REVIEW: Investigations of Molecular Mechanisms of Actin–Myosin Interactions in Cardiac Muscle
L. V. Nikitina*, G. V. Kopylova, D. V. Shchepkin, S. R. Nabiev, and S. Y. Bershitsky
Institute of Immunology and Physiology, Ural Division of the Russian Academy of Sciences, 620041 Ekaterinburg, Russia; E-mail: l.nikitina@iip.uran.ru* To whom correspondence should be addressed.
Received May 31, 2015
The functional characteristics of cardiac muscle depend on the composition of protein isoforms in the cardiomyocyte contractile machinery. In the ventricular myocardium of mammals, several isoforms of contractile and regulatory proteins are expressed – two isoforms of myosin (V1 and V3) and three isoforms of tropomyosin chains (α, β, and κ). Expression of protein isoforms depends on the animal species, its age and hormonal status, and this can change with pathologies of the myocardium. Mutations in these proteins can lead to cardiomyopathies. The functional significance of the protein isoform composition has been studied mainly on intact hearts or on isolated preparations of myocardium, which could not provide a clear comprehension of the role of each particular isoform. Present-day experimental techniques such as an optical trap and in vitro motility assay make it possible to investigate the phenomena of interactions of contractile and regulatory proteins on the molecular level, thus avoiding effects associated with properties of a whole muscle or muscle tissue. These methods enable free combining of the isoforms to test the molecular mechanisms of their participation in the actin–myosin interaction. Using the optical trap and the in vitro motility assay, we have studied functional characteristics of the cardiac myosin isoforms, molecular mechanisms of the calcium-dependent regulation of actin–myosin interaction, and the role of myosin and tropomyosin isoforms in the cooperativity mechanisms in myocardium. The knowledge of molecular mechanisms underlying myocardial contractility and its regulation is necessary for comprehension of cardiac muscle functioning, its disorders in pathologies, and for development of approaches for their correction.
KEY WORDS: cardiac myosin isoforms, tropomyosin, actin–myosin interaction, Ca2+ regulation, optical trap, in vitro motility assayDOI: 10.1134/S0006297915130106
Abbreviations: AOD, acousto-optical deflector; A7TmTn, a regulatory group consisting of seven G-actin molecules, one tropomyosin molecule, and one troponin molecule; CaTnC, calcium–troponin complex; CaTnC–CaTnC, troponin–troponin cooperativity; F-actin, filamentary actin; G-actin, globular actin; HMM, heavy meromyosin; LMM, light meromyosin; MHC, myosin heavy chains; NEM, N-ethylmaleimide; pCa, negative decimal logarithm of calcium concentration; pCa50, the calcium concentration corresponding to half-maximum of the sliding velocity or of the force (calcium sensitivity); S1, subfragment 1, or myosin molecule head; S2, subfragment 2, or rod of heavy meromyosin; Tm, tropomyosin; TnC, troponin C; TnI, troponin I; TnT, troponin T; Vmax, maximal velocity of muscle shortening; Xb, cross-bridge; Xb–CaTnC, bridge–troponin cooperativity.
The functional characteristics of cardiac muscle directly depend on the
composition of the protein isoforms of the cardiomyocyte contractile
machinery. In the ventricular myocardium of mammals, a whole pattern of
contractile and regulatory proteins is expressed, in particular, two
myosin isoforms, V1 and V3 [1], two α-actin
isoforms (cardiac and skeletal) [2], and three
isoforms of tropomyosin chains – α, β [3], and κ [4, 5]. The expression of protein isoforms depends on the
animal species, its age and hormonal status [1-3, 6]. The composition of protein
isoforms in the cardiomyocyte also does change with changes in the
functioning conditions, including those during experimental modeling
the myocardium pathologies. Thus, in the experiments with the left
ventricle hypertrophy caused by pressure overload in adult rats, a
renewed expression of myosin, actin, and tropomyosin isoforms specific
for intrauterine development occurs: isomyosin V1 becomes replaced by
V3, cardiac actin isoforms by the skeletal isoform, and the tropomyosin
α-chain by β-chain [7].
κ-Tropomyosin, which plays an important role in the genesis of
myofibrils, gets also re-expressed in dilated cardiomyopathy [4, 5]. In the 1990s, protein
mutations, which lead to development of pathologies, were found. Thus,
hereditary hypertrophic cardiomyopathy that leads to heart failure and
sudden heart arrest is caused by more than 300 mutations in 11 genes
that encode the sarcomere proteins. In 85% of these cases, mutations
affecting the heavy β-chain of myosin, the α-isoform of
tropomyosin, myosin-binding protein C, cardiac troponin I, and troponin
T were discovered [8-11].
The majority of studies of changes in protein isoform composition were performed either on intact heart or on isolated strips of myocardium, which cannot give a clear comprehension of the role of each of isoforms in the myocardium contraction. Using an optical trap an in vitro motility assay allows studying interactions of regulatory and contractile proteins at the thin filament level, avoiding effects associated with other features of the whole muscle, as well as molecular mechanisms and their roles in actin–myosin interaction by combining isoforms of the sarcomere proteins. In this review, the results of studies of the influence of cardiac isoforms of myosin on myocardium contractile function and its regulation at different levels of cardiac muscle organization are considered including our own. The attention is paid to results of investigations of calcium-dependent regulation of actin–myosin interaction and the role of myosin and tropomyosin isoforms in cooperativity mechanisms in the myocardium that were obtained using the in vitro motility assay with the regulated thin filament and the optical trap techniques.
ISOFORMS OF CARDIAC MYOSIN
Myosin, a contractile protein of striated and smooth muscles, is a biological motor transforming the chemical energy of ATP hydrolysis into mechanical work. The myosin molecule is a heterohexamer with molecular weight of 450-500 kDa and length of about 17 nm that consists of two heavy chains (200-240 kDa), two regulatory (17-21 kDa) and two essential light chains (16-22 kDa). These six polypeptide chains are kept together via noncovalent bonds and form a complex that is called monomeric myosin. The N-terminal part of each heavy chain of myosin forms a globular head consisting of two main domains – motor (catalytic) and regulatory. In the N-terminal motor domain, actin-binding and ATPase centers are located, whereas the C-terminal regulatory domain is an α-helical “lever” associated with myosin light chains responsible for regulatory and stabilizing functions during the generation of force by the myosin head. Chymotrypsin cleaves the myosin molecule into heavy and light meromyosin (HMM and LMM, respectively). HMM in turn can be cleaved by papain into the globular head (subfragment 1 or S1) and rod subfragment 2 (S2) [12].
In 1977, the Australian researcher J. Hoh was the first to separate cardiac myosin from rat ventricle in a native pyrophosphate gel and to obtain three bands, which were designated after their migration rates in the gel as V1, V2, and V3. Cardiac myosin extracted from the atrium was separated in the same gel into two bands, which were termed A1 and A2. The letters indicated the origin of myosin: V — ventricular and A – atrial. Thus, three ventricular isoforms of cardiac myosin V1, V2, and V3 and two atrial isoforms A1 and A2 [1] were identified. The ATPase activity of the ventricular isoforms measured in the gel was strongly different, having ratios 6.4 : 3.7 : 1. The ventricular isoforms of cardiac myosin were also different in compositions of the heavy chains (MHC), but not of the light chains. It was shown by peptide mapping that myosin isoform V1 is a homodimer α-MHC, V3 is a homodimer β-MHC, and V2 is a heterodimer consisting of α- and β-MHC [13]. Using a denaturing gel, the ventricular myocardium was shown to contain two isoforms of myosin heavy chains, which together with the ventricular myosin light chains produced isomyosins V1 and V3 [14]. In the atrial myocardium, α- and β-MHC together with the atrial light chain produce two isomyosins: A1 (α-α) and A2 (β-β) [15].
Amino acid sequences of the heart myosin isoforms V1 and V3 of studied mammals (mouse, rat, rabbit, and human) have 93% identity [16]. The majority of the non-identical amino acid residues are located in five different regions of the molecule: in the catalytic domain basis and at the junction with the essential light chain (amino acid residues 32-36); at the entrance into the ATP-binding pocket, and also in surface loop 1 (amino acid residues 210-214 and 349-351); in surface loop 2 constricting the actin-binding cleft (amino acid residues 619-641); in the region of the neck or “lever” (amino acid residues 800-810); in subfragment 2 (amino acid residues 1088-1094).
To elucidate the functional significance of these amino acid substitutions, studies were performed using hybrid and mutant myosins. Using hybrid myosins, it was shown with transgenic mice that surface loops 1 and 2 are not responsible for differences in the characteristics of cardiac myosin isoforms [17]. However, substitution in the catalytic domain of cardiac myosin, when the hybrid myosin contained the catalytic domain β-MHC and the other part present in the α-chain, resulted in significant consequences. The actin-activated ATPase activity of such hybrid myosin was very similar to that of V3 isomyosin, whereas its calcium-activated ATPase activity was comparable to the activity of V1. In the in vitro motility assay, the hybrid myosin moved the actin filament (F-actin) with the same velocity as V1. The maximal isometric force of papillary muscle containing such hybrid myosin was not different from the force developed by muscles containing native isoforms of cardiac myosin. These studies showed that the difference in the amino acid sequences between α- and β-MHC determines the difference in their mechanical and kinetic characteristics both in the catalytic domain and in the “lever” region [16, 18].
In experiments with mutant animals, point mutations located in the actin-binding region (R403Q and V606M) and in the nucleotide-binding region (R249Q, G256E, and R453C), as well as in the so-called “converter” subdomain (R719Y and R723G) of the myosin head, led to changes in the mechanical and kinetic properties of the myosin [19, 20]. Thus, the mutation R403Q increased the force generation by myosin, whereas the mutation R453C had the opposite effect.
Both α- and β-heavy chains of cardiac myosin are essentially homologous in various animal species [16]. Thus, the sequences of α-MHC have a 95% identity in rat, rabbit, hamster, and human. The amino acid sequence of β-MHC has 94% homology in various animals. Thus, the difference between isomyosins V1 of rat and rabbit is only in eight amino acid residues, five of which are located in the myosin head, mainly in the motor domain, including its actin-binding sites (residues 2, 210, 442, 452, and 801), whereas three amino acid residues are located in the rod region (residues 1092, 1637, and 1681). These amino acid substitutions determine the difference of hydrolytic and mechanical properties of these isomyosins. Cardiac isomyosins V3 of rabbit and rat are different by four amino acid residues in positions 424, 573, 1210, and 1368. Two of these residues (424 and 573) are located in the actin-binding and nucleotide-binding regions, respectively, and can be functionally significant. These differences in the amino acid sequence result in higher ATPase activity of the rat isomyosin and its ability to move the actin filament with higher velocity than the corresponding rabbit isomyosin [16].
Features of cardiac myosin isoforms. Cardiac myosin isoforms in the majority of mammalian species are significantly different in their hydrolytic and functional characteristics. It was shown in biochemical studies that the actin-activated ATPase activity of isoform V1 was 1.6-3.5 times higher than this activity of V3 [21-23]. The Ca2+-ATPase activity of V1 is about twofold higher than of V3 [16, 21, 24, 25]. The K+-EDTA-ATPase activity of cardiac myosin isoforms was not different [24, 25]. Banerjee and Morkin [26] showed that the actin-activated ATPase activity of cardiac myosin isolated from hyperthyroid rabbits, which mainly contained the isoform V1, was nearly 1.7-fold higher than such activity of cardiac myosin from euthyroid rabbits with the prevalent isoform V3.
For many muscle types, the relation between the maximal velocities of muscle shortening (Vmax) and the ATPase activity of the myosin isolated from the corresponding muscle was recorded in afterloaded twitches on trabeculae and papillary muscles [27]. Maughan et al. [28] compared the Vmax of a muscle preparation from the right ventricle of a rabbit with experimentally hypertrophic heart and the actin-activated ATPase activity of myosin from the same preparation with the corresponding characteristics of muscle of normal heart. In the case of the hypertrophic heart, they observed a comparable decrease in both parameters, and they supposed that the velocity of shortening should correspond to the rate of myosin cross-bridge cycling.
Schwartz et al. [29] compared the maximal velocities of shortening of rat papillary muscle mainly containing isomyosin V1 and of papillary muscle with the prevalent isomyosin V3 and found that Vmax of the first muscle was four or five times higher than Vmax of the second muscle. For rabbit papillary muscle, this ratio was ~6 [30]. These data suggest a positive correlation between the ATPase activity and Vmax; nevertheless, these characteristics are not proportional, in particular, because of differences in activation [31].
In the literature, there is no common opinion about the influence of the cardiac myosin content on the force-generating ability of the cardiac muscle. Maughan et al. [28] did not find differences between the maximal forces generated by chemically demembranated fibrils isolated from the right ventricles of euthyroid (V3) and hyperthyroid (V1) rabbits. Similar results were obtained on rats with hypertrophic hearts.
In experiments on myofibrils from ventricles of small rodents and of humans, it was shown that α-MHC-containing myofibrils of mice generated the same force as human β-MHC-containing myofibrils [32]. The myocardium preparations mainly containing α- or β-MHC did not differ in the isometric force value [33]. A few measurements of the myocardium force of big animals have shown that the cardiac muscle mainly containing β-MHC generates force twofold higher than muscle containing α-MHC [34].
CALCIUM REGULATION OF CARDIAC MUSCLE CONTRACTION
At the molecular level, force generation or muscle shortening occur due to the cyclic interaction of the heads of myosin molecules with actin of the thin filament, during which they attach, make a working step, and detach, hydrolyzing ATP. The regulation of contraction is activation/inactivation process of the thin filament by the calcium-regulatory troponin–tropomyosin system and is determined by changes in calcium concentration in cytosol, which in turn is regulated by the electric activation of the cell, i.e. by the action potential. The action potential arises in the working myocardium cells in response to either a pacemaker or an external electric impulse (in experimental conditions or using a cardiostimulator), which triggers calcium, potassium, and sodium transmembrane ionic currents. All these processes involve a set of feedbacks. In particular, mechanical conditions of contraction and formation and breakup of cross-bridges affect both calcium and electrical activation.
Calcium-induced structure conformational changes of the thin filament. In the sarcomere thin filament, seven G-actin molecules, one tropomyosin molecule, and one troponin complex form a structure-functional group A7TmTn called the regulatory unit [35]. Troponin consists of three interacting subunits: TnC, TnI, and TnT. Troponin C is responsible for binding of calcium by the complex, troponin I binds actin and inhibits the ATPase activity of myosin, and troponin T binds the troponin complex to tropomyosin [35]. When TnC binds calcium, some structural changes occur in the thin filament that promote the interactions of myosin with actin [36].
In the model by McKillop and Geeves [37], tropomyosin can be on the actin filament in one of three states: blocking, closed, and open states. In the absence of Ca2+, tropomyosin blocks the majority of myosin-binding sites (blocked state). The binding of calcium by TnC leads to structural changes in the troponin complex that allows troponin to displace azimuthally on the actin filament surface, opening the majority, although not all, myosin-binding sites on actin and thus allowing weak electrostatic binding of myosin with actin (closed state). This attachment of myosin heads to strongly binding hydrophobic regions on actin leads to further displacement of tropomyosin from the closed state into the open one, and all myosin-binding sites of actin become available to myosin heads [37, 38]. In this position of tropomyosin, force generation by the cross-bridges becomes feasible.
Cooperative effects in cardiac muscle. The function of striated muscles is implemented with the participation of a number of cooperative (allosteric) effects. To describe the dependence of force generated by muscle on Ca2+ concentration, Donaldson and Kerrick [39] used the Hill equation:
F = F0[Ca2+]h/(Kh + [Ca2+]h),
where h is the Hill coefficient of cooperativity, F0 is the maximal force, Kh is the Ca2+ concentration corresponding to the half of maximal force. This Ca2+ concentration is termed the calcium sensitivity. Because the Ca2+ concentration is usually expressed as pCa, i.e. –log[Ca2+], the equation can be written as follows:
F = F0/[1 + 10h(pCa50 – pCa)],
where pCa50 is the Ca2+ concentration necessary for the half-maximal activation of muscle. The Hill cooperativity coefficient characterizes the cooperativity degree of the calcium-induced activation. From the theoretical concepts, it results that calcium binding with one TnC site controls the force in the ratio of 1 : 1, i.e. h = 1. If calcium binds with two TnC sites like in skeletal muscle, h can increase to 1.2 [40] on conditions that the sites are independent and that the binding with both sites is required for the activation. However, experiments show other values: the Hill cooperativity coefficient of the isometric force is usually in the range from 2 to 5-6 in both skeletal and cardiac muscles [41, 42].
The cooperativity concept was formulated by Gordon et al. [35] on an example of skeletal muscle. The force generated by muscle in the isometric state depends on the number of strongly bound cross-bridges and on the force generated by each of them. In turn, this is determined by the number of actin-binding sites available for the strong binding of myosin. Structural features of myosin and actin allow no more than four myosin heads to be attached to seven actin monomers. During isometric contraction, only 20-40% of all cross-bridges (Xb) are attached [35, 43-46] that approximately corresponds to 1-2 myosin heads per seven actin monomers.
A simplified mechanochemical model of the cross-bridge cycle [35] describes the interaction of one myosin molecule with one actin molecule of the thin filament. It follows from specific features of the sarcomere structure that the activation involves a multiplicity of potential interactions of myosin with actin molecules along every thin filament. This activation has to involve the cooperativity inside and among the regulatory units (A7TnTm).
There are several cooperativity mechanisms that provide Ca2+ binding by TnC through conformational changes in the thin filament with subsequent formation of a cross-bridge, resulting in a cooperative increase in the number of Xbs along the actin filament. According to some authors, a key role in the regulation of cardiac muscle contraction belongs to two cooperativity mechanisms [36, 40, 47, 48]:
– Xb–CaTnC cooperativity, i.e. the more cross-bridges have formed on an actin filament, the higher TnC affinity for calcium along the filament;
– CaTnC–CaTnC cooperativity, i.e. the more calcium–troponin complexes have formed along the filament, the higher TnC affinity for calcium.
These mechanisms are shown to underlie the influence of mechanical conditions of the myocardium on the activation/inactivation of its contractile function [49-54]. It is known that an increase in the distance between the actin and myosin filaments during sarcomere shortening, or “lattice spacing”, lowers the probability of cross-bridge formation. This decreases the concentration of calcium–troponin complexes through the Xb–CaTnC cooperativity, whereas the CaTnC–CaTnC cooperativity strengthens this effect inactivating contractility. And vice versa, an increase in muscle length promotes the activation.
In the framework of the mathematical model of myocardial contractile activity, it has been established that just this chain of intracellular events determines a broad spectrum of experimentally found mechano-mechanical, mechano-calcium, and mechano-electrical feedbacks in the myocardial contraction-relaxation cycle [55, 56]. Analysis of the model reveals that this chain is the cause of such fundamental effects specific for cardiac muscle as its load-dependent relaxation and inactivation in response to short-term deformations during the isometric contraction-relaxation cycle. The model also shows that this event chain is responsible for mechano-dependent differences in the time course of changes in the Ca2+ concentration in cytosol and in the duration of action potentials that has been found in classical investigations on papillary muscles and trabeculae [57-59] and confirmed later in other works [60-65], including studies on isolated cell preparations [66, 67]. Thus, numerous experimental and theoretical studies have resulted in formation of a concept of the role of the above-mentioned cooperativity mechanisms in the regulation of the contractile cycle of cardiac muscle, as well as of the rhythm–inotropic phenomena in normal and pathologic conditions [68, 69].
The Xb–CaTnC cooperativity of the thin filament activation implicates the contractile and regulatory proteins of the myocardium by multiple feedbacks. At that, the two myosin isoforms existing in the heart of mammals have different hydrolytic, kinetic, and mechanical characteristics. A question arises whether mechanisms of thin filament activation can be modulated by myosin isoforms through this type of cooperativity. The contribution of the cardiac myosin isoforms to the calcium regulation of the myocardium contractions was traditionally studied on fragments of cardiac muscle tissue and on isolated cardiomyocytes [70-73]. In these experiments, the relationships pCa–force and pCa–velocity were recorded, and the cooperativity coefficient and calcium sensitivity were identified.
METHODS OF STUDYING THE MOLECULAR MECHANISMS OF CARDIAC MUSCLE
FUNCTION
The optical trap and the in vitro motility assay allow studying functional characteristics of isolated molecules of cardiac muscle sarcomeric proteins. With these methods, one can investigate the mechanical interaction of regulatory and contractile proteins directly at the level of the thin filament that avoids the complicacy of interpreting the results related to the properties of whole muscle or cardiomyocytes. The possibilities of these approaches are considered below.
Optical trap technique. The laser trap is used to measure the mechanical and kinetic characteristics of interaction of single molecules of myosin, in particular cardiac myosin, with filamentous actin. The principle of the approach is as follows. A laser beam focused by a high aperture objective forms the “optical trap” (or “optical tweezers”) capable to capture and keep in the focus microscopic objects with the forces of order 10–12-10–11 N [74, 75]. An experimental setup of optical trap for studying the mechanical characteristics of single molecules of myosin was built in the Laboratory of Biological Motility of the Institute of Immunology and Physiology, Ural Branch of the Russian Academy of Sciences [76]. In the setup, two beams of IR laser keep two polystyrene beads of 1-µm diameter coated with NEM-modified myosin that serves as “glue” irreversibly binding actin filament (Fig. 1). A segment of fluorescently labeled actin filament is glued to these beads forming measuring dumbbell-like probe and then pre-stretched by moving one of the beams with a 2D acousto-optical deflector (Fig. 2). Silica beads of 1.7 µm in diameter are fixed on the surface of the experimental flow cell and serve as pedestals for molecules of the studied myosin. With a piezoelectric nanomanipulator, the dumbbell is located above the pedestal that allows interaction of myosin molecule with actin filament of the probe in the presence of ATP. Single interaction between myosin molecule and actin filament, or “event” is accompanied by bead deviation from the beam focus that is registered by quadrant photodiodes. Depending on the operating mode, i.e. with or without feedback, either average force or step size of a single myosin molecule can be measured, as well as the event duration.
Fig. 1. Principal scheme of the optical trap technique. The ends of actin filament are connected to two polystyrene beads of 1 µm in diameter held in the focus of laser beams. (Images of the beads are projected onto quadrant photodiodes for recording their positions and movements.) A twofold greater silica bead, a pedestal, is fixed on the flow cell surface; this bead is coated with molecules of the investigated myosin at very low concentration. When the “dumbbell” probe is put over the pedestal, a single myosin molecule can interact with actin filament of the probe.
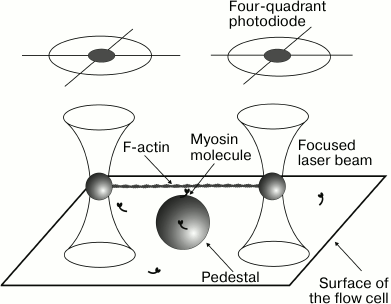
Fig. 2. Block-diagram of the two-beam optical trap setup. The setup is built on the base on an inverted fluorescent microscope (AxioObserver; Carl Zeiss, Germany). A half-wave plate rotates the polarization plane of IR laser beam (Nd:YLF, 1064 nm, 5 W; Inversion-Fiber, Russia) at 45º, and the beam splitter divides it into two polarizing components. One of the beams remains still and another one passes through 2D acousto-optical deflector (AOD) consisting of two perpendicularly oriented acousto-optical crystals that work as diffraction gratings with variable period controlled be a frequency generator. This changes position of the first order diffraction maximum of the transmitted beam for any of the two coordinates. Thus, the AOD enables the beam to rapidly (<10 µs) move in the focal plane. The second beam splitter collects the both components into one beam. The beam is expanded by two lenses to fill the back aperture of the objective. A custom-made dichroic mirror reflects the IR laser beam into the microscope objective and passes through visible light emitted by fluorescently labeled actin filament and the light from the halogen lamp. Passed through the objective, the beams form two optical traps that capture the polystyrene beads in experimental flow cell. Then the beams are collected by a high-aperture oil immersion condenser (NA 1.4), split again by the third polarized beam splitter and projected onto quadrant photodiodes (FD20KP, Russia), used for registration the positions of the beads. The photodiode signals are used in the AOD feedback control. A mercury lamp is used for excitation of fluorescence of tetramethylrhodamine-labeled (TRITC) actin and the beads with a set of filters (Filter Set 20; Carl Zeiss, Germany). The dumbbell is formed in the fluorescence mode. Pedestals coated with myosin are searched for in the bright field mode.
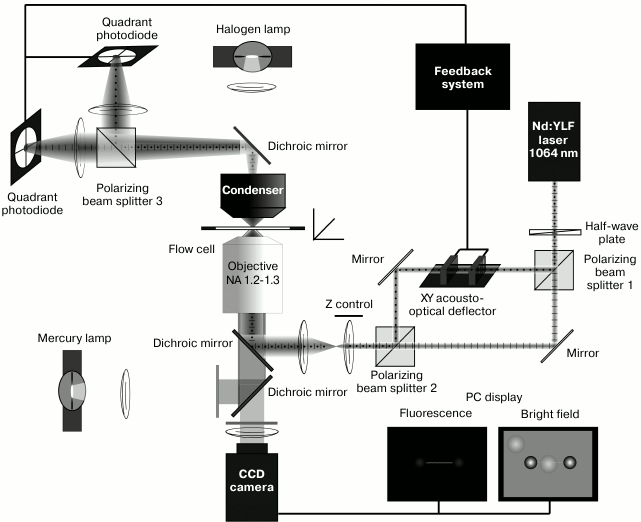
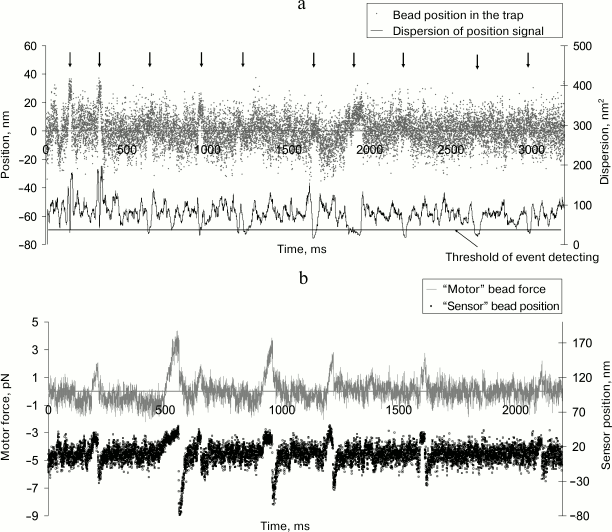
Figure 3 shows examples of experimental records of the step (a) and force (b) of a single myosin molecule.
Fig. 3. Examples of experimental records of the displacement (a) and force (b) generated by a single myosin molecule in our experiments. a) The “event”, i.e. attachment of a myosin molecule to actin filament, was determined by the decrease in dispersion of the bead position in the trap (lower trace) below a preset level [77]. The events are shown by vertical arrows. The displacement, or the step, was determined as a difference of the signal level in the attached and detached state of myosin. b) The force was measured in isometric force clamp mode [78]. The position of the movable bead (“motor”) was controlled by the position of the unmovable (“sensor”) bead via AOD feedback.
Investigation of mechanical and kinetic characteristics of cardiac myosin using the optical trap technique. The optical trap was used to measure the mechanical and kinetic characteristics of single molecules of cardiac myosin isoforms [79, 80]. At the single molecule level, the maximal velocity, Vmax, of sliding actin filaments on the surface covered by cardiac myosin molecules can be found as Vmax = d/ton, where d is the working stroke of the myosin head and ton is duration of the attached state of myosin head to the actin filament. The optical trap enables to measure the average force Favg generated by a molecule of myosin: Favg = F × f, where F is the force generated by a single myosin molecule, and f is the fraction of time of the ATPase cycle of myosin during which the molecule is bound to actin and generates force (duty ratio), i.e. f = ton/tcycle. Previously in optical trap experiments it was found that cardiac myosin isoforms have no differences in d and F, i.e. in the parameters describing mechanics of the myosin head, but differed in the kinetic parameters ton and f. It supposed that only kinetic parameters determine the difference in Vmax and Favg of cardiac myosin isoforms [79, 80].
In our optical trap experiments, we also have not found any differences in mechanical characteristics of rabbit cardiac myosin: the average step was 11.6 ± 2 and 12.5 ± 3 nm for isoforms V1 and V3, respectively [81]. However, the average step durations were different: the average actomyosin complex lifetime was 54.2 ± 1.3 ms for V1 and 62.1 ± 2.7 ms for V3. The average force values for isoforms did not differ significantly: 1.8 ± 0.7 and 2.1 ± 0.5 pN for V1 and V3, respectively. Values of the force generated by the cardiac myosin isoforms in our experiments were about two-fold higher than those obtained in the earlier work [80]. This difference can be explained by using different ways of loading the myosin molecule. Warshaw et al. [80] have measured force in static trap mode, while we used dynamic loading [78]. We have also found significant difference in the duration of actin–myosin interaction in the dynamic loading: isoform V1 was under load, on average, for 55.4 ± 5 ms, and isoform V3 was for 72.4 ± 4 ms. These results are in a good agreement with those obtained in the in vitro motility assay, indicating higher force-generating ability of isomyosin V3 as compared with V1 according to the above mentioned formula Favg = F × f [22, 82].
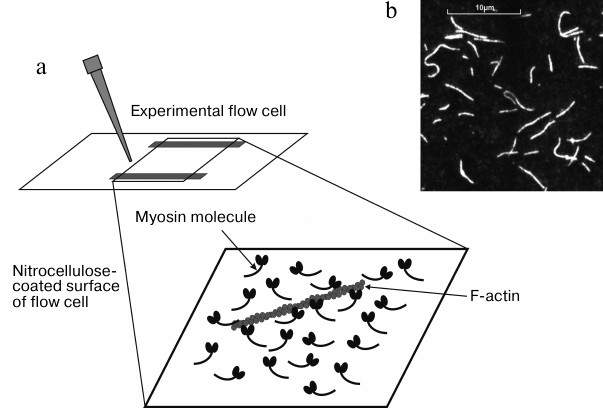
In vitro motility assay. Unlike the optical trap, the in vitro motility assay allows studying the mechanical and kinetic characteristics of an ensemble of myosin molecules immobilized on a glass surface. The principle of the technique is as follows. The studied motor protein (here, an isoform of cardiac myosin) is located on the surface of experimental flow cell coated with nitrocellulose, and then the flow cell is filled with a solution containing fluorescently-labeled actin filaments [83]. Actin filaments interact with myosin creating rigor complexes (Fig. 4). The flow cell is put on the stage of an inverted fluorescent microscope equipped with a CCD camera that provides visualization of actin filaments on the PC screen (Fig. 4). When ATP-containing solution is added into the flow cell, the filaments start to slide on the myosin-coated surface. Video of filament sliding is recorded on PC hard drive and then analyzed. Velocities of the filament sliding on the surface covered by myosin or its proteolytic fragments, such as HMM or S1, are measured using custom-made software [84].
Fig. 4. a) Sketch of the inner surface of flow cell covered with myosin molecules and actin filament on it. b) Image of fluorescently-labeled actin filaments on PC screen.
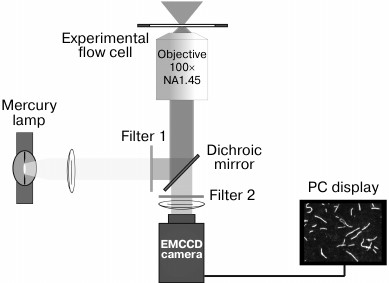
Experimental setup. The setup for recording the movements of fluorescently-labeled actin or regulated thin filaments and measuring their sliding velocity in vitro (Fig. 5) is built on an inverted fluorescence microscope (Axiovert 200 M; Carl Zeiss MicroImaging GmbH, Germany) equipped with a mercury lamp HBO 100 and a set of filters (Filter Set 20; Carl Zeiss) for tetramethylrhodamine (TRITC). Video of moving filaments is recorded to the PC hard drive and analyzed with GMimPro software [84].
Fig. 5. Scheme of the experimental setup for recording the movement of fluorescently-labeled actin filaments in the in vitro motility assay. The narrow-band green filter 1 transmits wavelength of 546 nm for excitation the TRITC fluorescence into the objective (Alpha Plan-Fluar 100× 1.45NA oil-immersion; Carl Zeiss) with use of the dichroic mirror. The TRITC fluorescent light at 575 nm passes through the dichroic mirror and is filtered by emission filter 2. The video of movement of fluorescently-labeled actin filaments is recorded with an iXon-897BV EMCCD camera (Andor Technology, Ireland).
Measurement of force generated by myosin using an in vitro motility assay. There are several approaches in the in vitro motility assay, which allow measuring either absolute or relative force. Absolute force is measured using a glass microneedle [22], a centrifuge microscope [85], or laser tweezers [80]. It should be noted that these approaches are rarely used because the experimental procedures are difficult and labor consuming. Relative force can also be measured using agents that prevent the movement of filaments on the myosin surface. These agents are actin-binding proteins, e.g. filamin, α-actinin [86], and NEM-modified myosin [87], which irreversibly binds actin. NEM-modified myosin is mixed at different ratios with unmodified studied myosin, and the dependence of filament sliding velocity on the fraction of NEM-myosin in this mixture is measured. The relative force of myosin is calculated from the slope of this dependence. Results of this approach are in a good agreement with those of direct measurements of the isometric force with glass microneedles [22].
Instead of chemically modified myosin, the actin-binding protein α-actinin can be used as an anchor to slow down the movements of actin filaments. α-Actinin is a structural component of the Z-line and forms crosslinks along the actin filament. The concentration of α-actinin necessary for braking the filament movement depends linearly on the concentration of myosin heads on the flow cell surface and force. Malmqvist et al. [23] have compared slopes of the dependencies of α-actinin concentration on the myosin concentration from chicken skeletal, smooth and rabbit cardiac muscles. The relative average isometric force was found to be equal to that measured by the microneedles. In the in vitro motility assay it has been shown [88] that α-actinin has no effect on properties of muscle proteins and their interaction. These studies have confirmed that α-actinin is a good tool for measurement the relative force of myosin in the in vitro motility assay.
In vitro motility assay with a regulated thin filament. The in vitro motility assay allows studying the mechanisms of the calcium-dependent regulation of contraction of skeletal and cardiac muscles using reconstructed thin filaments consisting of actin and regulatory proteins: troponin and tropomyosin [83, 86, 89-93]. Depending on aim of the study, thin filament can contain different isoforms of regulatory proteins and their mutants [94, 95].
Such system makes it possible to record dependencies of the myosin force and velocity of thin filament movement on the calcium concentration (relationships pCa–force and pCa–velocity, respectively), allowing investigation of characteristics of actin–myosin interaction and its regulation by calcium directly, avoiding the effects associated with mechanical properties and biochemical processes in muscles. The approach allows comparing characteristics of isoforms of contractile and regulatory proteins and their combinations.
In 1989, Honda et al. [93] were the first to observe a calcium-dependent movement of filamentous actin containing tropomyosin and troponin on a surface covered with myosin. Further, the approach was used for studying molecular mechanisms of calcium regulation of contractions in both skeletal and cardiac muscles. A method of enzymatic cleavage of thin filaments in a skinned muscle fiber and their reconstruction using actin and regulatory proteins has been developed [96, 97]. It was shown that native and reconstructed filaments have no functional differences [92]. Sata et al. [98] using double labeling showed that the regulatory proteins uniformly embedded into the thin filament structure. They also found that decrease in pH and temperature and increase in inorganic phosphate concentration reduce the sensitivity of contractile proteins to calcium in the same way as in muscle preparations. Thus, those results confirmed that the reconstructed thin filaments have the same structural and functional properties as native ones, and the in vitro motility assay with the reconstructed thin filament can be successfully used for studying interactions of proteins and their regulation.
The optical trap and of in vitro motility assay techniques have some limitations. The in vitro motility assay consisting only of contractile and regulatory proteins is deprived of modulators of connections existing in a cardiomyocyte in vivo. Nevertheless, using the optical trap and the in vitro motility assay allows studying other sarcomere proteins and their contribution to actin–myosin interaction and its regulation. In particular, using this approach we have investigated the modulating role of the myosin-binding protein C [99, 100]. Similar studies can be performed with other proteins.
STUDIES ON MOLECULAR MECHANISMS OF CALCIUM REGULATION OF CARDIAC
MUSCLE CONTRACTILITY
Experiments in the in vitro motility assay revealed that the sliding velocity of both filamentary actin and regulated thin filament on isomyosin V1 was about twofold higher than on V3 [22, 101]. We found this difference to be 1.7-fold. After addition of regulatory proteins to F-actin, its sliding velocity on both cardiac myosin isoforms increased 3.8-fold [82].
The difference in sliding velocities of F-actin and the regulated thin filament at saturating calcium concentration on the same motor protein in the in vitro motility assay is actively discussed. Some authors observed an increase in the sliding velocity of thin filament at saturating calcium concentration using skeletal [90, 91, 102] and cardiac regulatory proteins [82], whereas other researchers [93, 98] did not find such effect. The difference in the results can be explained by technical differences, e.g. in the composition of buffer used for the experiments [93].
Gordon et al. [91] believed that the sliding velocity of the regulated thin filament was higher than that of F-actin due to an increase in the actin-activated ATPase activity of myosin in the presence of the regulated thin filament as compared to the ATPase activity with pure actin. This can be associated with a possible influence of regulatory proteins on the actin–myosin interaction. The increase in the movement velocity of the thin filament can be explained by: 1) interaction of skeletal troponin and tropomyosin with actin that changes the structure of actin and thus influences the interaction of actin with myosin [103, 104]; 2) presence of a possible contact of the motor part of the myosin head with the central part of the tropomyosin molecule, which might be involved in the activation of thin filaments [105, 106].
Using the actin-binding protein α-actinin in experiments in the in vitro motility assay with both regulated and non-regulated thin filament, we measured the relative force at two concentrations of isomyosins V1 and V3 placed into the flow chamber: 300 µg/ml (saturating concentration) and 200 µg/ml, at saturating concentration of free calcium (pCa 4) [82]. The isometric force generated by isomyosin V3 was twofold higher than that generated by V1 at both myosin concentrations, which correlated with data of other authors obtained in the in vitro motility assay with both F-actin [22, 23] and regulated thin filament [107]. Note that for the two isoforms the amount of α-actinin stopping the movement of filaments was six times higher at myosin concentration of 300 µg/ml than at the concentration of 200 µg/ml.
At present, regulatory mechanisms of cardiac muscle contractility are intensively studied using the in vitro motility assay. These studies mainly concern the influence of different isoforms of regulatory proteins in the norm and pathology. However, molecular mechanisms of the contribution of cooperativity to the regulation of interaction of contractile proteins remain insufficiently studied.
The pCa–force relationship is the main characteristic of cooperative effects in muscles. Traditionally, the pCa–force relationship is recorded using skinned muscle fibers [70, 108] or cardiomyocytes [71]. Noguchi at al. [107] were the first to obtain in the in vitro motility assay the pCa–force relationship for isoforms of rabbit cardiac myosin. However, on analyzing their data, they failed to detect a contribution of different isomyosins to calcium-dependent regulation. Partially this failure was due to incompleteness of their analysis. Authors did not determine values of the Hill cooperativity coefficient, although Fig. 5 of their paper showed that slopes of the pCa–force relationship were different and, consequently, the cooperativity coefficients had to be different. Since authors did not present Hill coefficients, it is not known whether these differences were statistically significant.
We studied the contribution of cardiac myosin isoforms to the calcium-dependent activation of thin filaments. In experiments with regulated thin filament and using the actin-binding protein α-actinin, we obtained dependences of pCa–[α-actinin] for two concentrations (200 and 300 µg/ml) of isomyosins V1 and V3 placed into the flow chamber [82]. The pCa–[α-actinin] curves for V1 and V3 were sigmoidal.
Curves of pCa–force were obtained by normalizing the α-actinin concentration at the given pCa to its minimal concentration stopping the filament movement at saturating concentration of calcium. The comparison of characteristics of pCa–force relations of cardiac myosin isoforms V1 and V3 at different concentrations of myosin revealed that the discrepancies in the Hill coefficients and calcium sensitivity for different myosins occurred at the lower concentration of the protein. It was also shown that for each isomyosin the cooperativity coefficient increased with decrease in protein concentration. We analyzed changes in the Hill coefficient values at different concentrations of myosin and supposed that the cooperative dependence of kinetics of calcium–troponin complexes with V1 should be higher than with V3 [82]. The data obtained from the pCa–force [82] and pCa–velocity [89] relationships indicated that the myosin isoform V3 had higher sensitivity to calcium.
The force–velocity relation can give more comprehensive information about the cooperative influence of myosin on the calcium-dependent regulation of contractile activity. The calcium level and mechanical conditions [109, 110] can modulate the force–velocity relationship through cooperative mechanisms. We expected that the force–velocity ratio of the cardiac myosin isoforms recorded in the in vitro motility assay at different calcium concentrations should be different in the case of different contribution of these isoforms to the Xb–CaTnC cooperativity.
In experiments performed in the in vitro motility assay with the regulated thin filament and α-actinin as a load, we obtained force–velocity relationship for isomyosins V1 and V3 at two concentrations of calcium: saturating concentration (pCa 6.5) and non-saturating concentration (pCa 7.0). The resulting curves had a shape different from the classic hyperbola recorded by A. Hill on skeletal muscle [111]. At small loads, the curve was hyperbolic, whereas at higher loads it deflected from this shape as other authors also showed [85, 112].
The existence of a non-hyperbolic part of the force–velocity curve recorded on preparations of cardiac and skeletal muscles [30, 70, 112, 113] was thought to be associated with specific features of recording this curve, but not with behavior of cross-bridges. The presence of the non-hyperbolic part in the force–velocity curves obtained with the centrifugal microscope for rat cardiac myosin isoforms [85] and also in curves obtained by us on rabbit cardiac myosin isoforms [89] has indicated that the specific feature of such shape of the force–velocity curve manifests itself on the level of the actin–myosin interaction and depends on the composition of the contractile proteins.
Our study of the force–velocity relation revealed that the maximal sliding velocity of the thin filament on the V1 isoform was higher than on the V3 isoform, and that the maximal force generated by isomyosin V1 was lower than by the force of V3 at both saturating and non-saturating calcium concentrations. The force–velocity relations of myosin isoforms recorded at different calcium levels were different, and this difference could be associated with difference in the isoform contribution to Xb–CaTnC cooperativity. On change in the calcium concentration, the curvature of the force–velocity relation of the V3 isoform changed more markedly, which suggested its higher sensitivity to changes in the calcium concentration.
Study on contribution of tropomyosin isoforms to regulation of cardiac muscle contractile activity. Using the in vitro motility assay, we studied the possible contribution of tropomyosin isoforms to regulation of the myocardial contractile activity. In particular, we evaluated the regulatory influence of tropomyosin isoforms on the interaction of cardiac myosin isoforms with actin.
In mammalian myocardium, tropomyosin α- and β-chain isoforms are expressed, as well as an alternative splicing product – κ. The expression of tropomyosin isoforms, similarly to expression of myosin isoforms, depends on the animal species and changes during ontogenesis [3, 114]. The tropomyosin α-chain is prevalent in hearts of adult rats, mice, rabbits, and humans. In hearts of rats and mice, the tropomyosin β-chain is expressed mainly during gestation. Expression of the isoforms also changes in heart pathologies. Expression of the tropomyosin β-chain is increased in the hearts of adult rats and mice during hypertrophy caused by pressure overload. Experiments with artificial overexpression of tropomyosin β-chain up to 50-60% revealed in the hearts of adult rats and mice an increase in the calcium sensitivity of the pCa–force relation, a decrease in the maximal speed of relaxation, and, as a result, arising of diastolic dysfunction [115]. Further increase in the β-tropomyosin expression to 75-80% led to death of the animal soon after birth [114].
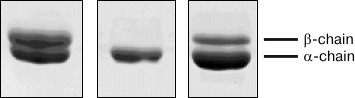
We used tropomyosin isoforms from muscles of various animals with different ratios of α- and β-chains (Fig. 6). The homodimer of tropomyosin α-chains (α-α)-Tm was prepared from rabbit myocardium and α-β-tropomyosin (90% α : 10% β)-Tm was isolated from bovine cardiac muscle. To model situations typical for some heart pathologies associated with increased expression of β-chain, tropomyosin with a high content of β-chain was isolated from rabbit m. psoas (60% α : 40% β)-Tm.
Fig. 6. SDS-PAGE of tropomyosin preparations obtained from various sources: 1) tropomyosin from rabbit m. psoas (60% α : 40% β)-Tm; 2) tropomyosin from rabbit myocardium (α-α)-Tm; 3) tropomyosin from bovine heart (90% α : 10% β)-Tm).
We found that tropomyosin with different contents of α- and β-chains differently influenced the velocity of movement of actin–tropomyosin filaments [116]. Tropomyosin with higher content of β-chain inhibited the sliding velocity of actin–tropomyosin filaments on both isoforms of cardiac myosin. The exact mechanism of this effect is unclear, but there are some hypotheses based on the literature data. It was found in studies with yeast [117] and skeletal [118] actin that different isoforms of tropomyosin bound with different regions of F-actin, and that the binding with actin depended not on the length of the tropomyosin molecule but on its amino acid sequence. In addition to the amino acid sequence of tropomyosin, the regulation of actin–tropomyosin filament can be influenced by amino acid sequence of the actin-binding domain of myosin. According to data of Ajtai et al. [119], an important role in the interaction of myosin with actin belongs to the C-loop of myosin. Substituting the native sequence of the C-loop in smooth muscle myosin by the sequence from cardiac and skeletal myosin, the authors showed that such chimerical myosins interacted differently with the actin–tropomyosin filament.
We studied the role of the tropomyosin β-chain in the calcium activation. We recorded the pCa–velocity relationships of thin filaments containing tropomyosins with different contents of α- and β-chains. The Hill cooperativity coefficients for the pCa–velocity relationship were not significantly different for all combinations of myosin isoforms with tropomyosin isoforms [120]. The calcium sensitivity of the pCa–velocity relation of the V1 isoform did not depend on the content of tropomyosin β-chain, whereas the sensitivity of the V3 isoform increased with increase in β-chain content in tropomyosin [120].
Our data suggest that the isoform composition of both myosin and tropomyosin is important for the interaction of cardiac myosin with actin–tropomyosin filament. In other words, myosin and tropomyosin reciprocally influence the actin–myosin interaction in cardiac muscle. The reciprocal influence of myosin and tropomyosin isoforms in cardiac muscle can be important for providing its efficiency during ontogenesis and in pathologies.
It is known that expression of cardiac myosin isoforms depends on the animal species and its age and hormonal status [121]. Thus, with hyperthyroidism expression of the V1 isoform is increased. In hypothyroidism and with heart pathologies caused by pressure overload at the aorta or mitral valve artificial or natural stenosis, the expression of the V3 isoform is increased [122]. The preferential expression of the gene encoding the slow β-chain of myosin leads to decrease in the maximal rate of the cardiac muscle contraction and has a negative inotropic effect. The preferential expression of myosin V3 with low ATPase activity under conditions of chronic heart overloading is a mechanism for energy economy, which promotes adaptation of the cardiac muscle to changes in conditions of its functioning [34, 122]. Studies on the pCa–force relation showed that V3 isomyosin has higher sensitivity to calcium than V1 isomyosin; therefore, the thin filament in the case of isomyosin V3 is activated at lower calcium concentration. Under conditions of heart failure, this can be another mechanism of cardiac muscle adaptation.
It is known that heart hypertrophy is accompanied by changes in expression of both myosin and tropomyosin isoforms [7]. Our investigations have shown that the highest calcium sensitivity of the pCa–velocity relation has on combination of myosin isoform V3 with tropomyosin with a high content of β-chain. This is a manifestation of the compensatory role of the elevated fraction of the tropomyosin β-chain.
Comprehension of cardiac muscle functioning, its disturbances in pathologies, as well as development of approaches to their correction are impossible without knowledge of the underlying molecular mechanisms. Such fundamental dependences as pCa–velocity, pCa–force, and force–velocity characterizing the functional state of muscle are determined by properties of contractile and regulatory proteins of sarcomere. Modern biological techniques, such as optical trap and in vitro motility assay, make it possible to study properties of isolated myosin molecules, in particular, their mechanical and kinetic characteristics and also regulatory mechanisms of their interaction with actin, which are critical for cardiac muscle. The optical trap allows characterizing properties of single molecules of myosin isoforms, whereas their ensemble behavior and effects of regulatory proteins can be evaluated using the in vitro motility assay.
This work was supported by the Russian Foundation for Basic Research (projects Nos. 13-04-96027, 13-04-40101, 15-34-20136, 15-04-01558) and by programs of the Russian Academy of Sciences (0401-2014-0002) and of the Government of Sverdlovsk Region.
REFERENCES
1.Hoh, J. F. Y., McGrath, P. A., and Hale, P. (1977)
Electrophoretic analysis of multiple forms of rat cardiac myosin:
effect of hypophysectomy and thyroxin replacement, J. Mol. Cell
Cardiol., 10, 1053-1076.
2.Vandekerckhove, J., Bugaisky, G., and Buckingham,
M. (1986) Simultaneous expression of skeletal muscle and heart actin
proteins in various striated muscle tissues and cells. A quantitative
determination of the two actin isoforms, J. Biol. Chem.,
261, 1838-1843.
3.Perry, S. V. (2001) Vertebrate tropomyosin:
distribution, properties and function, J. Muscle Res. Cell.
Motil., 22, 5-49.
4.Karam, C. N., Warren, C. M., Rajan, S., De Tombe,
P. P., Wieczorek, D. F., and Solaro, R. J. (2011) Expression of
tropomyosin-κ induces dilated cardiomyopathy and depresses
cardiac myofilament tension by mechanisms involving cross-bridge
dependent activation and altered tropomyosin phosphorylation, J.
Muscle Res. Cell. Motil., 31, 315-322.
5.Rajan, S., Jagatheesan, G., Karam, C. N., Alves, M.
L., Bodi, I., Schwartz, A., Bulcao, C. F., D’Souza, K. M.,
Akhter, S. A., Boivin, G. P., Dube, D. K., Petrashevskaya, N., Herr, A.
B., Hullin, R., Liggett, S. B., Wolska, B. M., Solaro, R. J., and
Wieczorek, D. F. (2010) Molecular and functional characterization of a
novel cardiac-specific human tropomyosin isoform, Circulation,
121, 410-418.
6.Boussouf, S. E., Maytum, R., Jaquet, K., and
Geeves, M. A. (2007) Role of tropomyosin isoforms in the calcium
sensitivity of striated muscle thin filaments, J. Muscle Res. Cell.
Motil., 28, 49-58.
7.Izumo, S., Nadal-Ginard, B., and Mahdavi, V. (1988)
Protooncogene induction and reprogramming of cardiac gene expression
produced by pressure overload, Proc. Natl. Acad. Sci. USA,
85, 339-343.
8.Richard, P., Charron, P., Carrier, L., Ledeuil, C.,
Cheav, T., Pichereau, C., Benaiche, A., Isnard, R., Dubourg, O.,
Burban, M., Gueffet, J. P., Millaire, A., Desnos, M., Schwartz, K.,
Hainque, B., and Komajda, M. (2003) EUROGENE Heart Failure Project.
Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of
mutations, and implications for a molecular diagnosis strategy,
Circulation, 107, 2227-2232.
9.Rajan, S., Ahmed, R. P., Jagatheesan, G.,
Petrashevskaya, N., Boivin, G. P., Urboniene, D., Arteaga, G. M.,
Wolska, B. M., Solaro, R. J., Liggett, S. B., and Wieczorek, D. F.
(2007) Dilated cardiomyopathy mutant tropomyosin mice develop cardiac
dysfunction with significantly decreased fractional shortening and
myofilament calcium sensitivity, Circ. Res., 101,
205-214.
10.Sheehan, K. A., Arteaga, G. M., Hinken, A. C.,
Dias, F. A., Ribeiro, C., Wieczorek, D. F., Solaro, R. J., and Wolska,
B. M. (2011) Functional effects of a tropomyosin mutation linked to FHC
contribute to maladaptation during acidosis, J. Mol. Cell.
Cardiol., 50, 442-450.
11.Konno, T., Chang, S., Seidman, J. G., and
Seidman, C. E. (2010) Genetics of hypertrophic cardiomyopathy, Curr.
Opin. Cardiol., 25, 205-209.
12.Lowey, C., and Cohen, C. (1962) Studies on the
structure of myosin, J. Mol. Biol., 4, 293-307.
13.Pope, B., Hoh, J. F. Y., and Weeds, A. (1980) The
ATPase activities of rat cardiac myosin isoenzymes, FEBS Lett.,
118, 205-208.
14.Narolska, N. A., Eiras, S., Van Loon, R. B.,
Boontje, N. M., Zaremba, R. S., Berg, S. R., Stooker, W., Huybregts, M.
A., Visser, F. C., Van der Velden, J., and Stienen, G. J. (2005) Myosin
heavy chain composition and the economy of contraction in healthy and
diseased human myocardium, J. Muscle Res. Cell. Motil.,
26, 39-48.
15.Chizzonite, R. A., Everett, A. W., Prior, G., and
Zak, R. (1984) Comparison of myosin heavy chains in atria and
ventricles from hyperthyroid, hypothyroid, and euthyroid rabbits, J.
Biol. Chem., 259, 15564-15571.
16.Alpert, N. R., Brosseau, C., Federico, A., Krenz,
M., Robbins, J., and Warshaw, D. M. (2002) Molecular mechanics of mouse
cardiac myosin isoforms, Am. J. Physiol. Heart Circ. Physiol.,
283, 1446-1454.
17.Krenz, M., Sanbe, A., Bouyer-Dalloz, F., Gulick,
J., Klevitsky, R., Hewett, T. E., Osinska, H. E., Lorenz, J. N.,
Brosseau, C., Federico, Alpert, N. R., Warshaw, D. M., Perryman, M. B.,
Helmke, S. M., and Robbins, J. (2003) Analysis of myosin heavy chain
functionality in the heart, J. Biol. Chem., 278,
17466-17474.
18.Krenz, M., Sadayappan, S., Osinska, H. E., Henry,
J. A., Beck, S., Warshaw, D. M., and Robbins, J. (2007) Distribution
and structure–function relationship of myosin heavy chain
isoforms in the adult mouse heart, J. Biol. Chem., 282,
24057-24064.
19.Schmitt, J. P., Debold, E. P., Ahmad, F.,
Armstrong, A., Frederico, A., Conner, D. A., Mende, U., Lohse, M. J.,
Warshaw, D., Seidman, C. E., and Seidman, J. G. (2006) Cardiac myosin
missense mutations cause dilated cardiomyopathy in mouse models and
depress molecular motor function, Proc. Natl. Acad. Sci. USA,
103, 14525-14530.
20.Tyska, M. J., Hayes, E., Giewat, M., Seidman, C.
E., Seidman, J. G., and Warshaw, D. M. (2000) Single-molecule mechanics
of R403Q cardiac myosin isolated from the mouse model of familial
hypertrophic cardiomyopathy, Circ. Res., 86, 737-744.
21.Banerjee, S. K., Kabbas, E. G., and Morkin, E.
(1977) Enzymatic properties of the heavy meromyosin subfragment of
cardiac myosin from normal and thyrotoxic rabbits, J. Biol.
Chem., 252, 6925-6929.
22.VanBuren, P., Harris, D. E., Norman, R. A., and
Warshaw, D. M. (1995) Cardiac V1 and V3 myosins differ in their
hydrolytic and mechanical activities in vitro, Circ.
Res., 77, 439-444.
23.Malmqvist, U. P., Aronsham, A., and Lowey, S.
(2004) Cardiac myosin isoforms from different species have unique
enzymatic and mechanical properties, Biochemistry, 43,
15058-15065.
24.Litten, R. Z., Martin, B. J., Low, R. B., and
Alpert, N. R. (1982) Altered myosin isozyme patterns from
pressure-overloaded and thyrotoxic hypertrophied rabbit hearts,
Circ. Res., 50, 856-864.
25.Yamashita, H., Sugiura, S., Serizawa, T.,
Sugimoto, T., Iizuka, M., Katayama, E., and Shimmen, T. (1992) Sliding
velocity of isolated rabbit cardiac myosin correlates with isozyme
distribution, Am. J. Physiol., 263, 464-472.
26.Banerjee, S. K., and Morkin, E. (1977)
Actin-activated adenosine triphosphatase activity of native and
N-ethylmaleimide-modified cardiac myosin from normal and thyrotoxic
rabbits, Circ. Res., 41, 630-634.
27.Barany, M. (1967) ATPase activity of myosin
correlated with speed of muscle shortening, J. Gen. Physiol.,
50, 197.
28.Maughan, D., Low, E., Litten, R., Brayden, J.,
and Alpert, N. (1979) Calcium-activated muscle from hypertrophied
rabbit hearts. Mechanical and correlated biochemical changes, Circ.
Res., 44, 279-287.
29.Schwartz, K., Lecarpentier, Y., Martin, J. L.,
Lompre, A. M., Mercadier, J. J., and Swynghedauw, B. (1981) Myosin
isozymic distribution correlates with speed of myocardial contraction,
J. Mol. Cell. Cardiol., 13, 1071-1075.
30.Pagani, E. D., and Julian, F. J. (1984) Rabbit
papillary muscle myosin isozymes and the velocity of muscle shortening,
Circ. Res., 54, 586-594.
31.Saeki, Y. (1995) Crossbridge dynamics under
various inotropic states in cardiac muscle: evaluation by perturbation
analysis, Jpn. J. Physiol., 45, 687-705.
32.Stehle, M., Kruger, P., Scherer, K., Brixius, R.
H., Schwinger, G., and Pfitzer, G. (2002) Isometric force kinetics upon
rapid activation and relaxation of mouse, guinea pig, and human heart
muscle studied on the subcellular myofibrillar level, Basic Res.
Cardiol., 97, 127-135.
33.Fitzsimons, D. P., Patel, J. R., and Moss, R. L.
(1999) Aging dependent depression in the kinetics of force development
in rat skinned myocardium, Am. J. Physiol., 276,
1511-1519.
34.Alpert, N. R., Mulieri, L. A., and Hasenfuss, G.
(1991) The Heart and Cardiovascular System, Raven Press, New
York, pp. 111-128.
35.Gordon, A. M., Homsher, E., and Regnier, M.
(2000) Regulation of contraction in striated muscle, Physiol.
Rev., 80, 853-924.
36.Gordon, A. M., Regnier, M., and Homsher, E.
(2001) Skeletal and cardiac muscle contractile activation: tropomyosin
“rocks and rolls”, News Physiol. Sci., 16,
49-55.
37.McKillop, D. F., and Geeves, M. A. (1993)
Regulation of the interaction between actin and myosin subfragment 1:
evidence for three states of the thin filament, Biophys.
J., 65, 693-701.
38.Pirani, A., Vinogradova, M. V., Curmi, P. M. G.,
King, W. A., Fletterick, R. J., Craig, R., Tobacman, L. S., Xu, C.,
Hatch, V., and Lehman, W. (2006) An atomic model of the thin filament
in the relaxed and Ca2+-activated states, J. Mol.
Biol., 357, 707-717.
39.Donaldson, S. K., and Kerrick, W. G. (1975)
Characterization of the effects of Mg2+ on Ca2+
and Sr2+-activated tension generation of skinned skeletal
muscle fibers, J. Gen. Physiol., 66, 427-444.
40.Grabarek, Z., Grabarek, J., Leavis, P. C., and
Gergely, J. (1983) Cooperative binding to the Ca-specific sites of
troponin C in regulated actin and actomyosin, J. Biol. Chem.,
258, 14098-14102.
41.Brandt, P. W., Diamond, M. S., Rutchik, J. S.,
and Schachat, F. H. (1987) Cooperative interactions between
troponin-tropomyosin units extend the length of the thin filament in
skeletal muscle, J. Mol. Biol., 195, 885-896.
42.Brandt, P. W., Colomo, F., Piroddi, N., Poggesi,
C., and Tesi, C. (1998) Force regulation by Ca2+ in skinned
single cardiac myocytes of frog, Biophys. J., 74,
1994-2004.
43.Tsaturyan, A. K., Bershitsky, S. Y., Koubassova,
N. A., Fernandez, M., Narayanan, T., and Ferenczi, M. A. (2011) The
fraction of myosin motors that participates in isometric contraction of
rabbit muscle fibers at near-physiological temperature, Biophys.
J., 101, 404-410.
44.Linari, M., Caremani, M., and Lombardi, V. (2007)
Stiffness and fraction of myosin motors responsible for active force in
permeabilized muscle fibers from rabbit psoas, Biophys. J.,
92, 2476-2490.
45.Linari, M., Dobbie, I., and Lombardi, V. (1998)
The stiffness of skeletal muscle in isometric contraction and rigor:
the fraction of myosin heads bound to actin, Biophys. J.,
74, 2459-2473.
46.Wu, S., Liu, J., Reedy, M. C., Tregear, R. T.,
Winkler, H., Franzini-Armstrong, C., Sasaki, H., Lucaveche, C.,
Goldman, Y. E., Reedy, M. K., and Taylor, K. A. (2010) Electron
tomography of cryofixed, isometrically contracting insect flight muscle
reveals novel actin–myosin interactions, PLoS One,
5, e12643.
47.Wang, Y., and Kerrick, W. G. L. (2002) The off
rate of Ca2+ from troponin C is regulated by
force-generating cross bridges in skeletal muscle, J. Appl.
Physiol., 92, 2409-2418.
48.Turtle, C. W., Korte, F. S., Razumova, M. V., and
Regnier, M. (2011) Reducing thin filament Ca2+ affinity with
a CaTnC variant (L57Q) reduces force but enhances cross-bridge
dependence of cooperative activation in demembranated rat trabeculae,
Biophys. J., 100, 453a-453a.
49.Godt, R. E., and Maughan, W. M. (1995) Influence
of osmotic compression on calcium activation and tension in skinned
muscle fibers of the rabbit, Pflugers Arch., 391,
334-337.
50.McDonald, K. S., and Moss, R. L. (1995) Osmotic
compression of single cardiac myocytes eliminates the reduction in
Ca2+ sensitivity of tension at short sarcomere length,
Circ. Res., 77, 199-205.
51.Fuchs, F., and Wang, Y. P. (1996) Sarcomere
length versus interfilament spacing as determinants of cardiac
myofilament Ca2+ sensitivity and Ca2+ binding,
J. Mol. Cell. Cardiol., 28, 1375-1383.
52.Smith, S. H., and Fuchs, F. (2002) Length
dependence of cardiac myofilament Ca2+ sensitivity in the
presence of substitute nucleoside triphosphates, J. Mol. Cell.
Cardiol., 34, 547-554.
53.Moss, R. L., Razumova, M., and Fitzsimons, D. P.
(2004) Myosin crossbridge activation of cardiac thin filaments:
implications for myocardial function in health and disease, Circ.
Res., 94, 1290-1300.
54.Fuchs, F., and Martyn, D. (2005) Length-dependent
Ca2+ activation in cardiac muscle: some remaining questions,
J. Muscle Res. Cell. Motil., 26, 199-212.
55.Izakov, V., Katsnelson, L. B., Blyakhman, F. A.,
Markhasin, V. S., and Shklyar, T. F. (1991) Cooperative effects due to
calcium binding by troponin and their consequences for contraction and
relaxation of cardiac muscle under various conditions of mechanical
loading, Circ. Res., 69, 1171-1184.
56.Solovyova, O., Katsnelson, L. B., Konovalov, P.,
Lookin, O., Moskvin, A. S., Protsenko, Yu. L., Vikulova, N., Kohl, P.,
and Markhasin, V. S. (2006) Activation sequence as a key factor in
spatio-temporal optimization of myocardial function, Phil. Transact.
R. Soc. London, 364, 1367-1383.
57.Allen, D. G., and Kurihara, S. (1982) The effects
of muscle length on intracellular calcium transients in mammalian
cardiac muscle, J. Physiol., 327, 79-94.
58.Lab, M. J. (1982) Contraction-excitation feedback
in myocardium. Physiological basis and clinical relevance, Circ.
Res., 50, 757-766.
59.Lab, M. J., Allen, D. G., and Orchard, C. (1984)
The effects of shortening on myoplasmic calcium concentration and on
the action potential in mammalian ventricular muscle, Circ.
Res., 55, 825-829.
60.Vahl, C. F., Timek, T., Bonz, A., Fuchs, H.,
Dillman, R., and Hagl, S. (1998) Length dependence of calcium- and
force-transients in normal and failing human myocardium, J. Mol.
Cell., 30, 957-966.
61.Ishikava, T., Kajiwara, H., and Kurihara, S.
(1999) Modulation of Ca2+ transient decay by tension and
Ca2+ removal in hyperthyroid myocardium, Am. J. Physiol.
Heart Circ. Physiol., 276, 289-299.
62.Wakayama, Y., Miura, M., Sugai, Y., Kagaya, Y.,
Watanabe, J., Ter Keurs, H. E. D. J., and Shirato, K. (2001) Stretch
and quick release of rat cardiac trabeculae accelerates Ca2+
waves and triggered propagated contractions, Am. J. Physiol. Heart
Circ. Physiol., 281, 2133-2142.
63.Luers, C., Fialka, F., Elgner, A., Zhu, D.,
Kockskampe, J., von Lewinski, D., and Pieske, B. (2005)
Stretch-dependent modulation of [Na+]i,
[Ca2+]i, and pHi in rabbit myocardium
- a mechanism for the slow force response, Cardiovasc. Res.,
68, 454-463.
64.Monasky, M. M., Varian, K. D., Davis, J. P., and
Janssen, P. M. L. (2008) Dissociation of force decline from calcium
decline by preload in isolated rabbit myocardium, Pflugers
Arch., 456, 267-276.
65.Ter Keurs, H. E. D. G. (2011) Electromechanical
coupling in the cardiac myocyte; stretch-arrhythmia feedback,
Pflugers Arch., 462, 165-175.
66.Ruwhof, C., Van Wamel, J. T., Noordzij, L. A.,
Aydin, S., Harper, J. C., and Van der Laarse, A. (2001) Mechanical
stress stimulates phospholipase C activity and intracellular calcium
ion levels in neonatal rat cardiomyocytes, Cell. Calcium,
29, 73-83.
67.Yasuda, S., Sugiura, S., Yamashita, H.,
Nishimura, S., Saeki, Y., Momomura, S., Katoh, K., Nagai, R., and Sugi,
H. (2003) Unloaded shortening increases peak of Ca2+
transients but accelerates their decay in rat single cardiac myocytes,
Am. J. Physiol. Heart Circ. Physiol., 285, 470-475.
68.Sulman, T., Katsnelson, L. B., Solovyova, O., and
Markhasin, V. S. (2008) Mathematical modeling of mechanically modulated
rhythm disturbances in homogeneous and heterogeneous myocardium with
attenuated activity of Na+-K+ pump, Bull.
Math. Biol., 70, 910-949.
69.Katsnelson, L. B., Solovyova, O., Balakin, A.,
Lookin, O., Konovalov, P., Protsenko, Yu., Sulman, T., and Markhasin,
V. S. (2011) Contribution of mechanical factors to arrythmogenesis in
calcium overloaded cardiomyocytes: model predictions and experiments,
Progr. Bioph. Mol. Biol., 107, 81-89.
70.Edman, K. A., and Nilsson, P. E. (1972)
Relationships between force and velocity of shortening in rabbit
papillary muscle, Acta Physiol. Scand., 85, 488-500.
71.Metzger, J. M., Wahr, P. A., Michele, D. E.,
Albayya, F., and Westfall, M. V. (1999) Effects of myosin heavy chain
isoform switching on Ca2+-activated tension development in
single adult cardiac myocytes, Circ. Res., 11,
1310-1317.
72.Fitzsimons, D. P., Patel, J. R., and Moss, R. L.
(1998) Role of myosin heavy chain composition in kinetics of force
development and relaxation in rat myocardium, J. Physiol.,
513, 171-183.
73.Rundell, V. L., Manaves, V., Martin, A. F., and
De Tombe, P. P. (2005) Impact of β-myosin heavy chain isoform
expression on cross-bridge cycling kinetics, Am. J. Physiol. Heart
Circ. Physiol., 288, 896-903.
74.Ashkin, A., and Dziedzic, J. M. (1987) Optical
trapping and manipulation of viruses and bacteria, Science,
235, 1517-1520.
75.Finer, J. T., Simmons, R. M., and Spudich, J. A.
(1994) Single myosin molecule mechanics: piconewton forces and
nanometre steps, Nature, 368, 113-118.
76.Nabiev, S. R., Ovsyannikov, D. A., Bershitsky, B.
Y., and Bershitsky, S. Y. (2008) Optical trap as a tool for studying
motor proteins, Biophysics, 53, 488-493.
77.Molloy, J. E., Burns, J. E., Kendrick-Jones, J.,
Tregear, R. T., and White, D. C. S. (1995) Movement and force produced
by a single myosin head, Nature, 378, 209-212.
78.Takagi, Y., Homsher, E. E., Goldman, Y. E., and
Shuman, H. (2006) Force generation in single conventional actomyosin
complexes under high dynamic load, Biophys. J., 90,
1295-1307.
79.Sugiura, S., Kobayakawa, N., Fujita, H.,
Yamashita, H., Momomura, S., Chaen, S., Omata, M., and Sugi, H. (1998)
Comparison of unitary displacements and forces between 2 cardiac myosin
isoforms by the optical trap technique: molecular basis for cardiac
adaptation, Circ. Res., 82, 1029-1034
80.Palmiter, K. A., Tyska, M. J., Dupius, D. E.,
Alpert, N. R., and Warshaw, D. M. (1999) Kinetic differences at the
single molecule level account for the functional diversity of rabbit
cardiac myosin isoforms, J. Physiol., 519, 669-678.
81.Nabiev, S. R., Schepkin, D. V., Kopylova, G. V.,
and Bershitsky, S. Y. (2012) Comparison of the characteristics of the
single interactions of rabbit muscle proteins isoforms, in
Biological Motility: Fundamental and Applied Science [in
Russian], Pushchino, pp. 138-140.
82.Nikitina, L. V., Kopylova, G. V., Shchepkin, D.
V., and Katsnelson, L. V. (2008) Study of the interaction between
rabbit cardiac contractile and regulatory proteins. An in vitro
motility assay, Biochemistry (Moscow), 73, 178-184.
83.Kron, S. J., and Spudich, J. A. (1986)
Fluorescent actin filaments move on myosin fixed to a glass surface,
Proc. Natl. Acad. Sci. USA, 83, 6272-6276.
84.Mashanov, G. I., and Molloy, J. E. (2007)
Automatic detection of single fluorophores in live cells, Biophys.
J., 92, 2199-2211.
85.Sugiura, S., Yamashita, H., Sata, M., Momomura,
S., Serizawa, T., Oiwa, K., Chaen, S., Shimmen, T., and Sugi, H. (1995)
Force–velocity relations of rat cardiac myosin isozymes sliding
on algal cell actin cables in vitro, Biochim. Biophys.
Acta, 1231, 69-75.
86.Bing, W., Knott, A., and Marston, S. (2000) A
simple method for measuring the relative force exerted by myosin on
actin filaments in the in vitro motility assay: evidence that
tropomyosin and troponin increase force in single thin filaments,
Biochem. J., 350, 693-699.
87.Haeberle, J. R., and Hemric, M. E. (1995) Are
actin filaments moving under unloaded conditions in the in vitro
motility assay? Biophys. J., 68, 306-310.
88.VanBuren, P., Alix, S. L., Gorga, J. A., Begin,
K. J., LeWinter, M. M., and Alpert, N. R. (2002) Cardiac troponin T
isoforms demonstrate similar effects on mechanical performance in a
regulated contractile system, Am. J. Physiol. Heart Circ.
Physiol., 282, 1665-1671.
89.Nikitina, L. V., Kopylova, G. V., Shchepkin, D.
V., and Katsnelson, L. B. (2008) Assessment of the mechanical activity
of cardiac myosins V1 and V3 by the in vitro motility assay with
regulated thin filament, Biophysics, 53, 510-514.
90.Kopylova, G. V., Katsnelson, L. B., Ovsyannikov,
D. A., Bershitsky, S. Yu., and Nikitina, L. V. (2006) Application of
in vitro motility assay to studying the calcium-mechanical
relationship in skeletal and cardiac muscles, Biophysics,
51, 687-691.
91.Gordon, A. M., LaMadrid, M. A., Chen, Y., Luo,
Z., and Chase, P. B. (1997) Calcium regulation of skeletal muscle thin
filament motility in vitro, Biophys. J., 72,
1295-1307.
92.Homsher, E., Kim, B., Bobkova, A., and Tobacman,
L. S. (1996) Calcium regulation of thin filament movement in an in
vitro motility assay, Biophys. J., 70, 1881-1892.
93.Honda, H., and Asakura, S. (1989)
Calcium-triggered movement of regulated actin in vitro. A
fluorescence microscopy study, J. Mol. Biol., 205,
677-683.
94.Dyer, E. C., Jacques, A. M., Hoskins, A. C.,
Ward, D. G., Gallon, C. E., Messer, A. E., Kaski, J. P., Burch, M.,
Kentish, J. C., and Marston, S. B. (2009) Functional analysis of a
unique troponin C mutation, GLY159ASP, that causes familial dilated
cardiomyopathy, studied in explanted heart muscle, Circ. Heart
Fail., 2, 456-464.
95.Song, W., Dyer, E., Stuckey, D., Leung, M. C.,
Memo, M., Mansfield, C., Ferenczi, M., Liu, K., Redwood, C., Nowak, K.,
Harding, S., Clarke, K., Wells, D., and Marston, S. (2010)
Investigation of a transgenic mouse model of familial dilated
cardiomyopathy, J. Mol. Cell. Cardiol., 49, 380-389.
96.Funatsu, T., Anazawa, T., and Ishiwata, S. (1994)
Structural and functional reconstitution of thin filaments in skeletal
muscle, J. Muscle Res. Cell Motil., 15, 158-171.
97.Fujita, H., Yasuda, K., Niitsu, S., Funatsu, T.,
and Ishiwata, S. (1996) Structural and functional reconstitution of
thin filaments in the contractile apparatus of cardiac muscle,
Biophys. J., 71, 2307-2318.
98.Sata, M., Yamashita, H., Sugiura, S., Fujita, H.,
Momomura, S., and Serizawa, T. (1995) A new in vitro motility
assay technique to evaluate calcium sensitivity of the cardiac
contractile proteins, Pflugers Arch., 429, 443-445.
99.Shaffer, J. F., Razumova, M. V., Tu, A. Y.,
Regnier, M., and Harris, S. P. (2007) Myosin S2 is not required for
effects of myosin binding protein-C on motility, FEBS Lett.,
581, 1501-1504.
100.Shchepkin, D. V., Kopylova, G. V., Nikitina, L.
V., Katsnelson, L. B., and Bershitsky, S. Y. (2010) Effects of cardiac
myosin binding protein-C on the regulation of interaction of cardiac
myosin with thin filament in an in vitro motility assay,
Biochem. Biophys. Res. Commun., 401, 159-163.
101.Sugiura, S., and Yamashita, H. (1998)
Functional characterization of cardiac myosin isoforms, Jpn. J.
Physiol., 48, 173-179.
102.Fraser, I. D., and Marston, S. B. (1995) In
vitro motility analysis of actin–tropomyosin regulation by
troponin and calcium, J. Biol. Chem., 270, 7836-7841.
103.Lu, X., Tobacman, L. S., and Kawai, M. (2006)
Temperature-dependence of isometric tension and cross-bridge kinetics
of cardiac muscle fibers reconstituted with a tropomyosin internal
deletion mutant, Biophys. J., 91, 4230-4240.
104.Landis, C., Back, N., Homsher, E., and
Tobacman, L. S. (1999) Effects of tropomyosin internal deletions on
thin filament function, J. Biol. Chem., 274,
1279-31285.
105.Matyushenko, A. M., Artemova, N. V., Shchepkin,
D. V., Kopylova, G. V., Bershitsky, S. Y., Tsaturyan, A. K., Sluchanko,
N. N., and Levitsky, D. I. (2014) Structural and functional effects of
two stabilizing substitutions, D137L and G126R, in the middle part of
α-tropomyosin molecule, FEBS J., 281,
2004-2016.
106.Shchepkin, D. V., Matyushenko, A. M., Kopylova,
G. V., Artemova, N. V., Bershitsky, S. Y., Tsaturyan, A. K., and
Levitsky, D. I. (2013) Stabilization of the central part of tropomyosin
molecule alters the Ca2+-sensitivity of actin–myosin
interaction, Acta Naturae, 5, 126-129.
107.Noguchi, T., Camp, P. Jr., Alix, S. L., Gorga,
J. A., Begin, K. J., Leavitt, B. J., Ittleman, F. P., Alpert, N. R.,
LeWinter, M. M., and Van Buren, P. (2003) Myosin from failing and
non-failing human ventricles exhibit similar contractile properties,
J. Mol. Cell. Cardiol., 35, 91-97.
108.Bottinelli, R., Coviello, D. A., Redwood, C.
S., Pellegrino, M. A., Maron, B. J., Spirito, P., Watkins, H., and
Reggiani, C. (1998) A mutant tropomyosin that causes hypertrophic
cardiomyopathy is expressed in vivo and associated with an
increased calcium sensitivity, Circ. Res., 82,
106-115.
109.De Clerck, N. M., Claes, V. A., and Brutsaert,
D. L. (1977) Force–velocity relations of single cardiac muscle
cells: calcium dependency, J. Gen. Physiol., 69,
221-241.
110.Katsnelson, L. B., Markhasin, V. S., Nikitina,
L. V., and Ryvkin, M. V. (1997) Analysis of force–velocity
relationship in cardiac muscle by means of mathematical modeling, J.
Muscle Res. Cell Motil., 8, 228.
111.Hill, A. V. (1938) The heat of shortening and
the dynamic constants of muscle, Proc. R. Soc. London,
126, 136-195.
112.Hennekes, R., Kaufmann, R., and Steiner, R.
(1978) Why does the cardiac force–velocity relationship not
follow a Hill hyperbola? Possible implications of feedback loops
involved in cardiac excitation–contraction coupling, Basic
Res. Cardiol., 73, 47-67.
113.Katsnelson, L. B., Nikitina, L. V., Chemla, D.,
Solovyova, O. E., Coirault, C., Lecarpentier, Y., and Markhasin, V. S.
(2004) Influence of viscosity on myocardium mechanical activity: a
mathematical model, J. Theor. Biol., 230, 385-405.
114.Muthuchamy, M., Boivin, G. P., Grupp, I. L.,
and Wieczorek, D. F. (1998) Beta-tropomyosin overexpression induces
severe cardiac abnormalities, J. Mol. Cell. Cardiol., 30,
1545-1557.
115.Muthuchamy, M., Grupp, I. L., Grupp, G.,
O’Toole, B. A., Kier, A. B., Boivin, G. P., Neumann, J., and
Wieczorek, D. F. (1995) Molecular and physiological effects of
overexpressing striated muscle β-tropomyosin in the adult murine
heart, J. Biol. Chem., 270, 30593-30603.
116.Shchepkin, D. V., Kopylova, G. V., and
Nikitina, L. V. (2011) Study of reciprocal effects of cardiac myosin
and tropomyosin isoforms on actin–myosin interaction with in
vitro motility assay, Biochem. Biophys. Res. Commun.,
415, 104-108.
117.Chen, W., Wen, K. K., Sens, A. E., and
Rubenstein, P. A. (2006) Differential interaction of cardiac, skeletal
muscle, and yeast tropomyosins with fluorescent (pyrene235) yeast
actin, Biophys. J., 90, 1308-1318.
118.Sliwinska, M., Zukowska, M., Borys, D., and
Moraczewska, J. (2011) Different positions of tropomyosin isoforms on
actin filament are determined by specific sequences of end-to-end
overlaps, Cytoskeleton (Hoboken), 68, 300-312.
119.Ajtai, K., Halstead, M. F., Nyitrai, M.,
Penheiter, A. R., Zheng, Y., and Burghardt, T. P. (2009) The myosin
C-loop is an allosteric actin contact sensor in actomyosin,
Biochemistry, 48, 5263-5275.
120.Nikitina, L. V., Shchepkin, D. V., and
Kopylova, G. V. (2014) Study of effects of tropomyosin isoforms on the
regulation of actin–myosin interaction in myocardium with in
vitro motility assay, J. Muscle Res. Cell. Motil.,
35, 147.
121.Lompre, A. M., Schwartz, K., D’Albis, A.,
Lacombe, G., Van Thiem, N., and Swynghedauw, B. (1979) Myosin isoenzyme
redistribution in chronic heart overload, Nature, 282,
105-107.
122.Katz, A. M. (2001) Physiology of the
Heart, Lippincott, Williams and Wilkins.