, in HepG2 hepatoma cells, glucose deprivation stimulated mitochondrial biogenesis and OXPHOS, associated with increases in mtDNA, mRNA, product proteins translation, mitochondrial transcription factor A, and a twofold increase of cyclooxygenase expression [105].
As mentioned above, HIF stabilization induces PDK expression and PDH suppression. Dichloroacetate (DCA), acting as a PDK inhibitor, reactivates PDH and stimulates pyruvate conversion to acetyl-CoA, thus restoring cell metabolic profile to normal cell metabolism (Fig. 2b). DCA is able to switch metabolism from aerobic glycolysis to OXPHOS, stimulating mitochondrial activity and, as a consequence, production of ROS. The latter in turn may cause mitochondrial destabilization and apoptosis induction [106]. It is assumed that mitochondria of several tumors are significantly more sensitive to metabolic alterations because of their inability to adapt to OXPHOS rapidly and this may trigger apoptosis. DCA is undergoing phase I clinical trials. Therefore, DCA can be considered a prospective anticancer drug that targets cancer cell metabolism modulation [5, 8]. Unfortunately, in some cases the metabolism of normal cells is also affected by the doses of DCA that inhibit the enzyme activity in tumors. This limits its use in anticancer therapy [107].
Lipoic acid, a fatty acid with antioxidant heterocyclic group containing a disulfide bond, acts in a similar way and inhibits PDK. Lipoic acid is conjugated with proteins and is an essential cofactor of both the PDH complex and Krebs cycle component 2-oxoglutarate dehydrogenase. Despite the antioxidant activity of lipoic acid demonstrated in normal tissues, it may act as a prooxidant in cancer cells, leading to ROS production, p53 upregulation, and stimulation of caspase activity [108, 109]. Combined treatment with lipoic acid and chemotherapeutic agents, such as doxorubicin or IL-2 and methoxyprogesterone, may serve as a prospective strategy in anticancer therapy [14, 110].
MITOCHONDRIA AS TARGETS OF CHEMOTHERAPY
The requirement of adequate mitochondrial functioning for cancer cell growth and development has been demonstrated in various studies, suggesting that this bioenergetic pathway is a prospective target of chemotherapy. For instance, the vitamin E analog α-TOS selectively induces apoptosis, accompanied by ROS production, in cancer cells (Fig. 2c). ROS production is accounted for by respiratory chain complex II inhibition caused by the interaction of α-TOS with proximal and distal ubiquinone binding sites [111], which leads to leakage of electrons and superoxide radical production [8]. Therefore, complex II of the respiratory chain complex appears to be a prospective target for anticancer chemotherapy. Analogously, suppression of succinate oxidation in complex II by acetoin led to suppression of mitochondrial activity in Ehrlich carcinoma without affecting normal cells [112]. Notably, α-TOS has a wide range of targets, for instance, it is able to stimulate Ca2+ uptake, which in the presence of ROS promotes MPT induction [113]. The addition of triphenylphosphonium groups to the α-TOS molecule leads to its accumulation in mitochondria, promoting induction of cell death by lower doses of the drug. Applying such mitochondria-targeted agent as MitoVES suppressed cancer cell proliferation by affecting mtDNA [114].
3-BP, described previously as an anticancer drug that targets hexokinase, is an alkylating agent that induces apoptosis through alternative mechanisms as well. Recent studies demonstrated that 3-BP affects mitochondria; furthermore, it stimulates ROS production, thus stimulating the mitochondrial pathway of apoptosis in breast cancer MDA-MB-231 cells. It was shown that the decrease of antiapoptotic protein Mcl-1 was involved in stimulation of cell death. 3-BP-induced apoptosis was accompanied by a decrease in the p-Akt level, suggesting that Mcl-1 decline was associated with PI3/Akt signal cascade [115]. In hepatoma HepG2, 3-BP suppressed respiratory chain complex II, and in the case of glucose deprivation, it inhibited complex I as well. In addition, 3-BP triggered uncoupling of mitochondrial oxidation and phosphorylation, stimulating oxygen consumption insensitive to oligomycin [116, 117]. The anticancer activity of 3-BP was enhanced by glutamine deprivation, which is probably associated with stabilization of monocarboxylate transporters [118].
As described previously, MOMP is a nonreversible process of apoptosis. In recent years, the synthetic analogs of BH3-only proteins, known as BH3 mimetics, appeared. One of them is ABT-737, which is able to stimulate cell death by Bax or Bak release from their complexes with antiapoptotic protein (Fig. 2d). ABT-263, an orally administered derivative of ABT-737, also known as navitoclax, has a similar activity. Both drugs are undergoing phase I/II clinical trials. ABT-737 functions similarly to the BH3-only family member Bad. ABT-737 and ABT-263 bind with high affinity (IC50 < 10 nM) to Bcl-2, Bcl-XL and Bcl-W, but not to Mcl-1 or A1, thus greatly limiting the cytotoxic effect of ABT-737 (for instance, breast cancer line MEF are highly resistant to ABT-737). Therefore, the combined application of ABT-737 with Mcl-1-inhibiting agent is necessary for successful treatment. Such co-treatment is currently considered a prospective strategy in anticancer therapy [119]. Thus, cancer cell sensitivity to ABT-737 as a single anticancer agent absolutely depends on the profile of expressed Bcl-2 family proteins. For instance, small cell carcinoma and various types of lymphoma and leukemia have significantly higher Bcl-2 protein level and are highly sensitive to ABT-737. Notably, ABT-737 binding to Bcl-2, Bcl-XL, and Bcl-W proteins leads to the release of the actual BH3-only proteins, which are able to bind to and thus inactivate other antiapoptotic proteins, in particular Mcl-1, which does not interact with ABT-737 [120].
Despite the established mechanism of BH3 mimetics activity, some of them induce cell death in the absence of Bax and Bak, indicating the presence of targets other than antiapoptotic proteins [119, 121]. Obatoclax, a synthetic derivative of prodigiosin, binds to all of the antiapoptotic proteins with low affinity (comparable to the affinity of ABT-737 to Mcl-1) and kills both wild-type cells and cells lacking Bax and Bak. Presumably, the cytotoxic effect of obatoclax is determined by caspase-independent or autophagy-mediated mechanisms by endoplasmic reticulum (ER) stress or upregulation of Noxa. The latter causes Mcl-1/Bak complex dissociation, which explains the anticancer activity of obatoclax and its role in cell sensitization to ABT-737 [119, 122, 123].
Gossypol, a polyphenolic aldehyde derived from plants, was originally used clinically not as an anticancer drug, until its BH3 mimetic properties were found. Gossypol binds to antiapoptotic proteins with modest affinity (on the order of a few µM) [123]. The gossypol derivatives apogossypol and apogossypolone, which also function as BH3 mimetics, are undergoing preclinical studies, whereas the benzoylsulfonide derivative TW37 has already reached phase I/II trials.
Zhang and colleagues discovered a new BH3-mimetic, S1. It binds to both Mcl-1 and Bcl-2, causing dissociation of the Bax/Bcl-2 and Bak/Mcl-1 complexes (dose and time-dependent effect) and apoptosis induction [124]. However, Eastman and colleagues suggested that S1 does not act as a true inhibitor of Bcl-2 proteins, but stimulates Noxa upregulation, which inhibits Mcl-1 and induces Mcl-1 degradation, leading to cancer cell sensitization to the cytotoxic therapy [122]. Furthermore, S1 stimulates ROS production followed by ER stress [125]. In addition, Zhong et al. have demonstrated that S1-induced cell death may be associated with autophagy stimulated by ER stress and with alterations of Beclin 1 and Bcl-2 interaction [51, 126]. Besides, Song et al. synthesized derivatives more potent than S1 and other new Mcl-1 inhibitors: a 2-cyanoacetamide and a 2-hydroxynicotinonitrile [127, 128].
Recently, a number of other low molecular weight compounds with different structure have been identified that have BH3-mimetic activity. However, the lack of a sufficient number of in vivo studies of the newly discovered and synthesized Mcl-1 inhibitors does not allow us to consider their further application. The newly discovered BH3-mimetic 072RB [129] decreased Bcl-XL and Mcl-1; however, whether its activity is determined by direct binding of these antiapoptotic proteins remains to be elucidated [130].
Affecting cell bioenergetics for cancer cell elimination and for reduction of toxicity of chemotherapeutic drugs is a rapidly growing area that appears to be a prospective strategy of future anticancer therapy. Thus, it has been demonstrated on a murine breast cancer xenografts model that 2-DG combined with mitochondrially targeted anticancer agents, such as Mito-CP and Mito-Q, led to significant tumor regression with no revealed side effects to normal tissues [131]. Most recently, it has been shown that the well-known antidiabetic drug metformin induces cancer cell death and suppresses the growth of xenografts. Combined therapy with 2-DG and metformin synergistically stimulated cell death, and 2-DG treatment lowered the required metformin concentration more than five-fold [132]. Analogously, the lactate dehydrogenase inhibitor oxamate significantly stimulated ionizing radiation-induced suppression of cell growth in murine xenograft [133]. Inhibition of glycolysis by 2-DG not only stimulated both ABT-737-induced apoptosis in lymphoma cells, but also prolonged survival of mice with lymphoma xenograft [134].
In summary, there are two general features of metabolic reprogramming in the majority of cancer cells: preferential use of glycolysis over OXPHOS for ATP production (even in the presence of ample oxygen) and stimulation of glutamine consumption as one of the energy sources. However, various types of tumors demonstrate significant genetic and phenotypic heterogeneity in metabolic profiles of cells. Thus, about 30% of tumors cannot be detected by FDG-PET (fludeoxyglucose-positron emission tomography) due to insufficient (below threshold) glycolytic shift or because cells utilize substrates other than glucose. Cancer cell metabolism depends on the presence of oncogene and/or oncosuppressor mutations and is regulated by the tumor microenvironment (including oxygen and nutrient concentrations). Tumor radio- and chemoresistance may be determined by abnormalities of cell metabolism, suggesting that reactivation of the “normal” metabolic profile can sensitize cancer cells to therapy. Investigation of cancer cell bioenergetics and its contribution to chemoresistance appears to be a prospective strategy in cancer cell elimination.
The part of this review “Targeting Cancer Cell Bioenergetics” was supported by the Russian Science Foundation (grant No. 14-15-00463 to PVM and VGG), the part “Mechanisms of Bioenergetic Reprogramming” – by the Russian Science Foundation (grant No. 14-25-00056 to BDZ and AVK). Work in the authors’ lab also was supported by grants from the Russian Foundation for Basic Research (Nos. 14-04-00963, 14-04-01660, and 14-04-31078-mol-a). AVK has been awarded a grant of the President of the Russian Federation for young scientists No. 14.120.2849-MK.
We apologize to authors whose primary references could not be cited due to space limitations.
REFERENCES
1.Hanahan, D., and Weinberg, R. A. (2011) Hallmarks
of cancer: the next generation, Cell, 144, 646-674.
2.Lunt, S. Y., and Heiden, M. V. (2011) Aerobic
glycolysis: meeting the metabolic requirements of cell proliferation,
Annu. Rev. Cell Dev. Biol., 27, 441-464.
3.Tennant, D. A., Duran, R. V., Boulahbel, H., and
Gottlieb, E. (2009) Metabolic transformation in cancer,
Carcinogenesis, 30, 1269-1280.
4.Funes, J. M., Quintero, M., Henderson, S.,
Martinez, D., Qureshi, U., Westwood, C., Clements, M. O., Bourboulia,
D., Pedley, R. B., Moncada, S., and Boshoff, C. (2007) Transformation
of human mesenchymal stem cells increases their dependency on oxidative
phosphorylation for energy production, Proc. Natl. Acad. Sci.
USA, 104, 6223-6228.
5.Moreno-Sanchez, R., Rodriguez-Enriquez, S.,
Marin-Hernandez, A., and Saavedra, E. (2007) Energy metabolism in tumor
cells, FEBS J., 274, 1393-1418.
6.Rodriguez-Enriquez, S., Carreno-Fuentes, L.,
Gallardo-Perez, J. C., Saavedra, E., Quezada, H., Vega, A.,
Marin-Hernandez, A., Olin-Sandoval, V., Torres-Marquez, M. E., and
Moreno-Sanchez, R. (2010) Oxidative phosphorylation is impaired by
prolonged hypoxia in breast and possibly in cervix carcinoma, Int.
J. Biochem. Cell Biol., 42, 1744-1751.
7.Barbosa, I. A., Machado, N. G., Skildum, A. J.,
Scott, P. M., and Oliveira, P. J. (2012) Mitochondrial remodeling in
cancer metabolism and survival: potential for new therapies,
Biochim. Biophys. Acta, 1826, 238-254.
8.Ralph, S. J., Rodriguez-Enriquez, S., Neuzil, J.,
and Moreno-Sanchez, R. (2010) Bioenergetic pathways in tumor
mitochondria as targets for cancer therapy and the importance of the
ROS-induced apoptotic trigger, Mol. Aspects Med., 31,
29-59.
9.Vaughn, A., and Deshmukh, M. (2008) Glucose
metabolism inhibits apoptosis in neurons and cancer cells by redox
inactivation of cytochrome c, Nat. Cell Biol., 10,
1477-1483.
10.Cairns, R. A., Harris, I. S., and Mak, T. W.
(2011) Regulation of cancer cell metabolism, Nat. Rev. Cancer,
11, 85-95.
11.Gao, P., Tchernyshyov, I., Chang, T., and Lee, Y.
(2009) c-Myc suppression of miR-23a/b enhances mitochondrial
glutaminase expression and glutamine metabolism, Nature,
458, 762-765.
12.Wise, D. R., Deberardinis, R. J., Mancuso, A.,
Sayed, N., Zhang, X., Pfeiffer, H. K., Nissim, I., Daikhin, E.,
Yudkoff, M., Mcmahon, S. B., and Thompson, C. B. (2008) Myc regulates a
transcriptional program that stimulates mitochondrial glutaminolysis
and leads to glutamine addiction, PNAS, 105,
18782-18787.
13.Dang, C. V. (2010) Rethinking the Warburg effect
with Myc micromanaging glutamine metabolism, Cancer Res.,
70, 859-862.
14.Jose, C., Bellance, N., and Rossignol, R. (2011)
Choosing between glycolysis and oxidative phosphorylation: a
tumor’s dilemma? Biochim. Biophys. Acta, 1807,
552-561.
15.Mazurek, S., Michel, A., and Eigenbrodt, E.
(1997) Effect of extracellular AMP on cell proliferation and metabolism
of breast cancer cell lines with high and low glycolytic rates, J.
Biol. Chem., 272, 4941-4952.
16.Rossignol, R., Gilkerson, R., and Aggeler, R.
(2004) Energy substrate modulates mitochondrial structure and oxidative
capacity in cancer cells, Cancer Res., 64, 985-993.
17.Moreadith, R., and Lehninger, A. (1984)
Purification, kinetic behavior, and regulation of NAD(P)+
malic enzyme of tumor mitochondria, J. Biol. Chem., 259,
6222-6227.
18.Mandella, R., and Sauer, L. (1975) The
mitochondrial malic enzymes. I. Submitochondrial localization and
purification and properties of the NAD(P)+-dependent enzyme
from adrenal cortex, J. Biol. Chem., 250, 5877-5884.
19.Brand, R. M., Lyons, R. H., and Midgley, A. R.
(1994) Understanding the dynamics of cellular responsiveness to
modifications of metabolic substrates in perifusion, J. Cell.
Physiol., 160, 10-16.
20.Zu, X. L., and Guppy, M. (2004) Cancer
metabolism: facts, fantasy, and fiction, Biochem. Biophys. Res.
Commun., 313, 459-465.
21.Vander Heiden, M. G., Cantley, L. C., and
Thompson, C. B. (2009) Understanding the Warburg effect: the metabolic
requirements of cell proliferation, Science, 324,
1029-1033.
22.Gorlach, A., and Acker, H. (1994) pO2-
and pH-gradients in multicellular spheroids and their relationship to
cellular metabolism and radiation sensitivity of malignant human tumor
cells, Biochim. Biophys. Acta, 1227, 105-112.
23.Sutherland, R. (1998) Tumor hypoxia and gene
expression, Acta Oncol., 37, 567-574.
24.Vaupel, P., Kallinowski, F., and Okunieff, P.
(1989) Blood flow, oxygen and nutrient supply, and metabolic
microenvironment of human tumors: a review, Cancer Res.,
49, 6449-6465.
25.Matsumoto, A., Matsumoto, S., Sowers, A.,
Koscielniak, J., Trigg, N., Kuppusamy, P., Mitchell, J., Subramanian,
S., Krishna, M., and Matsumoto, K. (2005) Absolute oxygen tension
(pO2) in murine fatty and muscle tissue as determined by
EPR, Magn. Reson. Med., 54, 1530-1535.
26.Schroeder, T., Yuan, H., Viglianti, B., Peltz,
C., Asopa, S., Vujaskovic, Z., and Dewhirst, M. (2005) Spatial
heterogeneity and oxygen dependence of glucose consumption in R3230Ac
and fibrosarcomas of the Fischer 344 rat, Cancer Res.,
65, 5163-5171.
27.Mason, M. G., Nicholls, P., Wilson, M. T., and
Cooper, C. E. (2006) Nitric oxide inhibition of respiration involves
both competitive (heme) and noncompetitive (copper) binding to
cytochrome c oxidase, Proc. Natl. Acad. Sci. USA,
103, 708-713.
28.Gnaiger, E., Lassnig, B., Kuznetsov, A., Riege,
A. G., and Margreiter, R. (1998) Mitochondrial oxygen affinity,
respiratory flux control and excess capacity of cytochrome c
oxidase, J. Exp. Biol., 201, 1129-1139.
29.Pecina, P., Gnaiger, E., Zeman, J., Pronicka, E.,
and Houstek, T. (2004) Decreased affinity for oxygen of cytochromec
oxidase in Leigh syndrome caused by SURF1 mutations, Am. J. Physiol.
Cell Physiol., 287, C1384-C1388.
30.Matoba, S., Kang, J.-G., Patino, W. D., Wragg,
A., Boehm, M., Gavrilova, O., Hurley, P. J., Bunz, F., and Hwang, P. M.
(2006) P53 regulates mitochondrial respiration, Science,
312, 1650-1653.
31.Pollard, P. J., Wortham, N. C., and Tomlinson, I.
P. M. (2003) The TCA cycle and tumorigenesis: the examples of fumarate
hydratase and succinate dehydrogenase, Ann. Med., 35,
632-639.
32.Robey, I. F., Lien, A. D., Welsh, S. J., Baggett,
B. K., and Gillies, R. J. (2005) Hypoxia-inducible factor-1α and
the glycolytic phenotype in tumors, Neoplasia, 7,
324-330.
33.Kroemer, G. (2006) Mitochondria in cancer,
Oncogene, 25, 4630-4632.
34.Yeunga, S. J., Pand, J., and Leec, M.-H. (2008)
Roles of p53, Myc and HIF-1 in regulating glycolysis – the
seventh hallmark of cancer, Cell. Mol. Life Sci., 65,
3981-3999.
35.Shaw, R. J. (2006) Glucose metabolism and cancer,
Curr. Opin. Cell Biol., 18, 598-608.
36.Parlo, R., and Coleman, P. (1984) Enhanced rate
of citrate export from cholesterol-rich hepatoma mitochondria. The
truncated Krebs cycle and other metabolic ramifications of
mitochondrial membrane cholesterol, J. Biol. Chem., 259,
9997-10003.
37.Briscoe, D., Fiskum, G., Holleran, A., and
Kelleher, J. (1994) Acetoacetate metabolism in AS-30D hepatoma cells,
Mol. Cell Biochem., 136, 131-137.
38.Dietzen, D., and Davis, E. (1993) Oxidation of
pyruvate, malate, citrate, and cytosolic reducing equivalents by AS-30D
hepatoma mitochondria, Arch. Biochem. Biophys., 305,
91-102.
39.Schmitt, S., Schulz, S., Schropp, E.-M.,
Eberhagen, C., Simmons, A., Beisker, W., Aichler, M., and Zischka, H.
(2014) Why to compare absolute numbers of mitochondria,
Mitochondrion, 19 (Pt. A), 113-123.
40.Pedersen, P. (1978) Tumor mitochondria and the
bioenergetics of cancer cells, Prog. Exp. Tumor Res., 22,
190-274.
41.LaNoue, K., Hemington, J., Ohnishi, T., Morris,
H., and Williamson, J. (1974) Defects in anion and electron transport
in Morris hepatoma mitochondria, Horm. Cancer, 131-167.
42.Lichtor, T., and Dohrmann, G. (1987) Oxidative
metabolism and glycolysis in benign brain tumors, Neurosurgery,
67, 336-340.
43.Melo, R., Stevan, F., Campello, A., Carnieri, E.,
and de Oliveira, M. (1998) Occurrence of the Crabtree effect in HeLa
cells, Cell Biochem. Funct., 16, 99-105.
44.Sauer, L. (1977) On the mechanism of the Crabtree
effect in mouse ascites tumor cells, J. Cell Physiol.,
93, 313-316.
45.Sussman, I., Erecinska, M., and Wilson, D. (1980)
Regulation of cellular energy metabolism: the Crabtree effect,
Biochim. Biophys. Acta, 591, 209-223.
46.Seshagiri, P., and Bavister, B. (1991) Glucose
and phosphate inhibit respiration and oxidative metabolism in cultured
hamster eight-cell embryos: evidence for the “Crabtree
effect”, Mol. Reprod. Dev., 30, 105-111.
47.Yang, X., Borg, L., and Eriksson, U. (1997)
Altered metabolism and superoxide generation in neural tissue of rat
embryos exposed to high glucose, Am. J. Physiol., E173-E180.
48.Rodriguez-Enriquez, S., Juarez, O.,
Rodriguez-Zavala, J. S., and Moreno-Sanchez, R. (2001) Multisite
control of the Crabtree effect in ascites hepatoma cells, Eur. J.
Biochem., 268, 2512-2519.
49.Covian, R., and Moreno-Sanchez, R. (2001) Role of
protonatable groups of bovine heart bc1 complex in
ubiquinol binding and oxidation, Eur. J. Biochem., 268,
5783-5790.
50.Tsujimoto, Y., Ikegaki, N., and Croce, C. M.
(1987) Characterization of the protein product of bcl-2, the
gene involved in human follicular lymphoma, Oncogene, 2,
3-7.
51.Belmar, J., and Fesik, S. W. (2014) Small
molecule Mcl-1 inhibitors for the treatment of cancer, Pharmacol.
Ther., 145, 76-84.
52.Dutta, S., Gulla, S., Chen, T. S., Fire, E.,
Grant, R. A., and Keating, A. E. (2010) Determinants of BH3 binding
specificity for Mcl-1 versus Bcl-xL, J. Mol. Biol., 398,
747-762.
53.Moldoveanu, T., Follis, A. V., Kriwacki, R. W.,
and Green, D. R. (2014) Many players in BCL-2 family affairs, Trends
Biochem. Sci., 39, 101-111.
54.Chen, L., Willis, S., Wei, A., Smith, B.,
Fletcher, J., Hinds, M., Colman, P., Day, C., Adams, J., and Huang, D.
(2005) Differential targeting of prosurvival Bcl-2 proteins by their
BH3-only ligands allows complementary apoptotic function, Mol.
Cell, 17, 393-403.
55.Biasutto, L., Dong, L.-F., Zoratti, M., and
Neuzil, J. (2010) Mitochondrially targeted anti-cancer agents,
Mitochondrion, 10, 670-681.
56.Gogvadze, V., Orrenius, S., and Zhivotovsky, B.
(2008) Mitochondria in cancer cells: what is so special about them?
Trends Cell Biol., 18, 165-173.
57.Hartman, M., and Czyz, M. (2012) Pro-apoptotic
activity of BH3-only proteins and BH3 mimetics: from theory to
potential cancer therapy, Anticancer Agents Med. Chem.,
12, 966-981.
58.Gogvadze, V., Robertson, J. D., Zhivotovsky, B.,
and Orrenius, S. (2001) Cytochrome c release occurs via
Ca2+-dependent and Ca2+-independent mechanisms
that are regulated by Bax, J. Biol. Chem., 276,
19066-19071.
59.Armstrong, J. S. (2006) The role of the
mitochondrial permeability transition in cell death,
Mitochondrion, 6, 225-234.
60.Narita, M., Shimizu, S., Ito, T., Chittenden, T.,
Lutz, R. J., Matsuda, H., and Tsujimoto, Y. (1998) Bax interacts with
the permeability transition pore to induce permeability transition and
cytochrome c release in isolated mitochondria, Proc. Natl.
Acad. Sci. USA, 95, 14681-14686.
61.Brenner, C., Cadiou, H., Vieira, H. L., Zamzami,
N., Marzo, I., Xie, Z., Leber, B., Andrews, D., Duclohier, H., Reed, J.
C., and Kroemer, G. (2000) Bcl-2 and Bax regulate the channel activity
of the mitochondrial adenine nucleotide translocator, Oncogene,
19, 329-336.
62.Crompton, M., Barksby, E., Johnson, N., and
Capano, M. (2002) Mitochondrial intermembrane junctional complexes and
their involvement in cell death, Biochimie, 84,
143-152.
63.Marzo, I., Brenner, C., Zamzami, N.,
Jurgensmeier, J. M., Susin, S. A., Vieira, H. L., Prevost, M. C., Xie,
Z., Matsuyama, S., Reed, J. C., and Kroemer, G. (1998) Bax and adenine
nucleotide translocator cooperate in the mitochondrial control of
apoptosis, Science, 281, 2027-2031.
64.Rostovtseva, T. K., Antonsson, B., Suzuki, M.,
Youle, R. J., Colombini, M., and Bezrukov, S. M. (2004) Bid, but not
Bax, regulates VDAC channels, J. Biol. Chem., 279,
13575-13583.
65.Green, D. R., and Kroemer, G. (2004) The
pathophysiology of mitochondrial cell death, Science,
305, 626-629.
66.Arora, K. K., and Pedersen, P. L. (1988)
Functional significance of mitochondrial bound hexokinase in tumor cell
metabolism. Evidence for preferential phosphorylation of glucose by
intramitochondrially generated ATP, J. Biol. Chem., 263,
17422-17428.
67.Vander Heiden, M. G., Li, X. X., Gottleib, E.,
Hill, R. B., Thompson, C. B., and Colombini, M. (2001) Bcl-xL promotes
the open configuration of the voltage-dependent anion channel and
metabolite passage through the outer mitochondrial membrane, J.
Biol. Chem., 276, 19414-19419.
68.Tan, W., and Colombini, M. (2007) VDAC closure
increases calcium ion flux, Biochim. Biophys. Acta, 29,
2510-2515.
69.Shulga, N. (2009) Hexokinase II detachment from
the mitochondria potentiates cisplatin induced cytotoxicity through a
caspase-2 dependent mechanism, Cell Cycle, 8,
3355-3364.
70.Mathupala, S., Ko, Y., and Pedersen, P. (2012)
Hexokinase II: cancer’s double-edged sword acting as both
facilitator and gatekeeper of malignancy when bound to mitochondria,
Oncogene, 25, 4777-4786.
71.Pastorino, J., and Hoek, J. (2008) Regulation of
hexokinase binding to VDAC, J. Bioenerg. Biomembr., 40,
171-182.
72.Schindler, A., and Foley, E. (2013) Hexokinase 1
blocks apoptotic signals at the mitochondria, Cell. Signal.,
25, 2685-2692.
73.Robey, R. B., and Hay, N. (2006) Mitochondrial
hexokinases, novel mediators of the antiapoptotic effects of growth
factors and Akt, Oncogene, 25, 4683-4696.
74.Shinohara, Y., Ishida, T., Hino, M., Yamazaki,
N., Baba, Y., and Terada, H. (2000) Characterization of porin isoforms
expressed in tumor cells, Eur. J. Biochem., 267,
6067-6073.
75.Kennedy, S. G., Kandel, E. S., Cross, T. K., and
Hay, N. (1999) Akt/protein kinase B inhibits cell death by preventing
the release of cytochrome c from mitochondria, Mol. Cell.
Biol., 19, 5800-5810.
76.Yamaguchi, A., Tamatani, M., Matsuzaki, H.,
Namikawa, K., Kiyama, H., Vitek, M. P., Mitsuda, N., and Tohyama, M.
(2001) Akt activation protects hippocampal neurons from apoptosis by
inhibiting transcriptional activity of p53, J. Biol. Chem.,
276, 5256-5264.
77.Del Peso, L., Gonzalez-Garcia, M., Page, C.,
Herrera, R., and Nunez, G. (1997) Interleukin-3-induced phosphorylation
of BAD through the protein kinase Akt, Science, 278,
687-699.
78.Franke, T. F., Hornik, C. P., Segev, L., Shostak,
G. A., and Sugimoto, C. (2003) PI3K/Akt and apoptosis: size matters,
Oncogene, 22, 8983-8998.
79.Gogvadze, V., Zhivotovsky, B., and Orrenius, S.
(2010) The Warburg effect and mitochondrial stability in cancer cells,
Mol. Aspects Med., 31, 60-74.
80.Majewski, N., Nogueira, V., Bhaskar, P., Coy, P.
E., Skeen, J. E., Gottlob, K., Chandel, N. S., Thompson, C. B., Robey,
R. B., and Hay, N. (2004) Hexokinase–mitochondria interaction
mediated by Akt is required to inhibit apoptosis in the presence or
absence of Bax and Bak, Mol. Cell, 16, 819-830.
81.Mookherjee, P., and Quintanilla, R. (2007)
Mitochondrial-targeted active Akt protects SH-SY5Y neuroblastoma cells
from staurosporine-induced apoptotic cell death, J. Cell
Biochem., 102, 196-210.
82.Weinberg, S. E., and Chandel, N. S. (2015)
Targeting mitochondria metabolism for cancer therapy, Nat. Publ.
Gr., 11, 9-15.
83.Neuzil, J., Dong, L. F., Rohlena, J., Truksa, J.,
and Ralph, S. J. (2013) Classification of mitocans, anti-cancer drugs
acting on mitochondria, Mitochondrion, 13, 199-208.
84.Lea, M. A., Qureshi, M. S., Buxhoeveden, M.,
Gengel, N., Kleinschmit, J., and DesBordes, C. (2013) Regulation of the
proliferation of colon cancer cells by compounds that affect
glycolysis, including 3-bromopyruvate, 2-deoxyglucose and biguanides,
Anticancer Res., 33, 401-407.
85.Loar, P., Wahl, H., Kshirsagar, M., Gossner, G.,
Griffith, K., and Liu, J. R. (2010) Inhibition of glycolysis enhances
cisplatin-induced apoptosis in ovarian cancer cells, Am. J. Obstet.
Gynecol., 202, 1-8.
86.Maher, J. C., Krishan, A., and Lampidis, T. J.
(2004) Greater cell cycle inhibition and cytotoxicity induced by
2-deoxy-D-glucose in tumor cells treated under hypoxic vs aerobic
conditions, Cancer Chemother. Pharmacol., 53,
116-122.
87.Sullivan, E. J., Kurtoglu, M., Brenneman, R.,
Liu, H., and Lampidis, T. J. (2014) Targeting cisplatin-resistant human
tumor cells with metabolic inhibitors, Cancer Chemother.
Pharmacol., 73, 417-427.
88.Birsoy, K., Wang, T., Possemato, R., Yilmaz, O.,
Koch, C., Chen, W., Hutchins, A., Gultekin, Y., Peterson, T., Carette,
J., Brummelkamp, T., Clish, C., and Sabatini, D. M. (2012)
MCT1-mediated transport of a toxic molecule is an effective strategy
for targeting glycolytic tumors, Changes, 29,
997-1003.
89.Urakami, K., Zangiacomi, V., Yamaguchi, K., and
Kusuhara, M. (2013) Impact of 2-deoxy-D-glucose on the target
metabolome profile of a human endometrial cancer cell line, Biomed.
Res., 34, 221-229.
90.Xu, R., Pelicano, H., and Zhou, Y. (2005)
Inhibition of glycolysis in cancer cells : a novel strategy to
overcome drug resistance associated with mitochondrial respiratory
defect and hypoxia, 65, 613-621.
91.Ralph, S. J., Low, P., Dong, L., Lawen, A., and
Neuzil, J. (2006) Mitocans: mitochondrial targeted anti-cancer drugs as
improved therapies and related patent documents, Recent Pat.
Anticancer Drug Discov., 1, 327-346.
92.Zu, X. L., and Guppy, M. (2004) Cancer
metabolism: facts, fantasy, and fiction, Biochem. Biophys. Res.
Commun., 313, 459-465.
93.Dang, C. V., and Semenza, G. L. (1999) Oncogenic
alterations of metabolism, Trends Biochem. Sci., 24,
68-72.
94.Griguer, C. E., Oliva, C. R., and Gillespie, G.
Y. (2005) Glucose metabolism heterogeneity in human and mouse malignant
glioma cell lines, J. Neurooncol., 74, 123-133.
95.Mathupala, S. P., Ko, Y. H., and Pedersen, P. L.
(2010) The pivotal roles of mitochondria in cancer: Warburg and beyond
and encouraging prospects for effective therapies, Biochim. Biophys.
Acta – Bioenergetics, 1797, 1225-1230.
96.Solaini, G., Sgarbi, G., and Baracca, A. (2011)
Oxidative phosphorylation in cancer cells, Biochim. Biophys.
Acta, 1807, 534-542.
97.Reitzer, L., Wice, B., and Kennell, D. (1979)
Evidence that glutamine, not sugar, is the major energy source for
cultured HeLa cells, J. Biol. Chem., 254, 2669-2676.
98.Fuchs, B. C., and Bode, B. P. (2006) Stressing
out over survival: glutamine as an apoptotic modulator, J. Surg.
Res., 131, 26-40.
99.Yuneva, M., Zamboni, N., Oefner, P.,
Sachidanandam, R., and Lazebnik, Y. (2007) Deficiency in glutamine but
not glucose induces MYC-dependent apoptosis in human cells, J. Cell
Biol., 178, 93-105.
100.Ladurner, A. G. (2006) Rheostat control of gene
expression by metabolites, Mol. Cell, 24, 1-11.
101.Jonas, E. A., Hickman, J. A., Chachar, M.,
Polster, B. M., Brandt, T. A., Fannjiang, Y., Ivanovska, I., Basanez,
G., Kinnally, K. W., Zimmerberg, J., Hardwick, J. M., and Kaczmarek, L.
K. (2004) Proapoptotic N-truncated BCL-xL protein activates endogenous
mitochondrial channels in living synaptic terminals, Proc. Natl.
Acad. Sci. USA, 101, 13590-13595.
102.Baggetto, L. G. (1992) Deviant energetic
metabolism of glycolytic cancer cells, Biochimie, 74,
959-974.
103.Newsholme, E., Crabtree, B., and Ardawi, M.
(1985) The role of high rates of glycolysis and glutamine utilization
in rapidly dividing cells, Biosci. Rep., 400,
393-400.
104.Kruspig, B., Zhivotovsky, B., and Gogvadze, V.
(2014) Mitochondrial substrates in cancer: drivers or passengers?
Mitochondrion, 19, Pt. A, 8-19.
105.Smolkova, K., Plecita-Hlavata, L., Bellance,
N., Benard, G., Rossignol, R., and Jezek, P. (2011) Waves of gene
regulation suppress and then restore oxidative phosphorylation in
cancer cells, Int. J. Biochem. Cell Biol., 43,
950-968.
106.Bonnet, S., Archer, S. L., Allalunis-Turner,
J., Haromy, A., Beaulieu, C., Thompson, R., Lee, C. T., Lopaschuk, G.
D., Puttagunta, L., Bonnet, S., Harry, G., Hashimoto, K., Porter, C.
J., Andrade, M. A., Thebaud, B., and Michelakis, E. D. (2007) A
mitochondria–K+ channel axis is suppressed in cancer
and its normalization promotes apoptosis and inhibits cancer growth,
Cancer Cell, 11, 37-51.
107.Cai, P., Boor, P. J., Khan, M., Kaphalia, B.
S., Ansari, G. A., and Konig, R. (2007) Immuno- and hepato-toxicity of
dichloroacetic acid in MRL+/+ and B6C3F1 mice, J.
Immunotoxicol., 4, 107-115.
108.Moungjaroen, J., Nimmannit, U., Callery, P. S.,
Wang, L., Azad, N., Lipipun, V., Chanvorachote, P., and Rojanasakul, Y.
(2006) Reactive oxygen species mediate caspase activation and apoptosis
induced by lipoic acid in human lung epithelial cancer cells through
Bcl-2 down-regulation, J. Pharmacol. Exp. Ther., 319,
1062-1069.
109.Simbula, G., Columbano, A., Ledda-Columbano, G.
M., Sanna, L., Deidda, M., Diana, A., and Pibiri, M. (2007) Increased
ROS generation and p53 activation in alpha-lipoic acid-induced
apoptosis of hepatoma cells, Apoptosis, 12, 113-123.
110.Feron, O. (2009) Pyruvate into lactate and
back: from the Warburg effect to symbiotic energy fuel exchange in
cancer cells, Radiother. Oncol., 92, 329-333.
111.Dong, L., Low, P., Dyason, J., and Wang, X.
(2008) α-Tocopheryl succinate induces apoptosis by targeting
ubiquinone-binding sites in mitochondrial respiratory complex II,
Oncogene, 27, 4324-4335.
112.Baggetto, L., and Testa-Parussini, R. (1990)
Role of acetoin on the regulation of intermediate metabolism of Ehrlich
ascites tumor mitochondria: its contribution to membrane cholesterol
enrichment modifying passive proton permeability, Arch. Biochem.
Biophys., 283, 241-248.
113.Kruspig, B., Nilchian, A., Bejarano, I.,
Orrenius, S., Zhivotovsky, B., and Gogvadze, V. (2012) Targeting
mitochondria by α-tocopheryl succinate kills neuroblastoma cells
irrespective of MycN oncogene expression, Cell. Mol. Life Sci.,
69, 2091-2099.
114.Truksa, J., Dong, L.-F., Rohlena, J., Stursa,
J., Vondrusova, M., Goodwin, J., Nguyen, M., Kluckova, K.,
Rychtarcikova, Z., Lettlova, S., Spacilova, J., Stapelberg, M.,
Zoratti, M., and Neuzil, J. (2015) Mitochondrially targeted vitamin E
succinate modulates expression of mitochondrial DNA transcripts and
mitochondrial biogenesis, Antioxid. Redox Signal., 22,
883-900.
115.Liu, Z., Zhang, Y., Zhang, Q., Zhao, S., Wu,
C., Cheng, X., Jiang, C., Jiang, Z., and Liu, H. (2014) 3-Bromopyruvate
induces apoptosis in breast cancer cells by downregulating Mcl-1
through the PI3K/Akt signaling pathway, Anticancer Drugs,
25, 447-455.
116.Macchioni, L., Davidescu, M., and Roberti, R.
(2014) The energy blockers 3-bromopyruvate and lonidamine: effects on
bioenergetics of brain mitochondria, J. Bioenerg. Biomembr.,
46, 389-394.
117.Pereira da Silva, A. P., El-Bacha, T., Kyaw,
N., dos Santos, R. S., da Silva, W. S., Almeida, F. C. L., Da Poian, A.
T., and Galina, A. (2009) Inhibition of energy-producing pathways of
HepG2 cells by 3-bromopyruvate, Biochem. J., 417,
717-726.
118.Cardaci, S., Rizza, S., Filomeni, G.,
Bernardini, R., Bertocchi, F., Mattei, M., Paci, M., Rotilio, G., and
Ciriolo, M. R. (2012) Glutamine deprivation enhances antitumor activity
of 3-bromopyruvate through the stabilization of monocarboxylate
transporter-1, Cancer Res., 72, 4526-4536.
119.Van Delft, M. F., Wei, A. H., Mason, K. D.,
Vandenberg, C. J., Chen, L., Czabotar, P. E., Willis, S. N., Scott, C.
L., Day, C. L., Adams, J. M., Roberts, A. W., and Huang, D. C. S.
(2006) The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and
efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized,
Cancer Cell, 10, 389-399.
120.Fulda, S., Galluzzi, L., and Kroemer, G. (2010)
Targeting mitochondria for cancer therapy, Nat. Rev. Drug.
Discov., 9, 447-464.
121.Lessene, G., Czabotar, P. E., and Colman, P. M.
(2008) BCL-2 family antagonists for cancer therapy, Nat. Rev. Drug
Discov., 7, 989-1000.
122.Albershardt, T. C., Salerni, B. L., Soderquist,
R. S., Bates, D. J., Pletnev, A. A., Kisselev, A. F., and Eastman, A.
(2011) Multiple BH3 mimetics antagonize antiapoptotic MCL-1 protein by
inducing the endoplasmic reticulum stress response and upregulating
BH3-only protein NOXA, J. Biol. Chem., 286,
24882-24895.
123.Billard, C. (2013) BH3 mimetics: status of the
field and new developments, Mol. Cancer Ther., 12,
1691-1700.
124.Zhang, Z., Song, T., Zhang, T., Gao, J., Wu,
G., An, L., and Du, G. (2010) A novel BH3 mimetic S1 potently induces
Bax/Bak-dependent apoptosis by targeting both Bcl-2 and Mcl-1, J.
Cancer, 128, 1724-1735.
125.Soderquist, R., Pletnev, A. A., Danilov, A. V.,
and Eastman, A. (2013) The putative BH3 amimetic S1 sensitizes leukemia
to ABT-737 by increasing reactive oxygen species, inducing endoplasmic
reticulum stress, and upregulating the BH3-only protein NOXA,
Apoptosis, 19, 201-209.
126.Zhong, J. T., Xu, Y., Yi, H. W., Su, J., Yu, H.
M., Xiang, X. Y., Li, X. N., Zhang, Z. C., and Sun, L. K. (2012) The
BH3 mimetic S1 induces autophagy through ER stress and disruption of
Bcl-2/Beclin 1 interaction in human glioma U251 cells, Cancer
Lett., 323, 180-187.
127.Song, T., Li, X., Chang, X., Liang, X., Zhao,
Y., and Wu, G. Y. (2013)
3-Thiomorpholin-8-oxo-8H-acenaphtho[1,2-b]pyrrole-9-carbonitrile (S1)
derivatives as pan-Bcl-2-inhibitors of Bcl-2, Bcl-xL and Mcl-1,
Bioorg. Med. Chem., 21, 11-20.
128.Song, T., Chen, Q., Li, X., Chai, G., and
Zhang, Z. C. (2013) Correction to
3-thiomorpholin-8-oxo-8H-acenaphtho[1,2-b]pyrrole-9-carbonitrile
(S1)-based molecules as potent, dual inhibitors of B-cell lymphoma 2
(Bcl-2) and myeloid cell leukemia sequence 1 (Mcl-1): structure-based
design and structure–activity relationship studies, J. Med.
Chem., 56, 9366-9367.
129.Ponassi, R., Biasotti, B., Tomati, V., Bruno,
S., Poggi, A., Malacarne, D., Cimoli, G., Salis, A., Pozzi, S.,
Miglino, M., Damonte, G., Cozzini, P., Spyraki, F., Campanini, B.,
Bagnasco, L., Castagnino, N., Tortolina, L., Mumot, A., Frassoni, F.,
Daga, A., Cilli, M., Piccardi, F., Monfardini, I., Perugini, M.,
Zoppoli, G., D’Arrigo, C., Pesenti, R., and Parodi, S. (2008) A
novel Bim-BH3-derived Bcl-XL inhibitor: biochemical characterization,
in vitro, in vivo and ex vivo anti-leukemic
activity, Cell Cycle, 7, 3211-3224.
130.Ghiotto, F., Fais, F., Tenca, C., Tomati, V.,
Morabito, F., Casciaro, S., Mumot, A., Zoppoli, G., Ciccone, E.,
Parodi, S., and Bruno, S. (2009) Apoptosis of B-cell chronic
lymphocytic leukemia cells induced by a novel BH3 peptidomimetic,
Cancer Biol. Ther., 8, 263-271.
131.Cheng, G., Zielonka, J., Dranka, B. P.,
McAllister, D., Mackinnon, A. C., Jr., Joseph, J., and Kalyanaraman, B.
(2012) Mitochondria targeted drugs synergize with 2-deoxyglucose to
trigger breast cancer cell death, Cancer Res., 72,
2634-2644.
132.Sahra, I. B., Laurent, K., Giuliano, S.,
Larbret, F., Ponzio, G., Gounon, P., Le Marchand-Brustel, Y.,
Giorgetti-Peraldi, S., Cormont, M., Bertolotto, C., Deckert, M.,
Auberger, P., Tanti, J. F., and Bost, F. (2010) Targeting cancer cell
metabolism: the combination of metformin and 2-deoxyglucose induces
p53-dependent apoptosis in prostate cancer cells, Cancer Res.,
70, 2465-2475.
133.Zhai, X., Yang, Y., Wan, J., Zhu, R., and Wu,
Y. (2013) Inhibition of LDH-A by oxamate induces G2/M arrest, apoptosis
and increases radiosensitivity in nasopharyngeal carcinoma cells,
Oncol. Rep., 30, 2983-2991.
134.Meynet, O., Beneteau, M., Jacquin, M. A.,
Pradelli, L. A., Cornille, A., Carles, M., and Ricci, J.-E. (2012)
Glycolysis inhibition targets Mcl-1 to restore sensitivity of lymphoma
cells to ABT-737-induced apoptosis, Leukemia, 26,
1145-1147.