REVIEW: Small Heat Shock Proteins and Distal Hereditary Neuropathies
V. V. Nefedova#, L. K. Muranova#, M. V. Sudnitsyna, A. S. Ryzhavskaya, and N. B. Gusev*
Lomonosov Moscow State University, Faculty of Biology, 119991 Moscow, Russia; E-mail: NBGusev@mail.ru# These authors contributed equally to this work.
* To whom correspondence should be addressed.
Received February 15, 2015
Classification of small heat shock proteins (sHsp) is presented and processes regulated by sHsp are described. Symptoms of hereditary distal neuropathy are described and the genes whose mutations are associated with development of this congenital disease are listed. The literature data and our own results concerning physicochemical properties of HspB1 mutants associated with Charcot–Marie–Tooth disease are analyzed. Mutations of HspB1, associated with hereditary motor neuron disease, can be accompanied by change of the size of HspB1 oligomers, by decreased stability under unfavorable conditions, by changes in the interaction with protein partners, and as a rule by decrease of chaperone-like activity. The largest part of these mutations is accompanied by change of oligomer stability (that can be either increased or decreased) or by change of intermonomer interaction inside an oligomer. Data on point mutation of HspB3 associated with axonal neuropathy are presented. Data concerning point mutations of Lys141 of HspB8 and those associated with hereditary neuropathy and different forms of Charcot–Marie–Tooth disease are analyzed. It is supposed that point mutations of sHsp associated with distal neuropathies lead either to loss of function (for instance, decrease of chaperone-like activity) or to gain of harmful functions (for instance, increase of interaction with certain protein partners).
KEY WORDS: small heat shock proteins, phosphorylation, chaperone-like activity, cytoskeleton, congenital diseasesDOI: 10.1134/S000629791513009X
Abbreviations: sHsp, small heat shock proteins.
Small heat shock proteins (sHsp) form a large family of closely related
proteins that are expressed in practically all Kingdoms including
viruses, bacteria, plants, and animals [1]. A key
distinguishing property of all these proteins is the presence of highly
conservative α-crystallin domain consisting of about 80-100
residues and located in the C-terminal end of the protein
(figure) [2, 3]. This domain is
flanked by highly variable in length and composition sequences forming
unordered and highly flexible N- and C-terminal domains
(figure) [2]. Small heat shock proteins obtained
their name since monomers of these proteins have low molecular weight,
which is of the order of 12-43 kDa [4-6]. As a rule, the small heat shock proteins tend to
form large oligomers consisting of more than twenty identical or
different subunits [7-9]. Thus,
formed homo- or heterooligomers [10, 11] are highly labile and easily undergo
association/dissociation leading to either increase or decrease in the
number of subunits in the oligomeric complex [9].
High flexibility and mobility strongly impedes structural investigation
of sHsp. At present, the literature data contain information on the
structure of sHsp from hyperthermophilic archaeon Methanocaldococcus
jannaschii [12], wheat Triticum
aestivum [13], parasitic worm Taenia
saginata [14], acidothermophilic archaeon
Sulfolobus tokodaii [15], and
proteobacterium Xanthomonas [16]. Up to
now, all attempts to crystallize full-size mammalian or human small
heat shock proteins have been unsuccessful. However, the structure of
isolated crystallin domains of αB-crystallin (HspB5), HspB1, and
HspB6 are described in the literature [17-21].
Scheme of the structure of small heat shock proteins in terms of HspB1. The N-terminal, α-crystallin, and the C-terminal domains as well as the sites of phosphorylation (Ser15, Ser78, Ser82) are indicated. Mutations of certain residues that are associated with neurodegenerative diseases are also indicated
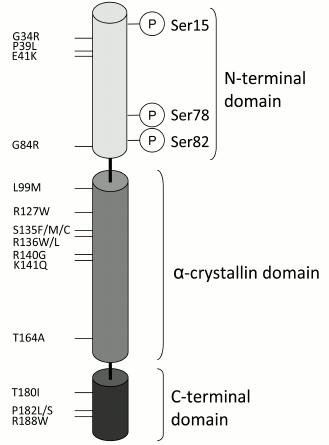
The human genome contains 10 genes of small heat shock proteins designated as HspB1-HspB10 [22, 23]. Certain members of the sHsp family (such as HspB1, HspB5, HspB6, and HspB8) are expressed ubiquitously [24]. The other members of this family (such as HspB2, HspB3, HspB4, HspB7, HspB9, and HspB10) are tissue-specific [4, 5, 19, 25]. The level of sHsp expression is tissue-dependent and can change in the course of ontogenesis [26-28]. Moreover, the level of sHsp expression is changed in response to different unfavorable conditions, such as denervation, hyperthermia, or ischemia [28, 29]. The sHsp content in different tissues can be very high, reaching as much as 0.3% of the total protein content [30].
The sHsp (together with other heat shock proteins) play important roles in keeping homeostasis and participate in the control of proper protein folding [5, 31]. Control of protein folding, often termed proteostasis, includes checking of proper folding of newly synthesized proteins and of already existing proteins. Therefore, different unfavorable conditions leading to accumulation of partially denatured proteins induce increased synthesis of different heat shock proteins and among them of certain members of the family of small heat shock proteins [4-6, 31]. Proteostasis is achieved in a number of different ways. First, the sHsp bind partially denatured proteins and by this means prevent their aggregation. In vitro, sHsp prevent aggregation of many different model substrate proteins [32, 33]. In vivo, the small heat shock proteins prevent aggregation of huntingtin [34] and β- and/or γ-crystallins [25]. In the cell, the sHsp not only bind denatured proteins, thus preventing their aggregation, but also are able to transmit denatured proteins to other heat shock proteins. These ATP-dependent proteins renature partially denatured proteins using ATP [35]. Moreover, the sHsp can promote selective elimination of denatured proteins in proteasomes and autophagosomes [36-38].
Second, sHsp prevent accumulation of denatured proteins by decreasing the effect of different unfavorable conditions. For instance, it is known that the sHsp are able to prevent effects induced by oxidative stress. HspB1 stabilizes and activates certain enzymes, participating in synthesis of reduced glutathione, and by this means prevents accumulation of reactive oxygen species [39, 40].
Third, sHsp interact with practically all proteins of cytoskeleton (such as actin, tubulin, or intermediate filament proteins) and are able to stabilize the cytoskeleton, thus preventing its damage induced by unfavorable conditions [41-46].
Fourth, data of the literature indicate that as a rule the small heat shock proteins such as HspB1, HspB5, and HspB6 have pronounced antiapoptotic activity [39, 47-49]. It is obvious that all these properties of sHsp are interconnected and therefore cannot be separated from each other. For instance, prevention of protein aggregation, stabilization of cytoskeleton, or protection against oxidative stress can effectively modulate apoptosis. At the same time, antiapoptotic activity can rely on very different mechanisms and can be due to the interaction of small heat shock proteins with certain protein kinases or by their ability to regulate cytochrome c release from mitochondria [47, 48]. Anyhow, the data presented clearly indicate that the small heat shock proteins play important roles in many different cellular processes. Therefore, any mutations of sHsp can lead to dramatic changes in the normal functioning of the cell and thus induce different diseases. Indeed, data from the literature indicate that mutations of sHsp correlate with different hereditary diseases such as cataract, myofibrillar myopathy, certain forms of cardiomyopathy, and hereditary neuropathies [50-52]. In this review, we will describe some properties of sHsp mutants associated with only one hereditary disease, namely distal neuropathy. We will also try to understand why these mutations are associated with these diseases.
HEREDITARY NEUROPATHIES
Hereditary motor and sensory neuropathy, also classified as Charcot–Marie–Tooth (CMT) disease, was first described in 1886 and is the most common hereditary peripheral neuropathy with prevalence of 1 : 2500 [53]. Onset age of the clinical symptoms varies from 10-20 up to 70 years and older. This disease is accompanied by slowly progressive degeneration of peripheral nerves resulting in muscle weakness or complete muscle atrophy. The disease usually starts in the legs and feet and later affects hands and arms. In the later stages, the disease leads to progressive inability to walk and manipulate small objects. In additions, these symptoms are sometimes associated with hand tremor, diaphragm palsy, optical nerve atrophy, and renal failure [53, 54].
CMT diseases are very heterogeneous, and a very complicated classification of disease is presented in the literature. There are two main groups within this disease. The first group is characterized by low nerve conduction (less than 38 m/s). This subtype of CMT is caused by abnormalities in myelin sheath and therefore is called demyelinating. There are autosomal dominant (the so-called AD CMT1) and autosomal recessive (the so-called AR CMT1 or CMT4) forms of the neuropathy. Each of these subgroups is divided in additional classes marked by a Latin letter depending on the affected gene. Among genes associated with CMT disease of the first type are genes encoding peripheral myelin protein 22, myelin protein zero, small integral membrane protein of lysosome/late endosome (SIMPLE), and many other proteins. Mutations of peripheral myelin protein 22 and myelin protein zero are especially often associated with CMT disease of the first type [55].
The second large group of CMT disease combines forms with unaltered conduction velocity and undamaged myelin sheath, but with alteration of axons of motor and/or sensory neurons. This group, designated as CMT of the second type (or CMT2), is divided into autosomal dominant and autosomal recessive diseases. As in the previous case, designation CMT2 is appended by the Latin letter indicating the affected protein. Axonal forms of CMT disease can be associated with mutation of certain motor proteins (such as kinesin, dynein heavy chains), proteins associated with mitochondrial dynamics (such as mitofusin), cytoskeletal proteins (such as neurofilament light chain or lamin A/C), small heat shock proteins (HspB1, HspB3, HspB8), and certain other proteins [53, 56].
The third and the fourth groups of CMT disease are characterized by intermediate nerve conduction (so-called dominant intermediate or DI-CMT, which is usually associated with autosomal recessive inheritance and designated as CMT4) and the forms linked to mutations in X-chromosome (so-called X-linked CMT or CMT5) [53, 57, 58]. In this group, symptoms of Charcot–Marie–Tooth disease are most often associated with mutations of gap junction protein β1 (connexin-32) [55].
The question arises – why are mutations of more than 30 different genes accompanied by similar symptoms? At present, it is complicated to answer this question. Let us restrict this problem and analyze the processes occurring in the axonal forms of CMT disease of the second type (CMT2). This disease is associated with impairment of different processes connected with intracellular traffic of vesicles and organelles [53, 56]. The length of motor neuron axons is more than 1 m [56], and therefore neurons should provide rapid and effective transport of organelles and vesicles over a very large distance. It is obvious that mutations of motor proteins such as dynein heavy chains [59] and p150 subunit of dynactin complex [60], participating in dynein-dependent organelle transport across microtubules, or of kinesin KIF1B [61], also participating in transport of cargoes across microtubules, can significantly affect both retrograde and anterograde transport, thus inducing CMT of the second type. Mutations of the small G-protein RAB7 responsible for the interaction of special adapter proteins with motor proteins and for translocation of endosomes inside the cell [62] are also associated with CMT2 [63]. Intermediate filaments are important components of neuron cytoskeleton, and their mutations are associated with CMT2 [64-66]. Moreover, short intermediate filaments are transported over microtubules by kinesin, and, therefore, mutations of intermediate filament proteins associated with their aggregation and interaction with protein motors can also affect trafficking in long axons [64-66].
Let us try to understand why mutations of the small heat shock proteins can affect CMT of the second type or distal hereditary motor neuropathy. Thinking mechanistically, we suppose that mutations can lead either to the loss of useful properties and function (so-called loss-of-function mutations), or to appearance of new negative properties and functions (so-called gain-of-function mutations). It is obvious that these processes may proceed both sequentially and simultaneously and cannot be separated from each other. Nevertheless, the gain of function can be accompanied by decrease of protein stability and increased tendency for aggregation, thus leading to association with other proteins and formation of insoluble aggregates inside the cell. Loss of sHsp function can be accompanied by inability to form functionally active homo- or heterooligomeric complexes with protein partners and decrease of chaperone-like activity. Let us try to analyze properties of certain sHsp mutants using this simplified approach.
MUTATIONS OF SMALL HEAT SHOCK PROTEIN HspB1 AND HEREDITARY DISTAL
MOTOR AND SENSORY NEUROPATHY
About twenty different mutations of HspB1 associated with CMT2 and/or hereditary motor neuropathy have been described in the literature [35, 51, 67] and presented in database HMGD Pro v.2014.2. Among these mutations, there are many missense point mutations, as well as mutations with frame shift and/or preliminary appearance of a stop-codon [51, 67]. Provisionally, all these mutations can be divided according to the place of mutation in the protein structure. As already mentioned, there are three domains in the structure of sHsp (figure). There is a variable and flexible N-terminal domain, a conservative α-crystallin domain predominantly containing β-sheets, and a variable C-terminal domain [2]. Mutations associated with hereditary neuropathies are located in any of the three domains of small heat shock proteins.
Three point mutations – G34R, P39L, and E41K – were detected in the very N-terminal part of the N-terminal domain of HspB1 (figure) [68, 69]. The first two mutations were associated with late (more than 50 years) onset of hereditary motor neuropathy and CMT2, whereas E41K mutation was associated with early onset of hereditary motor neuropathy. Residues G34 and P39 are highly conservative and are preserved in the primary structure of mammalian, bird, reptile, and amphibian HspB1. Residue E41 is slightly less conservative and can be replaced by Asp in the sequence of certain mammals, birds, and reptiles. However, this position is never occupied by positively charged residues [67]. Investigation performed in our group [70] indicates that the three indicated mutations lead to formation of large oligomers with size slightly larger than the corresponding oligomers of the wild-type protein. Oligomers of the mutated proteins are more resistant to limited chymotrypsinolysis and have lower thermal stability than the wild-type protein. The mutants with replacements in the N-terminal domain are phosphorylated by MAPKAP2 kinase with rate comparable with that of the wild-type protein. However, phosphorylation of the wild-type protein up to 1 mole of phosphate induces complete dissociation of large oligomers formed by this protein, whereas phosphorylation of mutant proteins even up to 2 moles of phosphate per mole of protein was not accompanied by significant dissociation of large oligomers. Chaperone-like activity of HspB1 mutants with replacements in the N-terminal domain is usually less than the corresponding activity of the wild-type protein. In summary, we conclude that the analyzed mutants form slightly larger oligomers containing tightly interacting with each other monomers. Because of this interaction, phosphorylation does not induce dissociation of large oligomers. The literature data indicate that phosphorylation-induced dissociation of large oligomers of HspB1 plays a crucial role in the interaction of this protein with different elements of cytoskeleton and in its chaperone-like activity [43, 47]. Thus, these mutations are accompanied by gain of functions (such as resistance to phosphorylation-induced dissociation), as well as by loss of certain functions (such as decrease of chaperone-like activity).
Two other mutants of HspB1 that were analyzed carry point mutations in the very C-terminal end of the N-terminal domain (G84R) and in the very beginning of α-crystallin domain (L99M) (figure). These mutations associate with hereditary distal neuropathy with dominant (G84R) and probably recessive (L99M) inheritance, and the onset of symptoms was detected in middle-aged patients [67, 68, 71]. Both residues are highly conservative and are conserved in the primary structure of mammalian, birds, reptiles, amphibians, and even fishes [67]. It is worthwhile to mention that these residues are so conservative that they are preserved even in the primary structure of human small heat shock proteins tending to form large oligomers, such as HspB4 and HspB5 [72]. At the same time, the small heat shock proteins unable to form large oligomers (HspB6 and HspB8) have replacements of either Gly in the position homologous to that of G84 or replacement of Leu in the position homologous to that of L99 [72]. Both mutants form oligomers, which are larger than the corresponding oligomers of the wild-type protein. However, oligomers formed by these mutants are less stable and easily dissociate to dimers and/or tetramers. Both mutants are phosphorylated by MAPKAP2 kinase with rate and efficiency comparable with those of the wild-type protein. Long incubation with protein kinase is accompanied by incorporation of ~3 moles of phosphate per mole of protein, and all potential sites (Ser15, Ser78, and Ser82) become phosphorylated. However, at low extent of phosphorylation (~0.6 mole of phosphate per mole of protein), large oligomers of the wild-type protein are only partially dissociated, whereas large oligomers of both mutants completely dissociate to small oligomers (probably dimers and tetramers) [72]. The wild-type HspB1 forms two types of heterooligomeric complexes with another small heat shock protein, HspB6. The apparent molecular weight of these heterooligomeric complexes is about 100-120 and about 300 kDa, respectively, whereas both mutants form only one type of heterooligomeric complexes with apparent molecular weight ~120 kDa. In vitro, both G84R and L99M have lower chaperone-like activity than the wild-type protein [72]. In summary, we conclude that as in the case with the N-terminal mutants, both G84R and L99M mutants form oligomers of larger size than oligomers formed by the wild-type protein. However, if mutations in the very N-terminal domain (G34R, P39L, and E41K) somehow stabilize the structure of oligomers, two other mutations, namely G84R and L99M, somehow destabilize the structure of homooligomers and provoke phosphorylation-induced dissociation. We suppose that replacement of small Gly84 by bulky positively charged Arg affects orientation and/or flexibility of the whole N-terminal domain, and this destabilizes the oligomer structure [72]. Molecular mechanisms underlying effects induced by L99M mutation remain incompletely understandable. However, we suppose that this point mutation may affect the interaction between two antiparallel β7 sheets belonging to the two neighboring HspB1 monomers, and in this way destabilize the structure of the whole oligomer [72].
The most detailed investigations were performed on mutants carrying replacements in the α-crystallin domain (figure). Mutations R127W, S135F, and R136W (R136L) are associated with hereditary distal neuropathy with dominant inheritance. The onset of symptoms is detected in early or middle age [73-76]. All three residues are highly conservative and are preserved in the corresponding positions in the primary structure of mammalian, avian, reptile, amphibian, and fish HspB1. Rather rarely, Arg in the position homologous to that of Arg127 is replaced by Lys [67]. Almeida-Souza et al. [77] analyzed the properties of these three HspB1 mutants. Unexpectedly, they found that mutations R127W and S135W induced increase rather than decrease of chaperone-like activity, and both mutants were more effective in protection of cells against heat shock than the wild-type protein. Moreover, mutants R127W, S135F, and R136W had higher affinity to denatured protein targets than the wild-type protein. According to Almeida-Souza et al. [77], increased affinity to potential substrates as well as increased chaperone-like activity and increased ability to protect cells against heat shock can be explained by the fact that all these mutations destabilize dimers of HspB1 without affecting ability of HspB1 monomers to form large oligomers. Thus, all these mutations induce HspB1 monomerization, and this process is accompanied by increase of chaperone-like activity.
If this explanation is correct, the question arises – why are R127W, S135F, and R136W mutants having higher chaperone-like activity and better protecting activity than the wild-type protein able to induce different forms of distal neuropathy? This apparent contradiction can be explained by the fact that certain HspB1 mutants (for instance, S135F) form a very tight complex with the light component of neurofilaments, and the complex of these two proteins forms amorphous aggregates and precipitates inside the cell [73]. This suggestion at least partially agrees with experimental data indicating that R127W and S135F affect interaction of neurofilaments with kinesin, and thus affect anterograde transport of neurofilaments in the cell [78]. This effect correlates with increased neurofilament phosphorylation by cyclin-dependent protein kinase Cdk5 and can be at least partially reversed by selective inhibition of Cdk5 [78]. Experiments performed on transgenic mice expressing R136W mutant indicate that these animals develop age-dependent axonopathy with impairment of neurofilament network and intracellular transport system. Impairments of axon–Schwann cell interaction were also detected in these transgenic animals [79].
Detailed investigation of the R127W and S135F mutants revealed that both mutant proteins tightly interact with isolated tubulin and microtubules, increasing stability of microtubules to different unfavorable factors such as addition of nocodazole [80]. Experiments performed on transgenic mice expressing S135F mutant indicate that in this case microtubules were less effectively acetylated, but had increased stability [81]. It was hypothesized that the analyzed HspB1 mutant tightly interacts with microtubules and significantly increases their stability. Over-stabilized microtubules affect normal transport processes in the cell, and this induces compensatory activation of deacetylases and deacetylation of tubulin, dramatically decreasing stability of microtubules, which cannot be overcome even by the presence of HspB1 mutants [82]. As a result, microtubules are depolymerized, thus leading to impairment of axonal transport and onset of symptoms of distal neuropathy. Thus, experiments performed on isolated proteins, on cultured cells, and with transgenic animals indicate that R127W, S135F, and R136W mutants form stable complexes with cytoskeletal proteins (intermediate filament proteins and tubulin), and this can be one of the reasons inducing neuropathy. Moreover, S135F mutant of HspB1 interacts with another small heat shock protein, HspB8, more tightly than the wild-type HspB1 [83]. This tight interaction can lead to predominant binding of HspB8 to mutated HspB1, thus preventing normal functioning of HspB8 in the cell.
Mutations R140G and K141Q are associated with comparably late (more than 30 years) onset of symptoms of hereditary distal motor neuropathy [68, 84]. Both residues are highly conservative and are preserved with minimal replacements in the primary structure of mammalian, avian, reptile, amphibian, and fish HspB1 [67]. In vitro, both mutants have lower thermal stability than the wild-type HspB1 [85]. Oligomeric structure of K141Q mutant is practically identical to that of the wild-type protein. At the same time, the R140G mutant is presented in the form of a mixture of small oligomers (dimers and tetramers) and very large oligomers (or aggregates), which are larger than the corresponding oligomers of the wild-type protein [44, 85]. The wild-type protein is able to prevent aggregation of R140G mutants either forming mixed oligomers or by its chaperone-like activity. Chaperone-like activity of the K141Q mutant is similar to that of the wild-type protein. In addition, it forms heterooligomers with another small heat shock protein, HspB6, which are similar to those formed by the wild-type HspB1. In contrast, the R140G mutant has much lower chaperone-like activity and much less effectively interacts with HspB6 than the wild-type HspB1 [85].
Summarizing, we conclude that mutations of the central part of the α-crystallin domain affect the region of intersubunit contacts formed by two antiparallel β7 strands belonging to two neighbor monomers of HspB1 (see the model structure presented in Almeida-Souza et al. [77] and Nefedova et al. [85]). Mutations R127W, S135F, and R136W destabilize intersubunit contacts, and according to the literature data induce HspB1 monomerization [77]. Monomers of HspB1 mutants have high affinity to certain cytoskeletal proteins and other small heat shock proteins, thus gaining negative functions leading to cytoskeleton damage and inactivation of other small heat shock proteins. Mutation R140G is located in a so-called hot spot, i.e. in a position that induces dramatic changes of the structure and properties of not only HspB1, but also of many other small heat shock proteins. Indeed, mutation of residues located in the position homologous to R140 of HspB1 is accompanied by a number of different congenital diseases. For instance, mutations R116C/H of HspB4 are associated with cataract [86, 87], mutation R120G of HspB5 is associated with cataract, myofibrillar myopathy and certain forms of cardiomyopathy [88, 89], and finally mutation K141E/N/T in HspB8 is associated with hereditary distal motor neuropathy or CMT disease [75, 90]. The corresponding residues of Arg (or Lys) participate in formation of salt bridges with negatively charged residues of the neighboring monomer and thus stabilize the whole structure of sHsp oligomers. This probably is the reason why the R140G mutation of HspB1 is accompanied by formation of equilibrium mixture of small and large oligomers tending to further aggregation. Significant changes induced by the R140G mutation are accompanied by decrease of chaperone-like activity and modification of interaction of HspB1 with other small heat shock proteins. Unexpectedly, mutation of the neighboring residue (K141Q) induces much less pronounced effects on the structure and properties of HspB1. This can be due to the fact that the K141 residue, although participating in formation of stabilizing intersubunit salt bridge, is located on the periphery of the interface, and therefore induces less pronounced effect than the neighboring R140 residue [85]. Nevertheless, mutation K141Q, as well as R140G mutation, is accompanied by decrease of thermal stability of HspB1 [85].
Recently, new data on the structure and properties of HspB1 mutants with replacements in the C-terminal end of the crystallin domain and in the C-terminal domain were published (figure). Mutation T164A is located in the last (ninth) β-strand of the crystalline domain. This residue is conserved in primary structure of HspB1 from many mammals, birds, reptiles, amphibians, and fishes [91]. Mutation T164A was detected in a Han Chinese patient living on Taipei [91]. This mutation was accompanied by the onset of symptoms at early age and was accompanied by sensitivity loss and atrophy of muscles of upper and lower extremities [92]. Mutations T180I, P182L/S, and R188W are localized in the flexible C-terminal domain of HspB1. The T180 residue is rather conservative and is conserved in the primary structure of mammals, birds, and reptiles, but it is replaced by Ser, Ala, Asn, Ile, or Val in certain amphibian and fish HspB1 [67]. Residue P182 is highly conservative and is preserved in the primary structure of practically all analyzed animal species [67]. Finally, residue R188 is not very conservative and is conserved in practically all analyzed mammalian HspB1, but is replaced in the primary structure of other animal species. Most of the C-terminal mutations are characterized by dominant inheritance, and the onset of the first symptoms is detected at early age (less than 18 years). Mutation T180I is associated with symptoms of distal hereditary neuropathy [69] or CMT disease of the second type [93]. Mutation P182L/S is accompanied by very early (less than 5 years) or early onset of symptoms of hereditary distal neuropathy [73, 75, 94]. Finally, mutation R188W is associated with early (less than 10 years) onset of symptoms of CMT2 disease [69].
Investigation of the physicochemical properties of the T164A and P182S mutants revealed significant decrease of their thermostability [91]. Oligomers formed by the T164A mutant are rather unstable and tend to dissociate. At the same time, mutation P182S leads to formation of very large oligomers tending to association. It is worthwhile to mention that the P182S mutant was able to form mixed oligomers with the wild-type HspB1, having higher thermal stability and lower tendency to aggregation than the isolated P182S mutant [91]. Chaperone-like activity of the T164A and T180I mutants was comparable, although slightly lower than that of the wild-type HspB1. The R188W and especially P182S mutant had significantly lower chaperone-like activity than the wild-type HspB1 [91].
In experiments performed on the cell level, the P182L mutant had chaperone-like activity comparable with that of the wild-type protein and was nearly as effective as the wild-type protein in cell protection against thermal shock [77]. At the same time, expression of the P182L mutant was accompanied by accumulation of insoluble aggregates, increased level of phosphorylation, and impairment of neurofilament transport and damages of the neurofilament network [78]. In addition, this mutation was accompanied by changes in the intracellular location of dynactin p150 component and synaptotagmin [95]. Japanese investigators also suppose that the P182S mutation is accompanied by damage of the neurofilament network [94]. Data on the effect of the P182L mutation on the microtubular system are rather contradictory. Experiments performed on the cell level indicate that this mutant does not interact with microtubules, and in the contrast to the S135F and R136W mutants does not affect extent of tubulin acetylation [80]. At the same time, experiments performed on transgenic animals indicate that the P182L mutation is accompanied by decrease of the level of tubulin acetylation, and symptoms associated with this mutation are partially or completely prevented by utilization of deacetylase inhibitors [81].
At present, it is rather complicated to give a detailed description of the molecular bases underlying neurodegenerative diseases associated with mutations in the C-terminal domain of HspB1. This is due to the fact that at present there are no detailed data concerning location of this part of the molecule in large HspB1 oligomers. However, the literature data [17, 96-98] indicate that the conservative tripeptide I-P-I(V) located in the unordered C-terminal domain of α-crystalline (and probable HspB1) can interact with the hydrophobic groove formed by β4 and β8 strands of the same or of the neighboring monomer. It is probable that due to this fact mutation of the P182 residue located in the middle of this conservative tripeptide induces dramatic changes in the oligomeric structure of HspB1, its chaperone-like activity, and its ability to interact with other protein targets and protein partners. It is possible that this explanation is also applicable for the T180I mutation. Indeed, the T180 residue borders with the conservative tripeptide I-P-V of HspB1, and replacement T180I leads to formation of a hydrophobic cluster consisting of three consecutive isoleucine residues. As already mentioned, the T164A mutation is located in the middle of the β9 strand and probably affects orientation and flexibility of the C-terminal domain. Perhaps this is the reason why the T164A mutation induces significant destabilization of HspB1 oligomeric structure. The residue R188 is located in the very C-terminal end of HspB1. This part of the molecule has high mobility [99] and plays an important role in the interaction of small heat shock proteins with target proteins [100, 101]. Therefore, it seems probable that replacement of arginine by bulky and hydrophobic tryptophan can change the interaction of HspB1 with different partners and protein targets.
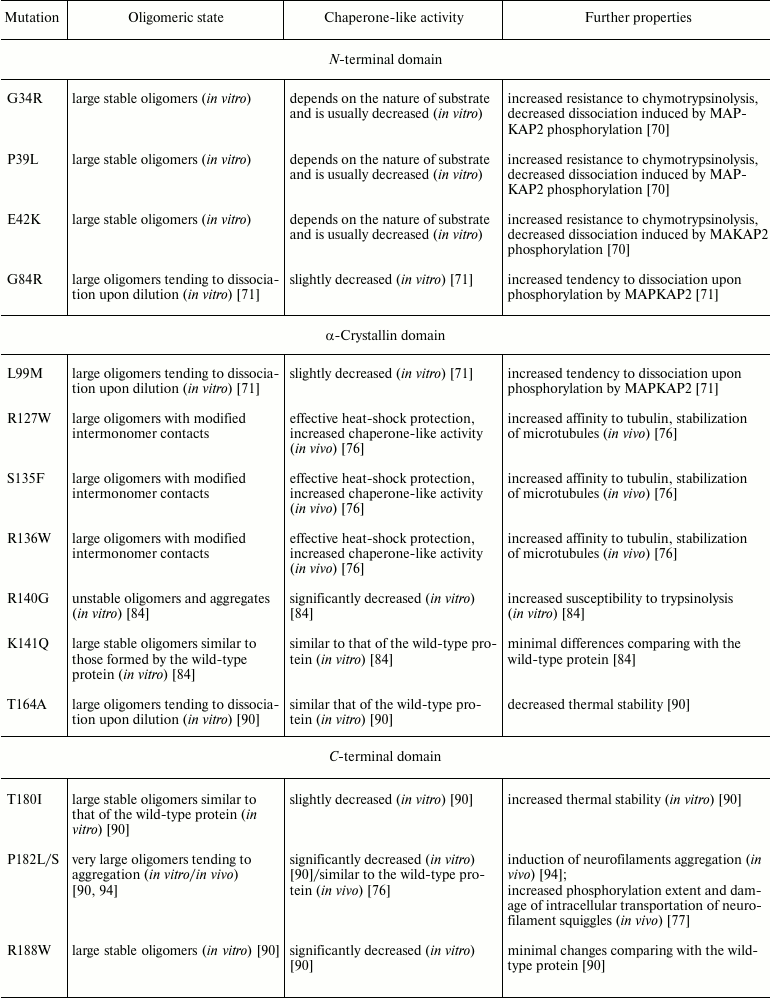
Let us try to compare experimental data obtained on different mutants of HspB1. Mutations in the N-terminal domain (G34R, P39L, E41K) are accompanied by increased stability of homooligomeric complexes formed by HspB1, resulting in hampering of phosphorylation-induced dissociation of large oligomers (table). These effects modify many important properties of HspB1 and can be one of the reasons leading to onset of symptoms characteristic for neuropathy. On the contrary, mutations in the C-terminal part of the N-terminal domain and in the beginning of the crystalline domain (G84R, L99M) induce destabilization of HspB1 oligomers and certain decrease of chaperone-like activity of HspB1 (table). These effects can also induce different types of neuropathy. Mutations in the central part of the crystalline domain (R127W, S135F, R136W) induce modification of intersubunit interactions inside of large oligomers and often lead to increase of affinity of HspB1 to certain target proteins (for instance, tubulin) (table). This affects the normal process of microtubule polymerization/depolymerization, thus leading to damage of axonal transportation and neuron death. The point mutation R140G affecting positively charged Arg residue involved in intersubunit interaction leads to dramatic changes of HspB1 structure and to significant decrease of chaperone-like activity (table). Mutation T164A in the C-terminal domain (like mutation L99M in the N-terminal end of the crystalline domain) is accompanied by destabilization of the quaternary structure of HspB1 (table). As already mentioned, the C-terminal domain plays an important role in stabilization of the quaternary structure and participates in the interaction of HspB1 with different protein targets and protein partners. Therefore, mutations in this domain lead to decrease of the chaperone-like activity (P182L/S, R188W) and to formation of large oligomers (aggregates) of very large size (P182L/S) (table). All these events affect cell homeostasis, leading to neuron death and neuropathy onset.
Domain location and some properties of HspB1 point mutants associated
with different forms of distal neuropathies
MUTATION OF SMALL HEAT SHOCK PROTEIN HspB3 AND AXONAL
NEUROPATHY
One HspB3 mutation associated with inherited axonal predominantly motor neuropathy is described in the literature [102]. In this case, Arg7 is replaced by Ser (R7S mutation). Most human small heat shock proteins (except of HspB8) contain positively charged residues in position homologous to that of R7 of HspB3 [67]. Therefore, one can suggest that replacement of positively charged arginine by the neutral polar serine residue somehow affects the structure and properties of this protein. Unfortunately, at present, the structure and properties of HspB3 have been investigated very superficially. It is known that in contrast to other human small heat shock proteins, HspB3 tends to form trimers, and although possessing general chaperone-like activity, it prevents aggregation of only certain (far from all) protein substrates [103]. Functions of this protein also remain poorly investigated; however, it is known that this protein can form heterooligomers consisting of 4, 8, 12, 16, 20, or 24 subunits with the small heat shock protein HspB2. In all cases, the stoichiometry HspB2/HspB3 is 3/1 [104]. HspB2 is also investigated very superficially [105], but it is supposed that this protein binds and regulates activity of myotonic dystrophy protein kinase [106]. We suppose that mutation R7S of HspB3 somehow affects its interaction with HspB2, and this modulates protein kinase activity or chaperone-like activity of HspB3 itself.
MUTATIONS OF SMALL HEAT SHOCK PROTEIN HspB8 AND HUMAN
NEUROPATHIES
Three point mutations – K141E, K141T, and K141N – have been described for HspB8, all these mutations being accompanied by replacement of the same K141 residue [75, 90, 107, 108]. It seems that all these mutations are dominantly inherited and are accompanied by comparatively early (14-15 years) onset of symptoms of hereditary distal muscle dystrophy or different forms of CMT disease [67]. Residue K141 of HspB8 is located in the center of the β7 strand in position homologous to R116 of αA-crystallin (HspB4), R120 of αB-crystallin (HspB5), or R140 of HspB1. As mentioned earlier, these mutations are associated with cataract (HspB4), cataract, myofibrillar myopathy, and certain forms of cardiomyopathy (HspB5), and distal neuropathy (HspB1). These residues play a key role in formation of the intersubunit contact forming a salt bridge with negatively charged residues of the neighboring subunit [85, 109]. Mutation K141E and especially double mutation K137E+K141E lead to destabilization of HspB8 structure and increases its susceptibility to proteolysis [110, 111]. In addition, mutation K141E is accompanied by decrease of chaperone-like activity with certain model substrates [111].
Expression of fluorescent chimeras of mutated forms of HspB8 in COS cells was accompanied by accumulation of protein aggregates [90]. However, the changes in solubility and increased tendency to aggregation are not the sole reasons inducing pathological phenomena. Expression of mutated HspB8 in motor neurons was not accompanied by accumulation of aggregates of mutated HspB8, but they lead to neurite degeneration [112]. This effect was highly specific and was not detected in primary glial cells and in sensory or cortical neurons [112]. These effects can be explained by specific recognition and interaction of mutated HspB8 with certain specific protein targets of the cell. It was found that HspB8 mutations are accompanied by decrease of mitochondria membrane potential and inhibition of autophagy, providing for selective proteolysis of improperly folded proteins [113-115]. There are at least two reasons explaining inhibition of autophagy. First, mutation of the K141 residue decreases interaction of HspB8 with adapter protein Bag3, participating in regulation of autophagy [116, 117]. Second, HspB8 mutations disturb transportation of lysosomes to autophagosomes [114], i.e. affect intracellular transport processes. Ddx20/Gemin3, RNA-helicase interacting with specific protein (so-called survival-of-motor-neurons protein, SMN protein) can be another protein partner of HspB8. Both proteins (Ddx20/Gemin3 and SMN) participate in formation of spliceosome and pre-mRNA processing [118]. Mutant forms of HspB8 especially actively interact with Ddx20/Gemin3. Interaction of the wild-type HspB8 with Ddx20/Gemin3 does not depend on the presence of RNase, whereas interaction of mutated HspB8 with Ddx20/Gemin3 is RNase-dependent [119]. This might indicate that the wild-type HspB8 in the complex with Ddx20/Gemin3 somehow protects RNA from RNase, whereas the HspB8 mutants are lacking this property. Other small heat shock proteins can also be potential partners of HspB8. The literature data [83] indicate that HspB8 mutants form tight complexes with HspB1 and HspB5 (αB-crystallin). These tight interactions can lead to sequestration of HspB8 in its complexes with two other small heat shock proteins, thus leading to HspB8 depletion, which being in the complex with other small heat shock proteins will not be able to participate in normal processes in the cell. This can induce different pathologies.
Different forms of CMT disease and hereditary distal neuropathy form a wide group of diseases with different etiology, different time of onset of symptoms, and different severity. The most probable reason leading to CMT2 is impairment of different intracellular transport processes, which can be due to damages caused by mutations of special motor or adapter proteins involved in organelle traffic inside the cell, by damages of cytoskeleton, or by impairment of energy supply for transport processes. Many proteins are involved in trafficking, and therefore certain changes of the structure of these proteins or their intracellular distribution can have severe consequences. Small heat shock proteins are involved in proteostasis, in other words, they control proper protein folding and participate both in renaturation of partially denatured proteins as well as in elimination of proteins that cannot be renatured. Since the small heat shock proteins interact with a huge number of different proteins and participate in the control of many intracellular processes, point mutations of the small heat shock proteins often are associated with different forms of neuropathy. Mutations can be associated with the loss of certain useful properties (for instance, decrease of chaperone-like activity, inability to interact with certain protein targets or protein partners) or with gaining of new detrimental properties (for instance, to increased interaction with old or new protein targets or formation of oligomers tending to aggregates). The quaternary structure of small heat shock proteins is rather complicated, and, therefore, mutations can evoke very different changes in the oligomeric state of these proteins as well as in their ability to interact with different protein partners. Therefore, the search for new methods for medical treatment of different congenital disease should be based on careful examination of each mutation, and success in treatment can be achieved only in the case of detailed investigation both on the level of isolated proteins and on the cell and tissue levels.
This investigation was supported by the Russian Science Foundation (project No. 14-35-00026).
REFERENCES
1.Maaroufi, H., and Tanguay, R. M. (2013) Analysis
and phylogeny of small heat shock proteins from marine viruses and
their cyanobacteria host, PLoS One, 8, e81207.
2.Kriehuber, T., Rattei, T., Weinmaier, T.,
Bepperling, A., Haslbeck, M., and Buchner, J. (2010) Independent
evolution of the core domain and its flanking sequences in small heat
shock proteins, FASEB J., 24, 3633-3642.
3.Kappe, G., Boelens, W. C., and De Jong, W. W.
(2010) Why proteins without an α-crystallin domain should not be
included in the human small heat shock protein family HSPB, Cell
Stress Chaperones, 15, 457-461.
4.Mymrikov, E. V., Seit-Nebi, A. S., and Gusev, N. B.
(2011) Large potentials of small heat shock proteins, Physiol.
Rev., 91, 1123-1159.
5.Basha, E., O’Neill, H., and Vierling, E.
(2012) Small heat shock proteins and α-crystallins: dynamic
proteins with flexible functions, Trends Biochem. Sci.,
37, 106-117.
6.Treweek, T. M., Meehan, S., Ecroyd, H., and Carver,
J. A. (2014) Small heat-shock proteins: important players in regulating
cellular proteostasis, Cell. Mol. Life Sci., 72,
429-451.
7.Peschek, J., Braun, N., Franzmann, T. M.,
Georgalis, Y., Haslbeck, M., Weinkauf, S., and Buchner, J. (2009) The
eye lens chaperone α-crystallin forms defined globular
assemblies, Proc. Natl. Acad. Sci. USA, 106,
13272-13277.
8.Peschek, J., Braun, N., Rohrberg, J., Back, K. C.,
Kriehuber, T., Kastenmuller, A., Weinkauf, S., and Buchner, J. (2013)
Regulated structural transitions unleash the chaperone activity of
αB-crystallin, Proc. Natl. Acad. Sci. USA, 110,
3780-3789.
9.Hochberg, G. K., and Benesch, J. L. (2014)
Dynamical structure of αB-crystallin, Prog. Biophys. Mol.
Biol., 115, 11-20.
10.Mymrikov, E. V., Seit-Nebi, A. S., and Gusev, N.
B. (2012) Heterooligomeric complexes of human small heat shock
proteins, Cell Stress Chaperones, 17, 157-169.
11.Arrigo, A. P. (2013) Human small heat shock
proteins: protein interactomes of homo- and hetero-oligomeric
complexes: an update, FEBS Lett., 587, 1959-1969.
12.Kim, K. K., Kim, R., and Kim, S. H. (1998)
Crystal structure of a small heat-shock protein, Nature,
394, 595-599.
13.Van Montfort, R. L., Basha, E., Friedrich, K. L.,
Slingsby, C., and Vierling, E. (2001) Crystal structure and assembly of
an eukaryotic small heat shock protein, Nat. Struct. Biol.,
8, 1025-1030.
14.Stamler, R., Kappe, G., Boelens, W., and
Slingsby, C. (2005) Wrapping the α-crystallin domain fold in a
chaperone assembly, J. Mol. Biol., 353, 68-79.
15.Hanazono, Y., Takeda, K., Yohda, M., and Miki, K.
(2012) Structural studies on the oligomeric transition of a small heat
shock protein, StHsp14.0, J. Mol. Biol., 422,
100-108.
16.Hilario, E., Martin, F. J., Bertolini, M. C., and
Fan, L. (2011) Crystal structures of Xanthomonas small heat
shock protein provide a structural basis for an active molecular
chaperone oligomer, J. Mol. Biol., 408, 74-86.
17.Laganowsky, A., Benesch, J. L., Landau, M., Ding,
L., Sawaya, M. R., Cascio, D., Huang, Q., Robinson, C. V., Horwitz, J.,
and Eisenberg, D. (2010) Crystal structures of truncated αA and
αB crystallins reveal structural mechanisms of polydispersity
important for eye lens function, Protein Sci., 19,
1031-1043.
18.Bagneris, C., Bateman, O. A., Naylor, C. E.,
Cronin, N., Boelens, W. C., Keep, N. H., and Slingsby, C. (2009)
Crystal structures of α-crystallin domain dimers of
αB-crystallin and Hsp20, J. Mol. Biol., 392,
1242-1252.
19.Clark, A. R., Lubsen, N. H., and Slingsby, C.
(2012) sHSP in the eye lens: crystallin mutations, cataract, and
proteostasis, Int. J. Biochem. Cell Biol., 44,
1687-1697.
20.Baranova, E. V., Weeks, S. D., Beelen, S.,
Bukach, O. V., Gusev, N. B., and Strelkov, S. V. (2011)
Three-dimensional structure of α-crystallin domain dimers of
human small heat shock proteins HSPB1 and HSPB6, J. Mol. Biol.,
411, 110-122.
21.Weeks, S. D., Baranova, E. V., Heirbaut, M.,
Beelen, S., Shkumatov, A. V., Gusev, N. B., and Strelkov, S. V. (2014)
Molecular structure and dynamics of the dimeric human small heat shock
protein HSPB6, J. Struct. Biol., 185, 342-354.
22.Kappe, G., Franck, E., Verschuure, P., Boelens,
W. C., Leunissen, J. A., and De Jong, W. W. (2003) The human genome
encodes 10 α-crystallin-related small heat shock proteins:
HspB1-10, Cell Stress Chaperones, 8, 53-61.
23.Fontaine, J. M., Rest, J. S., Welsh, M. J., and
Benndorf, R. (2003) The sperm outer dense fiber protein is the 10th
member of the superfamily of mammalian small stress proteins, Cell
Stress Chaperones, 8, 62-69.
24.Taylor, R. P., and Benjamin, I. J. (2005) Small
heat shock proteins: a new classification scheme in mammals, J. Mol.
Cell. Cardiol., 38, 433-444.
25.Slingsby, C., and Wistow, G. J. (2014) Functions
of crystallins in and out of lens: roles in elongated and post-mitotic
cells, Prog. Biophys. Mol. Biol., 115, 52-67.
26.Lutsch, G., Vetter, R., Offhauss, U., Wieske, M.,
Grone, H. J., Klemenz, R., Schimke, I., Stahl, J., and Benndorf, R.
(1997) Abundance and location of the small heat shock proteins HSP25
and αB-crystallin in rat and human heart, Circulation,
96, 3466-3476.
27.Verschuure, P., Tatard, C., Boelens, W. C.,
Grongnet, J. F., and David, J. C. (2003) Expression of small heat shock
proteins HspB2, HspB8, Hsp20, and cvHsp in different tissues of the
perinatal developing pig, Eur. J. Cell Biol., 82,
523-530.
28.Inaguma, Y., Hasegawa, K., Kato, K., and Nishida,
Y. (1996) cDNA cloning of a 20-kDa protein (p20) highly homologous to
small heat shock proteins: developmental and physiological changes in
rat hindlimb muscles, Gene, 178, 145-150.
29.Bartelt-Kirbach, B., and Golenhofen, N. (2013)
Reaction of small heat-shock proteins to different kinds of cellular
stress in cultured rat hippocampal neurons, Cell Stress
Chaperones, 19, 145-153.
30.Kato, K., Shinohara, H., Goto, S., Inaguma, Y.,
Morishita, R., and Asano, T. (1992) Copurification of small heat shock
protein with αB crystallin from human skeletal muscle, J.
Biol. Chem., 267, 7718-7725.
31.Hilton, G. R., Lioe, H., Stengel, F., Baldwin, A.
J., and Benesch, J. L. (2012) Small heat-shock proteins: paramedics of
the cell, Top. Curr. Chem., 328, 69-98.
32.Mymrikov, E. V., Bukach, O. V., Seit-Nebi, A. S.,
and Gusev, N. B. (2010) The pivotal role of the β7 strand in the
intersubunit contacts of different human small heat shock proteins,
Cell Stress Chaperones, 15, 365-377.
33.Bukach, O. V., Seit-Nebi, A. S., Marston, S. B.,
and Gusev, N. B. (2004) Some properties of human small heat shock
protein Hsp20 (HspB6), Eur. J. Biochem., 271,
291-302.
34.Carra, S., Sivilotti, M., Chavez Zobel, A. T.,
Lambert, H., and Landry, J. (2005) HspB8, a small heat shock protein
mutated in human neuromuscular disorders, has in vivo chaperone
activity in cultured cells, Hum. Mol. Genet., 14,
1659-1669.
35.Boncoraglio, A., Minoia, M., and Carra, S. (2012)
The family of mammalian small heat shock proteins (HSPBs): implications
in protein deposit diseases and motor neuropathies, Int. J. Biochem.
Cell Biol., 44, 1657-1669.
36.Carra, S., Seguin, S. J., Lambert, H., and
Landry, J. (2008) HspB8 chaperone activity toward poly(Q)-containing
proteins depends on its association with Bag3, a stimulator of
macroautophagy, J. Biol. Chem., 283, 1437-1444.
37.Zhang, H., Rajasekaran, N. S., Orosz, A., Xiao,
X., Rechsteiner, M., and Benjamin, I. J. (2010) Selective degradation
of aggregate-prone CryAB mutants by HSPB1 is mediated by
ubiquitin-proteasome pathways, J. Mol. Cell. Cardiol.,
49, 918-930.
38.Parcellier, A., Brunet, M., Schmitt, E., Col, E.,
Didelot, C., Hammann, A., Nakayama, K., Nakayama, K. I., Khochbin, S.,
Solary, E., and Garrido, C. (2006) HSP27 favors ubiquitination and
proteasomal degradation of p27Kip1 and helps S-phase re-entry in
stressed cells, FASEB J., 20, 1179-1181.
39.Arrigo, A. P. (2007) The cellular
“networking” of mammalian Hsp27 and its functions in the
control of protein folding, redox state, and apoptosis, Adv. Exp.
Med. Biol., 594, 14-26.
40.Wyttenbach, A., Sauvageot, O., Carmichael, J.,
Diaz-Latoud, C., Arrigo, A. P., and Rubinsztein, D. C. (2002) Heat
shock protein 27 prevents cellular polyglutamine toxicity and
suppresses the increase of reactive oxygen species caused by
huntingtin, Hum. Mol. Genet., 11, 1137-1151.
41.Ghosh, J. G., Houck, S. A., and Clark, J. I.
(2007) Interactive domains in the molecular chaperone human
αB-crystallin modulate microtubule assembly and disassembly,
PLoS One, 2, e498.
42.Clarke, J. P., and Mearow, K. M. (2013) Cell
stress promotes the association of phosphorylated HspB1 with F-actin,
PLoS One, 8, e68978.
43.Wettstein, G., Bellaye, P. S., Micheau, O., and
Bonniaud, P. (2012) Small heat shock proteins and the cytoskeleton: an
essential interplay for cell integrity? Int. J. Biochem. Cell
Biol., 44, 1680-1686.
44.Elliott, J. L., Der Perng, M., Prescott, A. R.,
Jansen, K. A., Koenderink, G. H., and Quinlan, R. A. (2013) The
specificity of the interaction between αB-crystallin and desmin
filaments and its impact on filament aggregation and cell viability,
Philos. Trans. R. Soc. Lond. B Biol. Sci., 368, doi:
10.1098/rstb.2012.0375.
45.Pivovarova, A. V., Chebotareva, N. A., Chernik,
I. S., Gusev, N. B., and Levitsky, D. I. (2007) Small heat shock
protein Hsp27 prevents heat-induced aggregation of F-actin by forming
soluble complexes with denatured actin, FEBS J., 274,
5937-5948.
46.Dreiza, C. M., Komalavilas, P., Furnish, E. J.,
Flynn, C. R., Sheller, M. R., Smoke, C. C., Lopes, L. B., and Brophy,
C. M. (2010) The small heat shock protein, HSPB6, in muscle function
and disease, Cell Stress Chaperones, 15, 1-11.
47.Bakthisaran, R., Tangirala, R., and Rao, C. M.
(2014) Small heat shock proteins: role in cellular functions and
pathology, Biochim. Biophys. Acta, 1854, 291-319.
48.Acunzo, J., Katsogiannou, M., and Rocchi, P.
(2012) Small heat shock proteins HSP27 (HspB1), αB-crystallin
(HspB5), and HSP22 (HspB8) as regulators of cell death, Int. J.
Biochem. Cell Biol., 44, 1622-1631.
49.Paul, C., Simon, S., Gibert, B., Virot, S.,
Manero, F., and Arrigo, A. P. (2010) Dynamic processes that reflect
anti-apoptotic strategies set up by HspB1 (Hsp27), Exp. Cell
Res., 316, 1535-1552.
50.Laskowska, E., Matuszewska, E., and
Kuczynska-Wisnik, D. (2010) Small heat shock proteins and
protein-misfolding diseases, Curr. Pharm. Biotechnol.,
11, 146-157.
51.Datskevich, P. N., Nefedova, V. V., Sudnitsyna,
M. V., and Gusev, N. B. (2012) Mutations of small heat shock proteins
and human congenital diseases, Biochemistry (Moscow), 77,
1500-1514.
52.Benndorf, R., Hayess, K., Ryazantsev, S., Wieske,
M., Behlke, J., and Lutsch, G. (1994) Phosphorylation and
supramolecular organization of murine small heat shock protein HSP25
abolish its actin polymerization-inhibiting activity, J. Biol.
Chem., 269, 20780-20784.
53.Bucci, C., Bakke, O., and Progida, C. (2012)
Charcot–Marie–Tooth disease and intracellular traffic,
Prog. Neurobiol., 99, 191-225.
54.Patzko, A., and Shy, M. E. (2011) Update on
Charcot–Marie–Tooth disease, Curr. Neurol. Neurosci.
Rep., 11, 78-88.
55.DiVincenzo, C., Elzinga, C. D., Medeiros, A. C.,
Karbassi, I., Jones, J. R., Evans, M. C., Braastad, C. D., Bishop, C.
M., Jaremko, M., Wang, Z., Liaquat, K., Hoffman, C. A., York, M. D.,
Batish, S. D., Lupski, J. R., and Higgins, J. J. (2014) The allelic
spectrum of Charcot–Marie–Tooth disease in over 17,000
individuals with neuropathy, Mol. Genet. Genom. Med., 2,
522-529.
56.Gentil, B. J., and Cooper, L. (2012) Molecular
basis of axonal dysfunction and traffic impairments in CMT, Brain
Res. Bull., 88, 444-453.
57.Timmerman, V., Strickland, A. V., and Zuchner, S.
(2014) Genetics of Charcot–Marie–Tooth (CMT) disease within
the frame of the human genome project success, Genes, 5,
13-32.
58.Jerath, N. U., and Shy, M. E. (2014) Hereditary
motor and sensory neuropathies: understanding molecular pathogenesis
could lead to future treatment strategies, Biochim. Biophys.
Acta, 1852, 667-678.
59.Weedon, M. N., Hastings, R., Caswell, R., Xie,
W., Paszkiewicz, K., Antoniadi, T., Williams, M., King, C., Greenhalgh,
L., Newbury-Ecob, R., and Ellard, S. (2011) Exome sequencing identifies
a DYNC1H1 mutation in a large pedigree with dominant axonal
Charcot–Marie–Tooth disease, Am. J. Hum. Genet.,
89, 308-312.
60.Puls, I., Jonnakuty, C., LaMonte, B. H.,
Holzbaur, E. L., Tokito, M., Mann, E., Floeter, M. K., Bidus, K.,
Drayna, D., Oh, S. J., Brown, R. H., Jr., Ludlow, C. L., and Fischbeck,
K. H. (2003) Mutant dynactin in motor neuron disease, Nat.
Genet., 33, 455-456.
61.Zhao, C., Takita, J., Tanaka, Y., Setou, M.,
Nakagawa, T., Takeda, S., Yang, H. W., Terada, S., Nakata, T., Takei,
Y., Saito, M., Tsuji, S., Hayashi, Y., and Hirokawa, N. (2001)
Charcot–Marie–Tooth disease type 2A caused by mutation in a
microtubule motor KIF1Bβ, Cell, 105, 587-597.
62.Wang, T., Ming, Z., Xiaochun, W., and Hong, W.
(2011) Rab7: role of its protein interaction cascades in endo-lysosomal
traffic, Cell. Signal., 23, 516-521.
63.Verhoeven, K., De Jonghe, P., Coen, K.,
Verpoorten, N., Auer-Grumbach, M., Kwon, J. M., FitzPatrick, D.,
Schmedding, E., De Vriendt, E., Jacobs, A., Van Gerwen, V., Wagner, K.,
Hartung, H. P., and Timmerman, V. (2003) Mutations in the small GTP-ase
late endosomal protein RAB7 cause Charcot–Marie–Tooth type
2B neuropathy, Am. J. Hum. Genet., 72, 722-727.
64.De Jonghe, P., Mersivanova, I., Nelis, E., Del
Favero, J., Martin, J. J., Van Broeckhoven, C., Evgrafov, O., and
Timmerman, V. (2001) Further evidence that neurofilament light chain
gene mutations can cause Charcot–Marie–Tooth disease type
2E, Ann. Neurol., 49, 245-249.
65.Jordanova, A., De Jonghe, P., Boerkoel, C. F.,
Takashima, H., De Vriendt, E., Ceuterick, C., Martin, J. J., Butler, I.
J., Mancias, P., Papasozomenos, S., Terespolsky, D., Potocki, L.,
Brown, C. W., Shy, M., Rita, D. A., Tournev, I., Kremensky, I., Lupski,
J. R., and Timmerman, V. (2003) Mutations in the neurofilament light
chain gene (NEFL) cause early onset severe
Charcot–Marie–Tooth disease, Brain, 126,
590-597.
66.Mersiyanova, I. V., Perepelov, A. V., Polyakov,
A. V., Sitnikov, V. F., Dadali, E. L., Oparin, R. B., Petrin, A. N.,
and Evgrafov, O. V. (2000) A new variant of
Charcot–Marie–Tooth disease type 2 is probably the result
of a mutation in the neurofilament-light gene, Am. J. Hum.
Genet., 67, 37-46.
67.Benndorf, R., Martin, J. L., Kosakovsky Pond, S.
L., and Wertheim, J. O. (2014) Neuropathy- and myopathy-associated
mutations in human small heat shock proteins: characteristics and
evolutionary history of the mutation sites, Mutat. Res., doi:
10.1016/j.mrrev.2014.02.004.
68.Houlden, H., Laura, M., Wavrant de Vrieze, F.,
Blake, J., Wood, N., and Reilly, M. M. (2008) Mutations in the HSP27
(HSPB1) gene cause dominant, recessive, and sporadic distal HMN/CMT
type 2, Neurology, 71, 1660-1668.
69.Capponi, S., Geroldi, A., Fossa, P., Grandis, M.,
Ciotti, P., Gulli, R., Schenone, A., Mandich, P., and Bellone, E.
(2011) HSPB1 and HSPB8 in inherited neuropathies: study of an Italian
cohort of dHMN and CMT2 patients, J. Peripher. Nerv. Syst.,
16, 287-294.
70.Muranova, L. K., Weeks, S. D., Strelkov, S. V.,
and Gusev, N. B. (2015) Characterization of mutants of human small heat
shock protein HspB1 carrying replacements in the N-terminal
domain and associated with hereditary motor neuron diseases, PLoS
One, 10, e0126248.
71.James, P. A., Rankin, J., and Talbot, K. (2008)
Asymmetrical late onset motor neuropathy associated with a novel
mutation in the small heat shock protein HSPB1 (HSP27), J. Neurol.
Neurosurg. Psychiatry, 79, 461-463.
72.Nefedova, V. V., Sudnitsyna, M. V., Strelkov, S.
V., and Gusev, N. B. (2013) Structure and properties of G84R and L99M
mutants of human small heat shock protein HspB1 correlating with motor
neuropathy, Arch. Biochem. Biophys., 538, 16-24.
73.Evgrafov, O. V., Mersiyanova, I., Irobi, J., Van
den Bosch, L., Dierick, I., Leung, C. L., Schagina, O., Verpoorten, N.,
Van Impe, K., Fedotov, V., Dadali, E., Auer-Grumbach, M.,
Windpassinger, C., Wagner, K., Mitrovic, Z., Hilton-Jones, D., Talbot,
K., Martin, J. J., Vasserman, N., Tverskaya, S., Polyakov, A., Liem, R.
K., Gettemans, J., Robberecht, W., De Jonghe, P., and Timmerman, V.
(2004) Mutant small heat-shock protein 27 causes axonal
Charcot–Marie–Tooth disease and distal hereditary motor
neuropathy, Nat. Genet., 36, 602-606.
74.Tang, B., Liu, X., Zhao, G., Luo, W., Xia, K.,
Pan, Q., Cai, F., Hu, Z., Zhang, C., Chen, B., Zhang, F., Shen, L.,
Zhang, R., and Jiang, H. (2005) Mutation analysis of the small heat
shock protein 27 gene in chinese patients with
Charcot–Marie–Tooth disease, Arch. Neurol.,
62, 1201-1207.
75.Dierick, I., Baets, J., Irobi, J., Jacobs, A., De
Vriendt, E., Deconinck, T., Merlini, L., Van den Bergh, P., Rasic, V.
M., Robberecht, W., Fischer, D., Morales, R. J., Mitrovic, Z., Seeman,
P., Mazanec, R., Kochanski, A., Jordanova, A., Auer-Grumbach, M.,
Helderman van den Enden, A. T., Wokke, J. H., Nelis, E., De Jonghe, P.,
and Timmerman, V. (2008) Relative contribution of mutations in genes
for autosomal dominant distal hereditary motor neuropathies: a
genotype-phenotype correlation study, Brain, 131,
1217-1227.
76.Stancanelli, C., Fabrizi, G. M., Ferrarini, M.,
Cavallaro, T., Taioli, F., Di Leo, R., Russo, M., Gentile, L., Toscano,
A., Vita, G., and Mazzeo, A. (2014) Charcot–Marie–Tooth 2F:
phenotypic presentation of the Arg136Leu HSP27 mutation in a
multigenerational family, Neurol. Sci., 36,
1003-1006.
77.Almeida-Souza, L., Goethals, S., De Winter, V.,
Dierick, I., Gallardo, R., Van Durme, J., Irobi, J., Gettemans, J.,
Rousseau, F., Schymkowitz, J., Timmerman, V., and Janssens, S. (2010)
Increased monomerization of mutant HSPB1 leads to protein hyperactivity
in Charcot–Marie–Tooth neuropathy, J. Biol. Chem.,
285, 12778-12786.
78.Holmgren, A., Bouhy, D., De Winter, V.,
Asselbergh, B., Timmermans, J. P., Irobi, J., and Timmerman, V. (2013)
Charcot–Marie–Tooth causing HSPB1 mutations increase
Cdk5-mediated phosphorylation of neurofilaments, Acta
Neuropathol., 126, 93-108.
79.Srivastava, A. K., Renusch, S. R., Naiman, N. E.,
Gu, S., Sneh, A., Arnold, W. D., Sahenk, Z., and Kolb, S. J. (2012)
Mutant HSPB1 overexpression in neurons is sufficient to cause
age-related motor neuronopathy in mice, Neurobiol. Dis.,
47, 163-173.
80.Almeida-Souza, L., Asselbergh, B.,
D’Ydewalle, C., Moonens, K., Goethals, S., de Winter, V., Azmi,
A., Irobi, J., Timmermans, J. P., Gevaert, K., Remaut, H., Van den
Bosch, L., Timmerman, V., and Janssens, S. (2011) Small heat-shock
protein HSPB1 mutants stabilize microtubules in
Charcot–Marie–Tooth neuropathy, J. Neurosci.,
31, 15320-15328.
81.D’Ydewalle, C., Krishnan, J., Chiheb, D.
M., Van Damme, P., Irobi, J., Kozikowski, A. P., Van den Berghe, P.,
Timmerman, V., Robberecht, W., and Van den Bosch, L. (2011) HDAC6
inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced
Charcot–Marie–Tooth disease, Nat. Med., 17,
968-974.
82.Almeida-Souza, L., Timmerman, V., and Janssens,
S. (2011) Microtubule dynamics in the peripheral nervous system: a
matter of balance, Bioarchitecture, 1, 267-270.
83.Fontaine, J. M., Sun, X., Hoppe, A. D., Simon,
S., Vicart, P., Welsh, M. J., and Benndorf, R. (2006) Abnormal small
heat shock protein interactions involving neuropathy-associated HSP22
(HSPB8) mutants, FASEB J., 20, 2168-2170.
84.Ikeda, Y., Abe, A., Ishida, C., Takahashi, K.,
Hayasaka, K., and Yamada, M. (2009) A clinical phenotype of distal
hereditary motor neuronopathy type II with a novel HSPB1 mutation,
J. Neurol. Sci., 277, 9-12.
85.Nefedova, V. V., Datskevich, P. N., Sudnitsyna,
M. V., Strelkov, S. V., and Gusev, N. B. (2013) Physico-chemical
properties of R140G and K141Q mutants of human small heat shock protein
HspB1 associated with hereditary peripheral neuropathies,
Biochimie, 95, 1582-1592.
86.Hansen, L., Yao, W., Eiberg, H., Kjaer, K. W.,
Baggesen, K., Hejtmancik, J. F., and Rosenberg, T. (2007) Genetic
heterogeneity in microcornea-cataract: five novel mutations in CRYAA,
CRYGD, and GJA8, Invest. Ophthalmol. Vis. Sci., 48,
3937-3944.
87.Litt, M., Kramer, P., LaMorticella, D. M.,
Murphey, W., Lovrien, E. W., and Weleber, R. G. (1998) Autosomal
dominant congenital cataract associated with a missense mutation in the
human α-crystallin gene CRYAA, Hum. Mol. Genet., 7,
471-474.
88.Vicart, P., Caron, A., Guicheney, P., Li, Z.,
Prevost, M. C., Faure, A., Chateau, D., Chapon, F., Tome, F., Dupret,
J. M., Paulin, D., and Fardeau, M. (1998) A missense mutation in the
αB-crystallin chaperone gene causes a desmin-related myopathy,
Nat. Genet., 20, 92-95.
89.Inagaki, N., Hayashi, T., Arimura, T., Koga, Y.,
Takahashi, M., Shibata, H., Teraoka, K., Chikamori, T., Yamashina, A.,
and Kimura, A. (2006) αB-crystallin mutation in dilated
cardiomyopathy, Biochem. Biophys. Res. Commun., 342,
379-386.
90.Irobi, J., Van Impe, K., Seeman, P., Jordanova,
A., Dierick, I., Verpoorten, N., Michalik, A., De Vriendt, E., Jacobs,
A., Van Gerwen, V., Vennekens, K., Mazanec, R., Tournev, I.,
Hilton-Jones, D., Talbot, K., Kremensky, I., Van den Bosch, L.,
Robberecht, W., Van Vandekerckhove, J., Van Broeckhoven, C., Gettemans,
J., De Jonghe, P., and Timmerman, V. (2004) Hot-spot residue in small
heat-shock protein 22 causes distal motor neuropathy, Nat.
Genet., 36, 597-601.
91.Chalova, A. S., Sudnitsyna, M. V., Strelkov, S.
V., and Gusev, N. B. (2014) Characterization of human small heat shock
protein HspB1 that carries C-terminal domain mutations
associated with hereditary motor neuron diseases, Biochim. Biophys.
Acta, 1844, 2116-2126.
92.Lin, K. P., Soong, B. W., Yang, C. C., Huang, L.
W., Chang, M. H., Lee, I. H., Antonellis, A., and Lee, Y. C. (2011) The
mutational spectrum in a cohort of Charcot–Marie–Tooth
disease type 2 among the Han Chinese in Taiwan, PLoS One,
6, e29393.
93.Luigetti, M., Fabrizi, G. M., Madia, F.,
Ferrarini, M., Conte, A., Del Grande, A., Tasca, G., Tonali, P. A., and
Sabatelli, M. (2010) A novel HSPB1 mutation in an Italian patient with
CMT2/dHMN phenotype, J. Neurol. Sci., 298, 114-117.
94.Kijima, K., Numakura, C., Goto, T., Takahashi,
T., Otagiri, T., Umetsu, K., and Hayasaka, K. (2005) Small heat shock
protein 27 mutation in a Japanese patient with distal hereditary motor
neuropathy, J. Hum. Genet., 50, 473-476.
95.Ackerley, S., James, P. A., Kalli, A., French,
S., Davies, K. E., and Talbot, K. (2006) A mutation in the small
heat-shock protein HSPB1 leading to distal hereditary motor
neuronopathy disrupts neurofilament assembly and the axonal transport
of specific cellular cargoes, Hum. Mol. Genet., 15,
347-354.
96.Delbecq, S. P., Jehle, S., and Klevit, R. (2012)
Binding determinants of the small heat shock protein,
αB-crystallin: recognition of the “IxI” motif,
EMBO J., 31, 4587-4594.
97.Delbecq, S. P., and Klevit, R. E. (2013) One size
does not fit all: the oligomeric states of αB crystallin, FEBS
Lett., 587, 1073-1080.
98.Hochberg, G. K., Ecroyd, H., Liu, C., Cox, D.,
Cascio, D., Sawaya, M. R., Collier, M. P., Stroud, J., Carver, J. A.,
Baldwin, A. J., Robinson, C. V., Eisenberg, D. S., Benesch, J. L., and
Laganowsky, A. (2014) The structured core domain of αB-crystallin
can prevent amyloid fibrillation and associated toxicity, Proc.
Natl. Acad. Sci. USA, 111, 1562-1570.
99.Treweek, T. M., Rekas, A., Walker, M. J., and
Carver, J. A. (2010) A quantitative NMR spectroscopic examination of
the flexibility of the C-terminal extensions of the molecular
chaperones, αA- and αB-crystallin, Exp. Eye Res.,
91, 691-699.
100.Lindner, R. A., Carver, J. A., Ehrnsperger, M.,
Buchner, J., Esposito, G., Behlke, J., Lutsch, G., Kotlyarov, A., and
Gaestel, M. (2000) Mouse Hsp25, a small shock protein. The role of its
C-terminal extension in oligomerization and chaperone action,
Eur. J. Biochem., 267, 1923-1932.
101.Morris, A. M., Treweek, T. M., Aquilina, J. A.,
Carver, J. A., and Walker, M. J. (2008) Glutamic acid residues in the
C-terminal extension of small heat shock protein 25 are critical
for structural and functional integrity, FEBS J., 275,
5885-5898.
102.Kolb, S. J., Snyder, P. J., Poi, E. J., Renard,
E. A., Bartlett, A., Gu, S., Sutton, S., Arnold, W. D., Freimer, M. L.,
Lawson, V. H., Kissel, J. T., and Prior, T. W. (2010) Mutant small heat
shock protein B3 causes motor neuropathy: utility of a candidate gene
approach, Neurology, 74, 502-506.
103.Asthana, A., Raman, B., Ramakrishna, T., and
Rao, C. M. (2012) Structural aspects and chaperone activity of human
HspB3: role of the “C-terminal extension”, Cell
Biochem. Biophys., 64, 61-72.
104.Den Engelsman, J., Boros, S., Dankers, P. Y.,
Kamps, B., Vree Egberts, W. T., Bode, C. S., Lane, L. A., Aquilina, J.
A., Benesch, J. L., Robinson, C. V., De Jong, W. W., and Boelens, W. C.
(2009) The small heat-shock proteins HSPB2 and HSPB3 form well-defined
heterooligomers in a unique 3 to 1 subunit ratio, J. Mol. Biol.,
393, 1022-1032.
105.Prabhu, S., Raman, B., Ramakrishna, T., and
Rao, Ch. M. (2012) HspB2/myotonic dystrophy protein kinase binding
protein (MKBP) as a novel molecular chaperone: structural and
functional aspects, PLoS One, 7, e29810.
106.Hu, Z., Yang, B., Lu, W., Zhou, W., Zeng, L.,
Li, T., and Wang, X. (2008) HSPB2/MKBP, a novel and unique member of
the small heat-shock protein family, J. Neurosci. Res.,
86, 2125-2133.
107.Tang, B. S., Zhao, G. H., Luo, W., Xia, K.,
Cai, F., Pan, Q., Zhang, R. X., Zhang, F. F., Liu, X. M., Chen, B.,
Zhang, C., Shen, L., Jiang, H., Long, Z. G., and Dai, H. P. (2005)
Small heat-shock protein 22 mutated in autosomal dominant
Charcot–Marie–Tooth disease type 2L, Hum. Genet.,
116, 222-224.
108.Nakhro, K., Park, J. M., Kim, Y. J., Yoon, B.
R., Yoo, J. H., Koo, H., Choi, B. O., and Chung, K. W. (2013) A novel
Lys141Thr mutation in small heat shock protein 22 (HSPB8) gene in
Charcot–Marie–Tooth disease type 2L, Neuromuscul.
Disord., 23, 656-663.
109.Clark, A. R., Naylor, C. E., Bagneris, C.,
Keep, N. H., and Slingsby, C. (2011) Crystal structure of R120G disease
mutant of human αB-crystallin domain dimer shows closure of a
groove, J. Mol. Biol., 408, 118-134.
110.Kasakov, A. S., Bukach, O. V., Seit-Nebi, A.
S., Marston, S. B., and Gusev, N. B. (2007) Effect of mutations in the
β5-β7 loop on the structure and properties of human small
heat shock protein HSP22 (HspB8, H11), FEBS J., 274,
5628-5642.
111.Kim, M. V., Kasakov, A. S., Seit-Nebi, A. S.,
Marston, S. B., and Gusev, N. B. (2006) Structure and properties of
K141E mutant of small heat shock protein HSP22 (HspB8, H11) that is
expressed in human neuromuscular disorders, Arch. Biochem.
Biophys., 454, 32-41.
112.Irobi, J., Almeida-Souza, L., Asselbergh, B.,
De Winter, V., Goethals, S., Dierick, I., Krishnan, J., Timmermans, J.
P., Robberecht, W., De Jonghe, P., Van den Bosch, L., Janssens, S., and
Timmerman, V. (2010) Mutant HSPB8 causes motor neuron-specific neurite
degeneration, Hum. Mol. Genet., 19, 3254-3265.
113.Irobi, J., Holmgren, A., Winter, V. D.,
Asselbergh, B., Gettemans, J., Adriaensen, D., Groote, C. C., Coster,
R. V., Jonghe, P. D., and Timmerman, V. (2012) Mutant HSPB8 causes
protein aggregates and a reduced mitochondrial membrane potential in
dermal fibroblasts from distal hereditary motor neuropathy patients,
Neuromuscul. Disord., doi.10.1016/j.nmd.2012.04.005.
114.Kwok, A. S., Phadwal, K., Turner, B. J.,
Oliver, P. L., Raw, A., Simon, A. K., Talbot, K., and Agashe, V. R.
(2011) HspB8 mutation causing hereditary distal motor neuropathy
impairs lysosomal delivery of autophagosomes, J. Neurochem.,
119, 1155-1161.
115.Vicario, M., Skaper, S. D., and Negro, A.
(2014) The small heat shock protein HspB8: role in nervous system
physiology and pathology, CNS Neurol. Disord. Drug Targets,
13, 885-895.
116.Carra, S., Boncoraglio, A., Kanon, B.,
Brunsting, J. F., Minoia, M., Rana, A., Vos, M. J., Seidel, K., Sibon,
O. C., and Kampinga, H. H. (2010) Identification of the
Drosophila ortholog of HSPB8: implication of HSPB8 loss of
function in protein folding diseases, J. Biol. Chem.,
285, 37811-37822.
117.Shemetov, A. A., and Gusev, N. B. (2011)
Biochemical characterization of small heat shock protein HspB8
(Hsp22)–Bag3 interaction, Arch. Biochem. Biophys.,
513, 1-9.
118.Charroux, B., Pellizzoni, L., Perkinson, R. A.,
Shevchenko, A., Mann, M., and Dreyfuss, G. (1999) Gemin3: a novel DEAD
box protein that interacts with SMN, the spinal muscular atrophy gene
product, and is a component of gems, J. Cell Biol., 147,
1181-1194.
119.Sun, X., Fontaine, J. M., Hoppe, A. D., Carra,
S., De Guzman, C., Martin, J. L., Simon, S., Vicart, P., Welsh, M. J.,
Landry, J., and Benndorf, R. (2010) Abnormal interaction of motor
neuropathy-associated mutant HspB8 (Hsp22) forms with the RNA helicase
Ddx20 (gemin3), Cell Stress Chaperones, 15, 567-582.