REVIEW: Sperm-Specific Glyceraldehyde-3-Phosphate Dehydrogenase – An Evolutionary Acquisition of Mammals
V. I. Muronetz1,2*, M. L. Kuravsky1, K. V. Barinova1,2, and E. V. Schmalhausen1
1Lomonosov Moscow State University, Belozersky Institute of Physico-Chemical Biology, 119991 Moscow, Russia2Lomonosov Moscow State University, Faculty of Bioengineering and Bioinformatics, 119991 Moscow, Russia; E-mail: vimuronets@belozersky.msu.ru
* To whom correspondence should be addressed.
Received September 7, 2015
This review is focused on the mammalian sperm-specific glyceraldehyde-3-phosphate dehydrogenase (GAPDS). GAPDS plays the major role in the production of energy required for sperm cell movement and does not perform non-glycolytic functions that are characteristic of the somatic isoenzyme of glyceraldehyde-3-phosphate dehydrogenase. The GAPDS sequence is composed of 408 amino acid residues and includes an additional N-terminal region of 72 a.a. that binds the protein to the sperm tail cytoskeleton. GAPDS is present only in the sperm cells of mammals and lizards, possibly providing them with certain evolutionary advantages in reproduction. In this review, studies concerning the problems of GAPDS isolation, its catalytic properties, and its structural features are described in detail. GAPDS is much more stable compared to the somatic isoenzyme, perhaps due to the necessity of maintaining the enzyme function in the absence of protein expression. The site-directed mutagenesis approach revealed the two GAPDS-specific proline residues, as well as three salt bridges, which seem to be the basis of the increased stability of this protein. As distinct from the somatic isoenzyme, GAPDS exhibits positive cooperativity in binding of the coenzyme NAD+. The key role in transduction of structural changes induced by NAD+ is played by the salt bridge D311–H124. Disruption of this salt bridge cancels GAPDS cooperativity and twofold increases its enzymatic activity instead. The expression of GAPDS was detected in some melanoma cells as well. Its role in the development of certain pathologies, such as cancer and neurodegenerative diseases, is discussed.
KEY WORDS: glyceraldehyde-3-phosphate dehydrogenase, sperm-specific glyceraldehyde-3-phosphate dehydrogenase, GAPDH, evolution of GAPDH, stability of GAPDH, sperm motility, glycolysis, melanoma cells, oncomarker, NAD-bindingDOI: 10.1134/S0006297915130040
Abbreviations: GAPD, somatic glyceraldehyde-3-phosphate dehydrogenase; GAPDS, sperm-specific glyceraldehyde-3-phosphate dehydrogenase; GdnHCl, guanidine hydrochloride; Km, Michaelis constant.
Glyceraldehyde-3-phosphate dehydrogenase (GAPD) is one of the most
common and well-studied enzymes. By now, the mechanism of oxidative
phosphorylation of glyceraldehyde-3-phosphate has been examined in
every detail, and so are the structures of glyceraldehyde-3-phosphate
dehydrogenases isolated from a number of organisms – from
bacteria to humans. The native GAPD protein molecule consists of four
identical subunits of 36 kDa and has four active centers, each
comprising the coenzyme- and the substrate-binding regions. GAPD
catalyzes the reaction of glycolytic oxidoreduction resulting in the
first macroergic compound of glycolysis – 1,3-diphosphoglycerate,
which is later metabolized to produce ATP. The GAPD-catalyzed reaction
also leads to the reduction of NAD+ to NADH, which is a
substrate of oxidative phosphorylation in eukaryotic mitochondria. The
present review centers on the animal GAPD, which has been demonstrated
to be a multifunctional protein. Moreover, some animals have acquired
an additional sperm-specific isoenzyme of GAPD, which will be the main
topic of discussion in this work. However, before we proceed to this
isoenzyme, we must review the properties of somatic GAPD first.
GAPD is known to be present in the cytoplasm of all somatic cells of animals in huge concentration – its content reaches from 5 to 15% of all soluble proteins. GAPD is a constitutively expressed protein encoded by a so-called “housekeeping gene”. For that reason, the GAPD protein and its mRNA are commonly used as standards in protein expression studies. For a long time, it was assumed that GAPD was not recruited for regulatory purposes and that there was no connection between the activity of this enzyme and the development of pathologies. However, data appeared on non-glycolytic functions of GAPD. It was found to participate in a number of processes that might be connected with various pathological states: endocytosis [1-3], plasmatic membrane fusion [4], microtubule assembly [5-7], transport of secretory vesicles [8, 9], protein phosphorylation [10, 11], control of gene expression at the levels of both transcription and translation [12-14], regulation of the structure of telomeres [15, 16], nuclear membrane fusion [17], mRNA translocation through the nuclear membrane [18], DNA excision repair [19, 20], and apoptosis induction [21-25]. GAPD can also be involved in the development of neurodegenerative diseases such as Alzheimer’s disease [26-28] and Huntington’s disease [28-30].
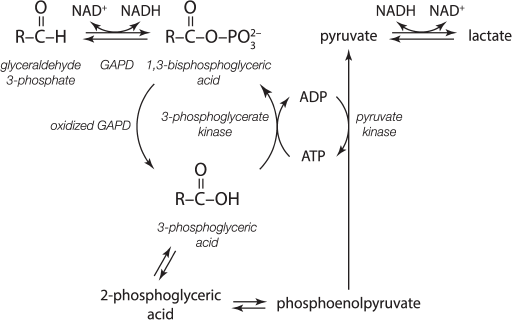
We must also point to the possibility of GAPD recruitment in the development of cancer. Disturbances in induction of apoptosis and regulation of glycolysis are among the other abnormalities characteristic for cancer cells. GAPD can be involved in both processes. For example, the Pasteur effect (suppression of glycolysis under aerobic conditions) is not characteristic for cancer cells. Glycolysis runs fast in cancer cells and is not slowed by oxygen. Such a change in metabolic regulation is known as the Warburg effect. Following Warburg, the switch to anaerobic-type metabolism results in autonomous uncontrolled living of a cell: it starts to behave as an independent organism aiming to reproduce itself [31, 32]. The acylphosphatase activity of partially oxidized GAPD, which was discovered in our laboratory, can play a certain role in coupling of glycolysis with oxidative phosphorylation. We showed that the partial oxidation of GAPD uncouples glycolytic oxidation and phosphorylation in normal cells, which allows coordination of the reactions of oxidative phosphorylation with the reactions of glycolysis (Fig. 1) [33, 34].
Fig. 1. Futile cycle of glycolysis – uncoupling of oxidation and phosphorylation [33, 34].
Moreover, in our studies, as well as in studies carried out in other laboratories, GAPD has been shown to form complexes with other glycolytic enzymes [35-38], as well as with structural elements of the cell, which can regulate the rate of glycolytic flux [5, 39-41]. Evidently, changes in GAPD properties or expression of its isoenzymes in cancer cells can alter the coupling of glycolysis and oxidative phosphorylation and GAPD-specific protein–protein interactions, leading to the Warburg effect or any other abnormality of energy metabolism.
During the last 15 years, many data about the participation of GAPD in the induction of apoptosis have been published [21, 42-45]. In normal cells, enzymatically active tetrameric molecules localized in the cytoplasm represent most of the GAPD. A certain amount of GAPD is adsorbed on structural elements of cells, such as the actin stress fibrils, microtubules, etc. Perhaps, a portion of the adsorbed GAPD is represented by dimers and monomers, which allows its identification by monoclonal antibodies specific to non-native forms of this protein. The situation changes upon exposure of cells to cytotoxic factors and induction of apoptosis by tumor necrosis factor or hydrogen peroxide. In these cases, the dimeric and monomeric GAPD molecules tend to translocate to the nucleus, as shown in Fig. 2.
Fig. 2. Hypothetical scheme representing the intracellular translocation of somatic GAPD forms in the case of oxidative stress.
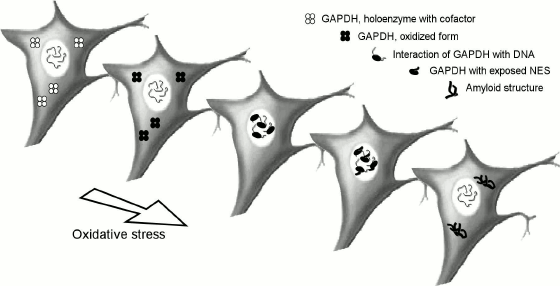
The scheme represents processes occurring on induction of apoptosis by reactive oxygen species. In the first step, the sulfhydryl groups in the active centers of GAPD are oxidized, which leads to a decrease in the coenzyme NAD+ binding affinity. The resulting apoenzyme of GAPD dissociates into subunits, which then are passively transported into the nucleus. The protein is further unfolded to expose the nuclear export signal (NES) upon its binding to intranuclear nucleic acids. The denatured polypeptide chains of GAPD are then transported to the cytoplasm, where they can undergo some further changes, e.g. aggregating, binding to chaperones, or becoming ubiquitinated. The transition of the denatured GAPD species from cytoplasm to nucleus is also characteristic for some cancer cells such as HeLa.
During the induction of apoptosis, GAPD evidently acts not as a glycolytic enzyme, but as a protein messenger, participating in a number of protein–protein and protein–ligand interactions. An important role is reserved for the strong and cooperative binding of the coenzyme NAD+, the possibility of regulating this binding by the modification of the active center sulfhydryl groups and, finally, to the nuclear export signal. Also, it should be pointed out that all these features are characteristic for the somatic isoenzyme of glyceraldehyde-3-phosphate dehydrogenase, which was the studied object in most of the studies. The absence of any of those features should considerably affect both energy metabolism and the GAPD-dependent apoptosis induction pathways.
As mentioned above, the multifunctionality of GAPD is known to be characteristic for the somatic isoenzyme of mammals, since the experiments were mostly carried out using either human or rabbit protein. In brief, in mammalian cells, a cytoplasmic protein is present, which performs not only glycolytic oxidoreduction, but also many other functions. Perhaps, some of the enzymatic features of the mammalian somatic GAPD (such as, e.g. negative cooperativity in coenzyme NAD+ binding, very high affinities for the first two NAD+ molecules, high reactivity of the active center sulfhydryl groups, etc.) are necessary not only for catalysis, but also for the additional functions. Unfortunately, data about the non-glycolytic GAPD functions in non-mammalian species are virtually absent. For that reason, it is not possible to follow the evolution of the non-glycolytic GAPD functions and their correlation with the protein structure.
However, mammals are known to possess another GAPD isoenzyme, which is the sperm-specific glyceraldehyde-3-phosphate dehydrogenase (GAPDS). Many of the functions of the somatic isoenzyme are not necessary for GAPDS (e.g. processes involving translocation of the protein through the nuclear membrane or its interactions with nucleic acids). Therefore, comparative analysis of the two isoenzymes would reveal the structural elements that are responsible for the functions specific to each of them. Such analysis could also facilitate the use of novel approaches for the enzyme rational design and help to obtain new biocatalysts with predicted characteristics for purposes of biotechnology by the use of the site-specific mutagenesis.
In this review, we would also like to pay attention to another aspect that concerns some pathological processes in human organism. GAPDS is known to have specialized to maintain glycolysis and provide energy for the sperm tail movement. This isoenzyme lacks most of the non-glycolytic functions of its somatic homolog. These functions are not necessary in sperm cells, but the expression of GAPDS in somatic cells must significantly affect their metabolism and survival. In our studies, we demonstrated GAPDS being expressed in certain cancer cells, which is likely to affect their energy metabolism and the induction of apoptosis [46]. Perhaps, the expression of GAPDS in neural cells has its part in the development of neurodegenerative diseases of amyloid nature [47].
Taken together, in this review we will focus on the basic properties of GAPDS, its role in energy supply of sperm cell movement, its evolution, and its possible participation in the development of pathological states when expressed in somatic cells.
SPERM-SPECIFIC GLYCERALDEHYDE-3-PHOSPHATE DEHYDROGENASE
GAPDS is a sperm-specific isoenzyme of glyceraldehyde-3-phosphate dehydrogenase whose expression is limited to sperm cells. GAPDS is encoded by a separate gene located on chromosome 19, and it is homologous to the gene of the somatic GAPD located on chromosome 12 [48, 49]. The sequences of GAPDS and the somatic GAPD are 68% identical. GAPDS is built up of 408 amino acid residues and has an additional N-terminal region of 72 a.a. that is responsible for binding to the cytoskeleton of the sperm tail [50]. The molecular mass of GAPDS predicted from its sequence is about 44.5 kDa, but due to the enrichment in proline residues, the GAPDS protein molecule moves anomalously slow in the SDS-PAGE gels and results in a band at the level of about 56 kDa [49].
Previously, we demonstrated that the sequence motifs responsible for glycolytic functions are common for both somatic GAPD and GAPDS. We also revealed some specific motifs responsible for the nuclear functions (DNA repair and replication) and a signal determining the intracellular localization in the sequence of the somatic GAPD. The absence of those motifs in the GAPDS sequence leads us to the conclusion that this isoenzyme does not perform the nuclear functions that are characteristic for the somatic enzyme. Additional evidence comes from the fact that GAPDS is tightly bound to the insoluble components of a cell via its N-terminal domain, which may prevent it from performing some of the additional functions as well. Perhaps GAPDS participates in the formation of mitotic spindle in spermatids, while this is not characteristic for the somatic GAPD [51].
GAPDS EVOLUTION
In our studies, the evolutionary history of the glyceraldehyde-3-phosphate dehydrogenase gene family was investigated. The GAPD-1 and GAPD-2 isoenzymes (corresponding to the mammalian somatic GAPD and GAPDS, respectively) were found to have orthologs in other lineages of vertebrates [51, 52]. The divergence of genes that encode those isoenzymes occurred in the course of the early evolution of chordates and was not due to either of the two vertebrate-specific full-genome duplications. The GAPD-1 gene encoding the somatic isoenzyme then underwent additional duplications in certain lineages, while the GAPD-2 gene was lost by many species and now is retained only by mammals, lizards, and fishes.
The main trend in the evolution of the vertebrate GAPD-1 and GAPD-2 was the functional divergence. The data provide evidence for the fish isoenzymes of glyceraldehyde-3-phosphate dehydrogenase, which are expressed in the same tissues, being specialized for performing the different non-glycolytic functions (the mammalian GAPDS isoenzyme is also likely not to perform at least some of the non-glycolytic functions of somatic GAPD). Moreover, the GAPD-2 isoenzyme of mammals and lizards evolved into a sperm-specific protein and acquired an additional proline-rich domain to attach to the sperm tail cytoskeleton. This attachment seems to be mediated by a nonspecific interaction that involves at least two different cytoskeletal proteins. In lizards, GAPD-2 is expressed in some somatic tissues as well (those with rapidly dividing cells), but the form lacking the proline-rich domain due to alternative splicing of its mRNA.
To conclude, the sperm-specific isoenzyme of glyceraldehyde-3-phosphate dehydrogenase is present in the sperm cells of mammals and lizards and supplies the sperm tail with the energy required for its movement. This acquisition should provide an evolutionary advantage for the reproduction of those organisms. It is interesting that the expression of GAPD-2 in the regenerating tissues of lizards has a side effect connected with the supply of rapidly dividing cells with glycolytic fuel. In turn, the expression of GAPDS in the somatic tissues of mammals might lead to pathologies, which will be discussed in the end of this review.
ROLE OF GLYCOLYSIS AND GAPDS IN ENERGY SUPPLY OF SPERM
CELLS
Sperm cell movement requires much energy of ATP hydrolysis, which is consumed by the motor protein dynein [53]. In animal cells, ATP is mostly generated in the course of the two processes: glycolysis and oxidative phosphorylation.
Localization of mitochondria in the mid piece of the sperm tail raised doubts about the ATP diffusion rate being fast enough to meet with the energy requirements of its distal part [54, 55]. It was calculated that the diffusion of ATP produced in the course of oxidative phosphorylation can cover ATP consumption in sperm cells with shorter tails, such as those of the sea urchin Strongylocentrotus purpuratus (40 µm) [56, 57]. However, the sperm cells of mammals generally have much longer tails. For example, the sperm tails of rodents can exceed 105 µm [58]. Moreover, the models used for the calculations treated ATP apart from ADP and inorganic phosphate. The use of energy accumulated in the form of ATP is known to be effective only when the reaction ATP ↔ ADP + Pi is shifted away from equilibrium. In the cytoplasm of a somatic animal cell, the Pi concentration is maintained at the level of 10 mM, while the ATP/ADP ratio is about 105 times higher than in equilibrium [59]. In the distal part of the sperm tail, the ATP concentration is lower than in the proximal part, where the ADP and Pi concentrations should be higher. Therefore, the reaction of the ATP hydrolysis should be shifted towards equilibrium, thus being less effective [55].
In the mid-1980s, the hypothesis of the creatine phosphate energy shuttle was proposed. The reaction ATP + creatine ↔ ADP + creatine phosphate can maintain the ATP/ADP ratio at a relatively constant level. Creatine phosphate is also able to provide a more efficient energy transfer along the sperm tail because of its higher diffusional mobility [60]. Creatine kinase, an enzyme catalyzing the transfer of the phosphate group between ATP and creatine, was later discovered in the sperm cells of the sea urchin [61]. However, in the sperm cells of all examined mammalian species, this enzyme either was not found or demonstrated decreased enzymatic activity levels [53, 62, 63]. The creatine phosphate production in mammalian sperms was also shown to be close to zero [64, 65], thus giving the evidence for the creatine phosphate shuttle playing an insignificant role or even not functioning. This conclusion meets with the data on knockout mice lacking creatine kinase: those mice were fertile, and their sperm cells were as motile as those of the wild type animals [62].
As distinct from the sperm cells of the sea urchin, which oxidize fatty acids [66], mammalian sperm cells utilize mainly carbohydrates [67]. Such a fuel makes it possible to generate ATP independently from the mitochondria in the cytoplasm of the sperm tail. The role of glycolysis and oxidative phosphorylation in the energy supply of the sperm tail was investigated by Mukai and colleagues [67]. They used the sperm tail beat frequency as an estimate of the efficacy of its energy supply. The following two indications for most ATP being generated in the course of glycolysis were obtained:
– in the presence of glucose, the addition of the oxidative phosphorylation inhibitors, such as antimycin A and CCCP (carbonylcyanide m-chlorophenylhydrazone), did not affect the beat frequency;
– addition of pyruvate did not increase the beat frequency when glycolysis was inhibited by 2-deoxy-D-glucose (a non-metabolizable glucose analog).
However, in the same study it was found that the sperm cells were able to maintain their motility when pyruvate was the only accessible energy source. To explain this observation, a hypothesis was proposed that pyruvate serves as a substrate for gluconeogenesis, a process known to be localized in mitochondria. The resulting glucose then diffuses along the sperm tail to be metabolized in the course of glycolysis. Since 2-deoxy-D-glucose inhibits not only glycolysis, but also gluconeogenesis [68, 69], it suppresses both pyruvate- and glucose-induced sperm tail beating.
The efficacy of glycolytic flux in sperm cells is secured by the presence of sperm-specific glycolytic isoenzymes that are not present in other tissues. Those isoenzymes are the sperm-specific forms of hexokinase, phosphofructokinase, aldolase, phosphoglycerate kinase, phosphoglycerate mutase, pyruvate kinase, lactate dehydrogenase, as well as the sperm-specific glyceraldehyde-3-phosphate dehydrogenase (GAPDS), whose role will be described below in detail.
The key role of GAPDS in providing energy for sperm cell movement was demonstrated in experiments on mice with GAPDS gene knockout [70]. In the absence of this isoenzyme, the sperm cells did not maintain the glycolytic flux. As the result, the ATP level constituted only 10.4% of normal and the incubation of the sperm cells for 4 h at 37°C decreased it to 2%. The sperm motility was also significantly decreased. The rotation of the sperm tail, which started from its proximal part, did not spread towards the distal part. Such sperm cells were not capable of progressive movement, and the male mice bearing the homozygous mutation were sterile. The described mutation did not affect the consumption of oxygen and the ATP production rate in the course of oxidative phosphorylation. The structure of the sperm tail cytoskeleton was morphologically undisturbed as well. Thus, GAPDS is the only active isoenzyme of glyceraldehyde-3-phosphate dehydrogenase in mature spermatozoa and plays the key role in the energy supply of the sperm cell movement.
We examined the correlation between the enzymatic activity of GAPDS and the motility of sperm cells [71]. The mean activity of GAPDS in low motility sperms was 2.5-3 times lower than in highly motile sperms. The sperm motility decreased in the presence of superoxide anion, hydroxyl radical, or hydrogen peroxide. The decrease in motility upon addition of hydrogen peroxide was proportional to its concentration and correlated with the GAPDS enzymatic activity (r = 0.96). Based on these data, as well as the literature data about the necessity of GAPDS for sperm cell movement, we concluded that the decrease in sperm motility was caused by the oxidation of GAPDS and the consequent inhibition of glycolysis.
The decrease in sperm cell motility upon oxidation of GAPDS is an indicator of the antioxidant defense system being not capable of removing the excess reactive oxygen species, which therefore could damage the genome. Thus, such a decrease can be considered as a natural biological barrier, which prevents fertilization of the egg cell by sperm cells with defective genomes. This means that treatments for enhanced sperm motility must be targeted not against reactive oxygen species, but against the reasons for their excessive production, such as avitaminosis, intoxication of the organism, or inflammatory diseases of the urogenital system.
CATALYTIC PROPERTIES OF GAPDS
Data on the catalytic, regulatory, and physicochemical properties of GAPDS are virtually absent because the purification of sperm-specific enzymes is rather complicated. GAPDS was once purified from boar spermatozoa, but its catalytic characteristics were not examined [72]. Purification of recombinant GAPDS did not succeed because of the formation of the heterotetrameric complexes composed of both the recombinant protein subunits and the E. coli glyceraldehyde-3-phosphate dehydrogenase subunits. Even the crystal structure of GAPDS was first resolved for such a hybrid tetramer [73].
We began our studies with the purification of GAPDS from human sperms [74]. At this stage, we investigated the properties of the enzyme with truncated N-terminus. Later, comparison of its characteristics with those of the recombinant protein lacking the N-terminal domain gave us evidence for the recombinant protein being in the native state.
The recombinant protein was expressed with plasmid DNAs encoding the full-length GAPDS and its truncated version lacking the N-terminal domain (dN-GAPDS). Neither contained any tags, and we used the traditional method of ammonium sulfate fractionation combined with ion-exchange chromatography for their isolation. The extraction of the full-length GAPDS failed because the protein accumulated in insoluble inclusion bodies during expression. Those inclusion bodies did not solubilize even upon addition of detergents. In contrast, we managed to isolate and study the properties of the dN-GAPDS protein. The specific dehydrogenase activity, its pH-dependence, and the Km values for both NAD+ and glyceraldehyde-3-phosphate determined for the recombinant dN-GAPDS do not significantly differ from those of the protein purified from human sperm (Table 1). Both proteins lacked the N-terminal domain. Considering that in vivo the hydrophobic N-terminal domain is bound to the fibrous sheath of the sperm flagellum and does not interact with the catalytic part of the protein, dN-GAPDS seems to be an adequate model for our investigations. It should also be noted that three of the four N-terminal domains of the GAPDS tetramer in sperm cells are likely to be truncated as well due to the limited proteolysis, since the purification of the full-length polypeptide chains did not succeed even after adding detergents. Perhaps, in human sperms, GAPDS exists as a tetramer of a subunit bound to the cytoskeleton and three other subunits without the N-terminal domains. Therefore, dN-GAPDS should resemble the protein that functions in sperms even better.
Table 1. Enzymatic properties of dN-GAPDS
isolated from human sperms, recombinant dN-GAPDS, and the somatic
GAPD
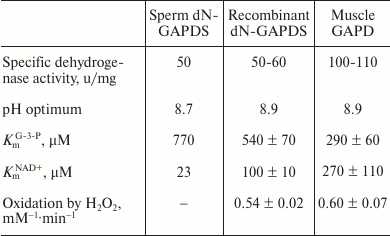
The catalytic characteristics of GAPDS from human sperms, recombinant dN-GAPDS, and the somatic GAPD are present in Table 1. According to these data, the catalytic properties of the somatic and the sperm-specific isoenzymes are different. First, the specific dehydrogenase activity of dN-GAPDS is almost 2-fold lower than the activity of the somatic GAPD, while the optimal pH values are the same. The Km value for glyceraldehyde-3-phosphate determined for dN-GAPDS 2-fold exceeds the value determined for GAPD. In contrast, the Km value for NAD+ determined for dN-GAPDS is about 2.5-fold lower than the value determined for GAPD. The two enzymes exhibited the same sensitivity towards hydrogen peroxide oxidation.
The cooperativity characteristics of the two isoenzymes were the most interesting. The somatic GAPD protein is known to exhibit a quite rare negative cooperativity in NAD+ binding: the first two coenzyme molecules are bound extremely tightly (Kd is equal 10–11-10–7 M), while the last two molecules are bound much weaker (Kd is equal 10–5-10–3 M) [75, 76]. This effect is considered to stabilize the enzyme at the expense of the constantly bound two NAD+ molecules and at the same time enhance its catalytic activity due to the active sites with weaker affinities to the coenzyme. We suppose that negative cooperativity of the somatic GAPD plays a key role in non-glycolytic functions. In our previous work, the oxidation of the active site sulfhydryl groups was shown to decrease the affinity to the cofactor and increase the affinity to nucleic acids. This process is probably linked with the dissociation of somatic GAPD, which precedes its denaturation and participation in the induction of apoptosis under oxidative stress.
According to our data and in contrast to the somatic GAPD, dN-GAPDS exhibited positive cooperativity, which is characteristic for the glyceraldehyde-3-phosphate dehydrogenase from baker’s yeast [77]. The structural basis for this effect was studied by the site-specific mutagenesis approach (see below).
QUARTERNARY STRUCTURE OF GAPDS
During the purification of GAPDS from sperm cells, the enzymatically active protein accumulated in the insoluble fraction of cells after sonication. In our previous work, we demonstrated that the treatment of human sperm debris with trypsin released an active GAPDS fragment of about 150 kDa (by Native Blue PAGE) into the solution because of elimination of the N-terminal domain. At the same time, the molecular mass of the protein subunit determined by SDS-PAGE constituted about 40 kDa. According to our investigations, GAPDS is probably a tetramer composed of four identical subunits [74]. Similar results were obtained for the recombinant dN-GAPDS.
Along with electrophoresis, dynamic light scattering analysis was applied for the detection of oligomeric structure of dN-GAPDS [78]. Purified dN-GAPDS was shown to be a protein 9.1 nm in diameter, which is close to the size of the somatic tetrameric glyceraldehyde-3-phosphate dehydrogenase (8.7 nm). Similar dimensions of the protein indicate that dN-GAPDS, as well as GAPD, is a tetramer.
ROLE OF CHAPERONIN Tric/CCT IN FOLDING OF dN-GAPDS
The cytoplasmic chaperonin Tric/CCT was considered necessary for the folding of structural proteins. However, the spectrum of its substrates is gradually widening. We supposed that Tric/CCT could take part in GAPDS folding because of following information. First, the highest expression of Tric/CCT is observed in mammalian reticulocytes and testes, so this chaperonin may take part in the folding of other proteins specific for these tissues. Second, GAPDS is bound to the fibrous sheath of the sperm flagellum through the hydrophobic N-terminal domain. Therefore, Tric/CCT is likely to be one of the main proteins providing the correct embedding of GAPDS. According to our previous work, the chaperonin Tric/CCT from testes was shown to enhance the reactivation of dN-GAPDS after denaturation in the presence of guanidine hydrochloride [79].
In addition, the efficiency of chaperonin-assisted reactivation of dN-GAPDS was studied using site-directed mutagenesis. Substitution of proline residues P111, P157, and P326 by alanine residues was shown not to influence the chaperonin-assisted reactivation of the mutant proteins in comparison with wild-type dN-GAPDS. The Tric/CCT-assisted reactivation was ATP-dependent in all examined cases.
In contrast, Tric/CCT did not influence the refolding of the muscle isoform of glyceraldehyde-3-phosphate dehydrogenase. At the same time, the acceleration of muscle lactate dehydrogenase refolding was ATP-independent. Thus, GAPDS turned out to be a rather specific substrate of the chaperonin Tric/CCT.
STABILITY OF dN-GAPDS
During the purification and investigation of diverse characteristics of sperm-specific GAPD, we noticed that the sperm-specific enzyme exhibited enhanced stability compared to somatic GAPD. The sensitivity of dN-GAPDS towards denaturing conditions was investigated by three methods: differential scanning calorimetry; measurement of stationary ratio of native and denatured forms of the protein at different concentrations of guanidine hydrochloride; rate of the protein inactivation in the presence of 4 M guanidine hydrochloride (GdnHCl). Each of these methods revealed a certain aspect of protein stability: thermodynamic stability towards the enhanced temperature; kinetic stability of the active site in the presence of GdnHCl; and thermodynamic stability in the presence of GdnHCl, respectively.
GAPDS was shown to be more stable compared to the somatic GAPD according to the data from all these methods [78]. The biological role of the enhanced stability of the sperm-specific enzyme may be associated with its tissue-specificity. Inactivation of nuclear DNA and elimination of a major part of the cytoplasm, which contains molecules of the translation apparatus, are known to occur at the end of spermatogenesis. Therefore, the same GAPDS molecules should remain active over the whole lifespan of a sperm cell (up to two weeks in humans) to generate the energy required for its movement. For that reason, GAPDS should exhibit enhanced stability compared to the somatic GAPD, whose half-life is a few hours. This hypothesis is supported by the fact that lactate dehydrogenase C, another sperm-specific glycolytic enzyme, is also known to have enhanced stability compared to its somatic homologs [80].
BASIS FOR ENHANCED STABILITY OF dN-GAPDS TOWARDS DENATURING
CONDITIONS
Formation of cross-linked structures due to disulfide bonding or noncovalent interactions (salt bridges or hydrogen bonds) is supposed to be the most widespread mechanism of protein stabilization. According to X-ray structures of the somatic GAPD and GAPDS, both proteins do not contain any disulfide bonds; therefore, the enhanced stability of dN-GAPDS should have another explanation.
Both isoenzymes contain 20 salt bridges per subunit, sharing 12 of them. The sperm-specific isoenzyme contains fewer solvent-exposed salt bridges than the somatic GAPD, but more salt bridges buried in the protein molecule. If the exposed salt bridges have destabilizing effect, the enhanced stability of dN-GAPDS could be explained by the decrease in their number. Contrariwise, the stabilizing effect might be attributed to the effect of the additional buried salt bridges.
Decrease in conformational entropy of a polypeptide chain can also be achieved by increasing its rigidity. Depending on their position, proline residues are supposed to stabilize proteins by fixing their native conformation, whereas glycine residues seem to increase the flexibility of a polypeptide chain [81]. Alignment of the somatic GAPD and GAPDS sequences revealed seven additional proline residues specific for GAPDS, as well as nine glycine residues that are present only in the somatic GAPD.
The enhanced stability of an enzyme taking part in the production of energy required for the sperm cell movement is likely to be crucial for efficient fertilization. Perhaps this is the reason for the necessity of a special sperm-specific isoenzyme of glyceraldehyde-3-phosphate dehydrogenase. Consequently, we have a convenient object for studying the role of structural elements in the stabilization of the protein structure. We decided to identify those sequence positions that are important for the stabilization of GAPDS but do not affect its enzymatic activity. Such data could be helpful for the engineering of the novel thermostable enzymes to be used in biotechnological applications.
The GAPDS sequence was shown to contain seven additional proline residues compared to the somatic GAPD (Fig. 3).
Fig. 3. Alignment of sperm-specific GAPDS and somatic GAPD. The N-terminal region of GAPDS, which binds the isoenzyme to the sperm tail cytoskeleton and which was truncated in recombinant dN-GAPDS protein, is highlighted by color. The points in the GAPD sequence correspond to the conserved residues. The gaps are shown by dashes. The GAPDS-specific proline residues are highlighted by rectangles.
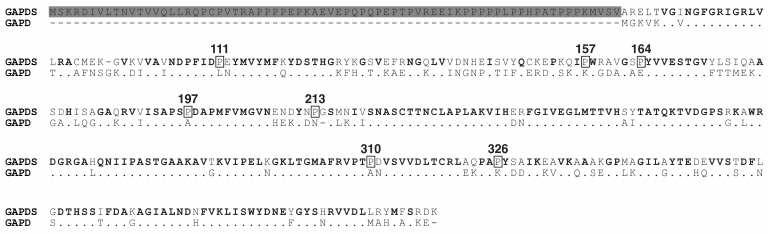
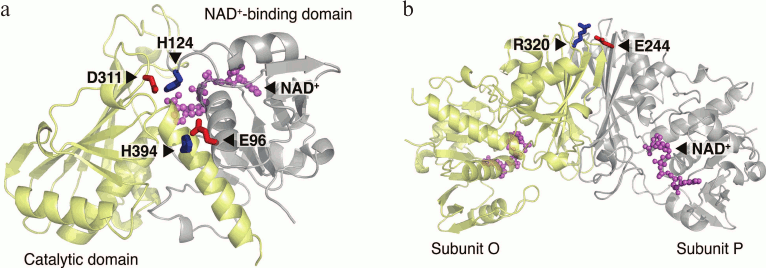
Proline is an imino acid differing from amino acid residues in its conformational properties, because its side chain is covalently bound to the nitrogen atom of the preceding peptide bond. The pyrrolidine ring of a proline residue significantly restricts the rotation around the N-Cα-bonds. Due to this characteristic, proline residues can stabilize proteins by fixing their native conformation. Comparison of enzymes from mesophilic and thermophilic organisms showed that thermophilic proteins contained more proline residues in non-structured regions, β-turns, and α-helices compared to their analogs from mesophilic organisms [81]. There are several examples of increased stability of proteins with a replacement of specific amino acid residues with proline residues. Introduction of prolines was shown to stabilize the lysozyme of T4 phage [82], protease from Bacillus sp. [83], thermolysin-like protease of Bacillus stearothermophilus [84], and oligo-1,6-glucosidase from Bacillus cereus [85]. The authors of the last work formulated an empirical so-called proline rule according to which proline residues important for protein stability are preferably located in the (i+1) position of β-turns or in the first position of α-helices. The stabilizing effects of these residues are independent and additive. In the sequence of thermostable GAPDS, six of seven additional proline residues were shown to correspond to this proline rule: three of them fall on the first position of α-helices (P111, P157, and P326), and the other three (P164, P197, and P213) fall on the second position of β-turns [78]. To test whether these proline substitutions in GAPDS are related to its enhanced stability, we obtained six mutant forms of dN-GAPDS, each containing a substitution of one of the indicated prolines by an alanine residue (P111A, P157A, P326A, P164A, P197A, and P213A). Then we investigated the thermostability and the stability of mutant proteins towards guanidine hydrochloride in comparison with wild-type dN-GAPDS. Besides this, comparative analysis of the X-ray structures of dN-GAPDS and GAPD isoenzymes revealed three additional buried salt bridges that might be significant for the stability of GAPDS (interdomain salt bridges D311–H124, E96–H394, and intersubunit salt bridge E244–R320) [78] (Fig. 4). To test this assumption, the contribution of amino acid residues in thermostability of dN-GAPDS was studied. The residues of aspartic and glutamic acids (D311, E96, and E244) involved in the formation of salt bridges in the molecule of dN-GAPDS were replaced with asparagine and glutamine residues, respectively. Stability of the resulting mutant proteins (D311N, E96Q, and E244Q) was investigated and compared with the wild-type dN-GAPDS.
Fig. 4. Interdomain (a) and intersubunit (b) salt bridges specific to dN-GAPDS. One subunit (a) or two subunits (b) of the tetramer are shown.
EFFECT OF POINT MUTATIONS ON STABILITY OF dN-GAPDS
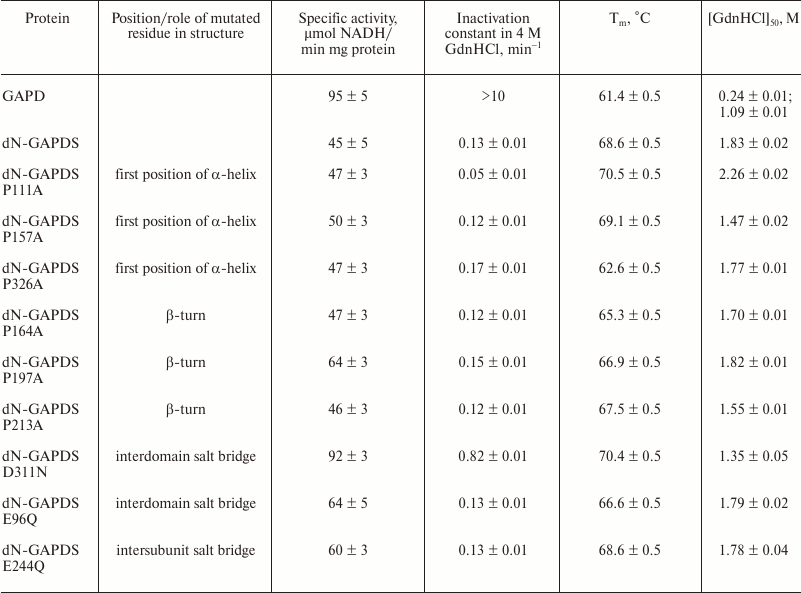
We obtained six mutant forms of dN-GAPDS with the additional prolines substituted by alanines (P111A, P157A, P326A, P164A, P197A, and P213A) and three mutant forms with the disrupted additional salt bridges (E96Q, D311N, E244Q).
All these mutants were enzymatically active (Table 2). The maximums of the particle size distributions and the sedimentation coefficients determined by dynamic light scattering and analytical ultracentrifugation, respectively, were very close in the cases of the wild-type dN-GAPDS and its mutant forms, which means that the latter are supposed to be tetramers too.
Table 2. Properties of dN-GAPDS mutant forms
compared to wild-type dN-GAPDS and somatic GAPD
The stabilities of the dN-GAPDS mutants were examined by the above-described methods in parallel with wild-type dN-GAPDS and somatic GAPD.
The Tm values obtained for the somatic GAPD and the wild-type dN-GAPDS differed by approximately 7°C. The proline mutations (P326A) decreased the Tm value of dN-GAPDS by 6°C (Table 2). The mutations P164A and P197A resulted in a moderate (3 and 2°C, respectively) decrease in the Tm value.
Among the mutations affecting the additional salt bridges (E96Q, E244Q, and D311N), E96Q breaking the interdomain salt bridge was the only mutation to significantly decrease the Tm value compared to the wild-type protein (by 2°C).
These data suggest that proline residues P326, P164, and P197 along with the interdomain salt bridge E96–H394 contribute to the thermostability of GAPDS.
The 4 M GdnHCl inactivation assay showed that two of the examined proline mutations affected the active center stability. The mutation P326A slightly increased the inactivation constant value (from 0.13 to 0.17 min–1), whereas the mutation P111A decreased the inactivation constant almost two-fold (from 0.13 to 0.05 min–1). The mutation D311N breaking the salt bridge D311–H124 significantly (6-fold) increased the inactivation rate compared to the wild-type protein (Table 2). The other mutations did not affect the rate of inactivation. Thus, the intersubunit salt bridge D311–H124 stabilizes the active site of the sperm-specific protein dN-GAPDS.
Another method used for evaluation of the protein stability was the determination the GdnHCl concentration [GdnHCl]50 corresponding to the half-denatured state of the protein. The native/denatured protein concentration ratios were estimated by the shift of the tryptophan fluorescence peak maximum from 335 to 355 nm [86]. The values of [GdnHCl]50 calculated from the denaturation curves are presented in Table 2. The mutations P157A, P164A, P213A, and D311N decreased the [GdnHCl]50 value compared to the wild-type protein, indicating that those mutant forms were destabilized. On the contrary, a pronounced stabilizing effect was observed in the case of the mutation P111A, which increased the value of [GdnHCl]50 by 0.43 M. In the other cases, the [GdnHCl]50 value was less affected or did not change.
As seen from Table 2, the largest effect on the thermostability of dN-GAPDS was observed in the case of the mutations P326A (first position of an α-helix) and P164A (β-turn): the Tm values of the heat-absorption curves decreased by 6.0 and 3.3°C, respectively, compared to the wild-type protein. The largest effect on the resistance of dN-GAPDS towards GdnHCl was observed for the mutation D311N disrupting the interdomain salt bridge between D311 and H124: the inactivation rate constant increased approximately 6-fold and the [GdnHCl]50 value decreased from 1.83 to 1.35 M. Besides, this mutation increased the enzymatic activity of the protein approximately 2-fold.
Thus, residues P326 and P164 appear to be the most important for enhanced thermostability of dN-GAPDS, exhibiting no effect on the catalytic activity of the enzyme. The salt bridge D311–H124 between the catalytic and NAD+-binding domains increases the stability of both the active center and the whole protein molecule towards GdnHCl, reducing the catalytic activity of the enzyme two-fold, but does not contribute into the thermostability.
EFFECT OF SUBSTITUTIONS DISRUPTING GAPDS-SPECIFIC SALT BRIDGES ON
COENZYME BINDING
The effects of the mutations disrupting the GAPDS-specific salt bridges E96–H394, D311–H124, and E244–R320 on the coenzyme-binding characteristics of dN-GAPDS were investigated [77]. The GAPD tryptophan fluorescence was quenched by the addition of NAD+, which makes it possible to study the interactions between the protein and the coenzyme by the use of fluorescence titrations. In contrast to GAPD, which had been previously reported to exhibit negative cooperativity in coenzyme binding [75, 76], dN-GAPDS was found to exhibit strong positive cooperativity (Table 3). We suggested that such a difference could be accounted for the additional salt bridges E96–H394, D311–H124, and E244–R320 (Fig. 4).
Table 3. Dissociation constants
characterizing NAD+ binding to the wild-type dN-GAPDS and
its mutant forms

The binding curves obtained for the dN-GAPDS mutant forms E96Q, D311N, and E244Q are shown at Fig. 5. The fluorescence intensities were assumed to conform to the equation derived by B. Kurganov and colleagues [87]:
[NAD+]bound = 4[E]T·(1 – F/F0)/(1 – F∞/F0),
where [NAD+]bound is the concentration of the bound coenzyme NAD+, [E]T is the total concentration of enzyme, F is the fluorescence intensity, F0 is the starting value of fluorescence intensity (at zero concentration of NAD+), and F∞ is the final value of fluorescence (at the saturating concentration of NAD+). The data were in perfect correspondence with the concerted model proposed by Monod and colleagues and the sequential model for both tetrahedral and square cases proposed by Koshland and colleagues.
Fig. 5. NAD+ binding to somatic GAPD (1), wild-type dN-GAPDS (2), and its mutant forms E96Q (3), E244Q (4), and D311N (5).
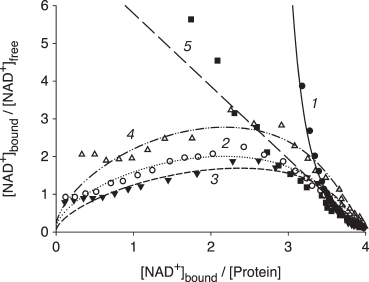
As seen from the binding curves represented in Scatchard coordinates, the wild-type dN-GAPDS, as well as the E96Q and E244Q mutants, exhibit a well-pronounced positive cooperativity in coenzyme binding (Fig. 5, curves 2-4). The somatic isoenzyme exhibits negative cooperativity (Fig. 5, curve 1). The D311N substitution resulted in complete elimination of positive cooperativity (Fig. 5, curve 5).
The calculated dissociation constants for all mutant forms of dN-GAPDS are shown in Table 3. According to these data, the E96Q and E244Q substitutions did not alter the NAD+-binding behavior of dN-GAPDS: in all cases, the dissociation constants of the first subunits were 20-50-fold higher than the dissociation constant of the last subunits. In contrast, the D311N substitution was found to alter the coenzyme binding behavior of dN-GAPDS: the mutant form bound NAD+ noncooperatively, and the dissociation constants for all subunits were the same (9.3·10–7 M).
Thus, the interdomain salt bridge D311–H124 not only stabilizes the active center of GAPDS, but it also provides positive cooperativity in coenzyme binding.
EXPRESSION OF GAPDS IN MELANOMA CELLS
Expression of GAPDS mRNA in melanoma cells. Sperm-specific proteins (or, to be exact, testis-specific proteins) can be markers of certain oncological disorders. Before our investigations, data about the expression of GAPDS mRNA in cancer cells were virtually absent. However, there were evidences for the presence of some unusual forms of glyceraldehyde-3-phosphate dehydrogenase in several types of malignant tumors [88].
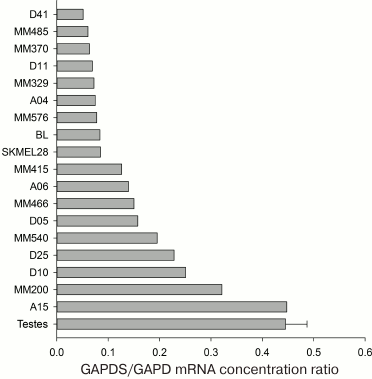
We analyzed the information about the expression of the GAPDS mRNA in 9765 samples available in the ArrayExpress Database (www.ebi.ac.uk/arrayexpress, accession numbers E-TABM-185, E-GEOD-2109, E-MTAB-37, E-GEOD-7127, E-GEOD-10843, and E-GEOD-7307). According to these data, GAPDS is not expressed in the somatic tissues, as well as in most tumor cell lines: the contents of its mRNA are two orders of magnitude lower than in testes. Nevertheless, we observed high GAPDS expression levels in some melanoma cells, such as, e.g. A15 and MM200 (Fig. 6).
Fig. 6. GAPDS mRNA levels in various melanoma cell lines and in testes (according to data available in the ArrayExpress database, www.ebi.ac.uk/arrayexpress, accession numbers E-TABM-185, E-GEOD-7127, E-GEOD-7307, and E-GEOD-10843).
Expression of GAPDS protein in melanoma cells. We supposed that the GAPDS protein could be detected in those cells with high levels of its mRNA. Therefore, we assayed various melanoma cell lines for the presence of GAPDS. For that purpose, antibodies were required against both native (for immunoprecipitation) and denatured species of the protein (for immunoblotting).
We developed a novel method for polyclonal antibody production that allowed us to obtain antibodies specific to either native and denatured species of GAPDS [89]. These antibodies were used for immunoprecipitation experiments, immunofluorescent staining of cells, and detection of GAPDS after SDS-PAGE.
The above-described antibodies were used to detect GAPDS in melanoma cells by immunoblotting. Melanoma cell lines MelIL, MelKor, and MelP were obtained previously during collaborative work with Blokhin Cancer Research Center of the Russian Academy of Medical Sciences and Petrov Research Institute of Oncology [90, 91]. As positive controls, we used sperm cells and a preparation of the recombinant sperm-specific dN-GAPDS. Teratocarcinoma cell lysates, as well as a preparation of somatic GAPD from rabbit muscles, were used as negative controls. The sperm cells yielded a set of protein bands: a band of approximately 56 kDa corresponding to the full-length GAPDS [49] and shorter bands corresponding to the products of its proteolysis (37 and 36 kDa). A preparation of recombinant dN-GAPDS yielded a band of 37 kDa only. All the examined melanoma cells showed a band of 37 kDa as well [46].
Thus, the analysis of three different lines of melanoma cells revealed a protein with subunit molecular weight of approximately 37 kDa that interacted with the antibodies against GAPDS. The revealed protein must be a fragment of the full-length GAPDS (56 kDa). Such a fragment can be a result of proteolysis of the full-length protein, which occurred either in the course of sample preparation or in the cells. It is also possible that, in the melanoma cells, GAPDS is expressed without the N-terminal domain.
We searched the mRNA section of GenBank and found that GAPDS without the N-terminal domain can present in actively reproducing somatic cells, such as embryonic or regenerating tissue of the lizard Anolis carolinensis [52]. In these cases, the lack of the N-terminal domain seems to be explained by alternative splicing. Since the N-terminal domain is required for the attachment of GAPDS to the cytoskeleton of the sperm tail, its presence should not be essential for the catalytic activity of the enzyme in somatic cells.
The presence of GAPDS in the lysates of the MelKor and MelP cells was also shown by immunoprecipitation [46]. For this, we used rabbit polyclonal antibodies against the native species of GAPDS, which were linked to the protein G-Sepharose. The complexes were analyzed by SDS-PAGE. They resulted in a protein band of approximately 37 kDa, which corresponds to the molecular weight of dN-GAPDS. This band was identified as GAPDS by MALDI mass spectrometry. It should also be mentioned that the content of GAPDS in the MelKor cells was significantly less than in the MelP cells.
The isolated protein complexes from the MelP cell lysate also contained the somatic GAPD, and its content was close to that of GAPDS. Since the antibodies used for the immunoprecipitation are known to be specific to the native GAPDS species and not interact with the somatic GAPD, these data suggest that these protein complexes represent heterooligomeric forms of the enzyme composed of both somatic GAPD and GAPDS subunits. Since dimer–dimer interactions in the GAPD molecule are weaker than those between monomers, and the ratio of the isoenzyme contents was close to one, the heterotetramers are likely to be composed of a dimer of the somatic GAPD and a dimer of GAPDS. Such hybrid tetramers had been already described in a previous study: the expression of rat GAPDS in E. coli cells resulted in the formation of tetramers composed of recombinant rat GAPDS subunits and host GAPD subunits [73]. Presumably, in the melanoma cells, we observed a similar situation.
We also demonstrated that the synthesis of the GAPDS protein is specific for the melanoma cells only and does not occur in cancer cell lines of other origins. For this, we estimated the levels of the GAPDS mRNA transcription in a number of non-melanoma cancer cells. The presence of the GAPDS protein in the cell lysates was also examined by SDS-PAGE with subsequent immunoblotting. The data are present in Table 4. The GAPDS-encoding mRNA was found to be virtually absent in all the examined non-melanoma cells, and its content was two orders of magnitude lower than in the testes. The GAPDS protein product was not detected as well.
Table 4. The content of the somatic GAPD and
GAPDS mRNAs in different tissues and cell lines
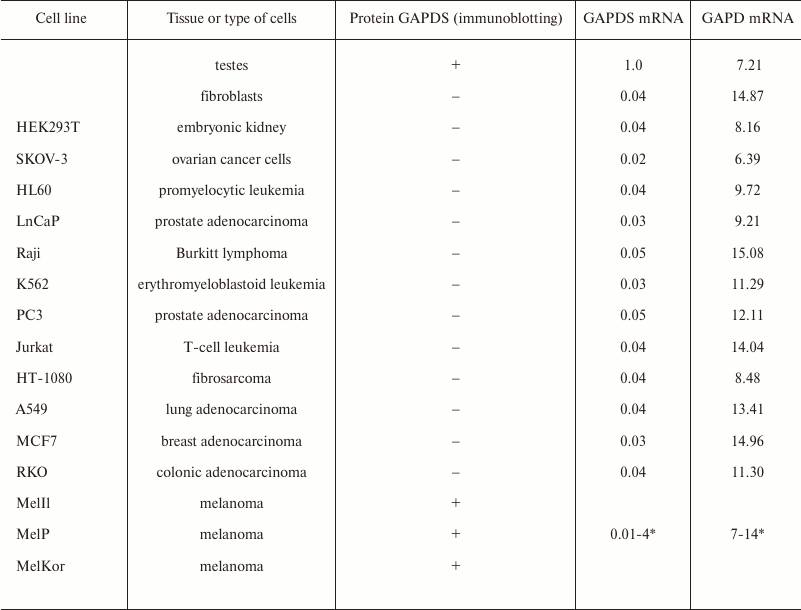
* Data spread are given for 63 melanoma cell lines (accession number
GEOD-7127 in the ArrayExpress database).
Intracellular localization of GAPDS in melanoma cells. We demonstrated that some lines of melanoma cells express the sperm-specific isoenzyme GAPDS. As mentioned above, using mass spectrometry, we confirmed that the protein detected in melanoma cell lysates, which interacted with antibodies against GAPDS, was the sperm-specific glyceraldehyde-3-phosphate dehydrogenase truncated at the N-terminal domain. Because the N-terminal domain is necessary for the attachment of GAPDS to the cytoskeleton of the sperm tail, it is obvious that GAPDS without this domain is to be found in the cytosolic fraction together with the cytoplasmic somatic GAPD. To confirm this, we performed immunochemical staining of MelP and MelKor melanoma cell lines with rabbit polyclonal antibodies against native GAPDS. Fibroblasts were used as the negative control, in which the GAPDS protein already had been shown to be completely absent (Table 4). According to the results, GAPDS is localized in the cytoplasm of melanoma cells (fibroblasts used as the negative control did not stain). For both types of cells, no staining was observed after treatment with only the secondary antibodies against rabbit IgG conjugated with the fluorescent dye. Similar results were obtained for MelKor melanoma cell line, but the staining of GAPDS was less intensive.
Cytoplasmic localization of GAPDS might induce the formation of heterooligomeric complexes composed of the sperm-specific and the somatic isoenzymes. On the one hand, formation of such hybrid complexes is likely to abolish some non-glycolytic functions of GAPD (e.g. the induction of apoptosis). On the other hand, such complexes may have enhanced stability because of the GAPDS subunits. Thereby, such new characteristics of the hybrid oligomers are supposed to alter metabolism of tumor cells leading to increased survival of these cells and enhanced extent of malignancy of the disease. We suppose that the presence and the expression level of the GAPDS gene may serve as a marker for the stage of tumor progression along with E-cadherin, N-cadherin, MITF, and S100A4/MTS1 gene transcripts. According to our data and the literature background, the GAPDS production in melanoma cells can be a result of enhanced expression of MITF transcriptional factor, but data about the relationships between MITF expression level and tumor aggression are contradictory, so this matter needs further investigation.
It is worth noting that GAPDS without the N-terminal domain can also be present in the actively reproducing somatic cells (embryonic and regenerating tissue cells) of some vertebrates (Anolis carolinensis) [52]. This means that the expression of GAPDS in a somatic cell does not always mean that the cell is malignant. Probably, expression of this protein gives some advantages for the rapidly dividing cells in terms of the glycolytic energy production. As we have shown, the recombinant dN-GAPDS protein exhibits the specific glyceraldehyde-3-phosphate dehydrogenase activity characteristic to the somatic isoenzyme, but it is much more stable [78]. Therefore, its expression may affect the metabolism of cells.
Taken together, we demonstrated that the extracts of some melanoma cell lines contain a protein of about 37 kDa that interacts with antibodies against GAPDS. Since not all analyzed melanoma cell lines contained mRNA encoding the full-length GAPDS, we supposed that the GAPDS protein without the N-terminal domain was expressed. The presence of GAPDS in melanoma cell lines was shown by immunoprecipitation with subsequent SDS-PAGE and mass-spectrometry analyses. The immunochemical staining of MelP and MelKor melanoma cell line lysates with rabbit polyclonal antibodies against native GAPDS demonstrated the cytoplasmic localization of GAPDS. We examined a number of melanoma cell lines corresponding to different stages of the disease. The GAPDS mRNA and its protein product were detected only in those corresponding to the moderate stages, whereas there was neither the mRNA nor the protein in the cell lines corresponding to the final stages (such as MelSi and MelME).
The average annual increase in melanoma disease is known to be one of the most considerable among all malignant tumors [92, 93]. The cure rate directly depends on the stage of the disease. Unfortunately, despite the possibility of visual localization, only 62-65% of patients were diagnosed in the initial stages, while the others had advanced malignancies [94]. Therefore, the survival rate is rather low. Obviously, the expression of GAPDS protein at least in some melanoma cell lines gives hope for it being used as a new specific oncomarker. GAPDS is not expressed in other types of malignant tumor cells, and its presence is typical of only a defined stage of the disease. Of course, the development of a method of melanoma diagnostics based on the detection of either GAPDS or antibodies against it in patient’s blood requires more investigations and clinical trials.
Participation of GAPD and GAPDS in metabolic regulation and induction of apoptosis in normal and cancer cells is considered a major requisite for generating antitumor drugs. Development of antitumor therapy needs detailed comprehension of molecular and biochemical processes underlying melanoma progression. The high rate of glycolysis as the main source of energy in cancer cells does not slow in the presence of oxygen. This is known as the Warburg effect. According to the Warburg hypothesis, switching to anoxic generation of energy leads to autonomous uncontrolled cell life. Therefore, the cell behaves like an independent organism aiming to reproduce itself [31, 32]. Some isoforms of different glycolytic enzymes, particularly GAPDS, are likely to take part in this effect. The expression of GAPDS in cancer cells may alter the induction of apoptosis as well because the somatic GAPD is involved in this process. In contrast to it, GAPDS was not shown to participate in the complicated intracellular translocations because of its enhanced stability and the absence of specific motifs in protein structure. In addition, GAPDS may prevent the participation of somatic GAPD in induction of apoptosis by interacting with its subunits and forming stable heterotetramers. If GAPDS is in fact involved in these metabolic and apoptotic features, ligands capable of specific binding to GAPDS could be considered as possible antitumor drugs.
CONCLUSION
Sperm-specific glyceraldehyde-3-phosphate dehydrogenase is an interesting and convenient model for studies of evolution, catalytic mechanisms, and stability of enzymes. The comparative analysis approach, which was a common tool half a century ago, makes it possible to obtain some novel information on the structural basis of glyceraldehyde-3-phosphate dehydrogenase functioning when combined with methods of bioinformatics and site-directed mutagenesis. As recently found that some properties of GAPDS are similar to those of the enzymes from microorganisms. For example, GAPDS does not show negative cooperativity in NAD+ binding, which is characteristic for the somatic isoenzyme of mammals. We plan to compare the structures and catalytic parameters of human somatic GAPD and GAPDS with those of GAPD from pathogenic microorganisms (particularly from mycobacteria) and, based on the findings, develop inhibitors of GAPDS and GAPD from microorganisms that would have only slight effect on the somatic GAPD. The experimental examination of such inhibitors would be performed using recombinant proteins and, later, on sperm cells. The well-established methods for sperm storage and the motility assessment, combined with the availability of the sperms of domestic animals, would make this approach easy and convenient.
However, in conclusion, we wish to focus on the medical aspects of sperm-specific glyceraldehyde-3-phosphate dehydrogenase studies.
The connection between GAPDS and sperm motility is the most obvious. We demonstrated a correlation between decrease in GAPDS enzymatic activity (e.g. upon exposure to reactive oxygen species) and decrease in sperm motility. Of course, this does not mean that antioxidants should be used to enhance the fertilization capacity of spermatozoa. On the contrary, oxidative stress can be used to select sperm cells with the best antioxidant protection. Low concentrations of antioxidants (10 µM hydrogen peroxide) activate antioxidative protection and thus increase sperm motility through increase in GAPDS enzymatic activity [95]. Moreover, a recent study demonstrated that GAPDS molecules localized at the acrosome surface participate in penetration through the membrane of the egg cell [96].
The correlation between the enzymatic activity of GAPDS and the motility of sperms motivated scientists to carry out a search for novel male contraceptive agents. For that purpose, several research groups attempted to isolate GAPDS and resolve its spatial structure. The X-ray analysis of heterotetrameric construct composed of rat GAPDS and E. coli GAPD subunits and, later, the recombinant human GAPDS homotetramer, showed that the structures of the sperm-specific and somatic isoenzymes are quite similar, but not the same [73, 97]. However, a GAPDS-specific inhibitor has not been created yet. Perhaps, the reason for this failure is that the desired inhibitor should be specific, not affecting the somatic isoenzyme.
Our findings on GAPDS expression in melanoma cells raise again the problem of the GAPDS-specific inhibitors. The suppression of glycolytic flux in melanoma cells by such inhibitors may slow or even stop the disease, especially in the case of their local application. Fragmentary data on the participation of GAPDS in the development of neurodegenerative diseases might also give grounds for the use of such inhibitors for either treatment or prophylactics.
This work was supported by the Russian Foundation for Basic Research (project No. 13-04-00823-a) and by the Russian Science Foundation (project No. 15-14-00069 for E. V. Schmalhausen).
REFERENCES
1.Glaser, P. E., and Gross, R. W. (1995) Rapid
plasmenylethanolamine-selective fusion of membrane bilayers catalyzed
by an isoform of glyceraldehyde-3-phosphate dehydrogenase:
discrimination between glycolytic and fusogenic roles of individual
isoforms, Biochemistry, 34, 12193-12203.
2.Robbins, A. R., Ward, R. D., and Oliver, C. (1995)
A mutation in glyceraldehyde 3-phosphate dehydrogenase alters
endocytosis in CHO cells, J. Cell Biol., 130,
1093-1104.
3.Raje, C. I., Kumar, S., Harle, A., Nanda, J. S.,
and Raje, M. (2007) The macrophage cell surface
glyceraldehyde-3-phosphate dehydrogenase is a novel transferrin
receptor, J. Biol. Chem., 282, 3252-3261.
4.Hessler, R. J., Blackwood, R. A., Brock, T. G.,
Francis, J. W., Harsh, D. M., and Smolen, J. E. (1998) Identification
of glyceraldehyde-3-phosphate dehydrogenase as a
Ca2+-dependent fusogen in human neutrophil cytosol, J.
Leukoc. Biol., 63, 331-336.
5.Muronetz, V. I., Wang, Z. X., Keith, T. J., Knull,
H. R., and Srivastava, D. K. (1994) Binding constants and
stoichiometries of glyceraldehyde 3-phosphate
dehydrogenase–tubulin complexes, Arch. Biochem. Biophys.,
313, 253-260.
6.Volker, K. W., Reinitz, C. A., and Knull, H. R.
(1995) Glycolytic enzymes and assembly of microtubule networks,
Comp. Biochem. Physiol. B Biochem. Mol. Biol., 112,
503-514.
7.Cueille, N., Blanc, C. T., Riederer, I. M., and
Riederer, B. M. (2007) Microtubule-associated protein 1B binds
glyceraldehyde-3-phosphate dehydrogenase, J. Proteome Res.,
6, 2640-2647.
8.Bryksin, A. V., and Laktionov, P. P. (2008) Role of
glyceraldehyde-3-phosphate dehydrogenase in vesicular transport from
Golgi apparatus to endoplasmic reticulum, Biochemistry (Moscow),
73, 619-625.
9.Tisdale, E. J., Azizi, F., and Artalejo, C. R.
(2009) Rab2 utilizes glyceraldehyde-3-phosphate dehydrogenase and
protein kinase Cι to associate with microtubules and to recruit
dynein, J. Biol. Chem., 284, 5876-5884.
10.Engel, M., Seifert, M., Theisinger, B., Seyfert,
U., and Welter, C. (1998) Glyceraldehyde-3-phosphate dehydrogenase and
Nm23-H1/nucleoside diphosphate kinase A. Two old enzymes combine for
the novel Nm23 protein phosphotransferase function, J. Biol.
Chem., 273, 20058-20065.
11.Duclos-Vallee, J. C., Capel, F., Mabit, H., and
Petit, M. A. (1998) Phosphorylation of the hepatitis B virus core
protein by glyceraldehyde-3-phosphate dehydrogenase protein kinase
activity, J. Gen. Virol., 79, 1665-1670.
12.Dai, R.-P., Yu, F. X., Goh, S. R., Chng, H. W.,
Tan, Y. L., Fu, J. L., Zheng, L., and Luo, Y. (2008) Histone 2B (H2B)
expression is confined to a proper NAD+/NADH redox status,
J. Biol. Chem., 283, 26894-28901.
13.Li, Y., Huang, T., Zhang, X., Wan, T., Hu, J.,
Huang, A., and Tang, H. (2009) Role of glyceraldehyde-3-phosphate
dehydrogenase binding to hepatitis B virus posttranscriptional
regulatory element in regulating expression of HBV surface antigen,
Arch. Virol., 154, 519-524.
14.Kondo, S., Kubota, S., Mukudai, Y., Nishida, T.,
Yoshihama, Y., Shirota, T., Shintani, S., and Takigawa, M. (2011)
Binding of glyceraldehyde-3-phosphate dehydrogenase to the
cis-acting element of structure-anchored repression in ccn2
mRNA, Biochem. Biophys. Res. Commun., 405, 382-387.
15.Sundararaj, K. P., Wood, R. E., Ponnusamy, S.,
Salas, A. M., Szulc, Z., Bielawska, A., Obeid, L. M., Hannun, Y. A.,
and Ogretmen, B. (2004) Rapid shortening of telomere length in response
to ceramide involves the inhibition of telomere binding activity of
nuclear glyceraldehyde-3-phosphate dehydrogenase, J. Biol.
Chem., 279, 6152-6162.
16.Demarse, N. A., Ponnusamy, S., Spicer, E. K.,
Apohan, E., Baatz, J. E., Ogretmen, B., and Davies, C. (2009) Direct
binding of glyceraldehyde 3-phosphate dehydrogenase to telomeric DNA
protects telomeres against chemotherapy-induced rapid degradation,
J. Mol. Biol., 394, 789-803.
17.Nakagawa, T., Hirano, Y., Inomata, A., Yokota,
S., Miyachi, K., Kaneda, M., Umeda, M., Furukawa, K., Omata, S., and
Horigome, T. (2003) Participation of a fusogenic protein,
glyceraldehyde-3-phosphate dehydrogenase, in nuclear membrane assembly,
J. Biol. Chem., 278, 20395-20404.
18.Singh, R., and Green, M. R. (1993)
Sequence-specific binding of transfer RNA by glyceraldehyde-3-phosphate
dehydrogenase, Science, 259, 365-368.
19.Meyer-Siegler, K., Mauro, D. J., Seal, G.,
Wurzer, J., De Riel, J. K., and Sirover, M. A. (1991) A human nuclear
uracil DNA glycosylase is the 37-kDa subunit of
glyceraldehyde-3-phosphate dehydrogenase, Proc. Natl. Acad. Sci.
USA, 88, 8460-8464.
20.Azam, S., Jouvet, N., Jilani, A., Vongsamphanh,
R., Yang, X., Yang, S., and Ramotar, D. (2008) Human
glyceraldehyde-3-phosphate dehydrogenase plays a direct role in
reactivating oxidized forms of the DNA repair enzyme APE1, J. Biol.
Chem., 283, 30632-30641.
21.Arutyunova, E. I., Danshina, P. V., Domnina, L.
V., Pleten, A. P., and Muronetz, V. I. (2003) Oxidation of
glyceraldehyde-3-phosphate dehydrogenase enhances its binding to
nucleic acids, Biochem. Biophys. Res. Commun., 307,
547-552.
22.Hara, M. R., Cascio, M. B., and Sawa, A. (2006)
GAPDH as a sensor of NO stress, Biochim. Biophys. Acta,
1762, 502-509.
23.Hara, M. R., and Snyder, S. H. (2006) Nitric
oxide-GAPDH-Siah: a novel cell death cascade, Cell. Mol.
Neurobiol., 26, 527-538.
24.Sen, N., Hara, M. R., Kornberg, M. D., Cascio, M.
B., Bae, B. I., Shahani, N., Thomas, B., Dawson, T. M., Dawson, V. L.,
Snyder, S. H., and Sawa, A. (2008) Nitric oxide-induced nuclear GAPDH
activates p300/CBP and mediates apoptosis, Nat. Cell Biol.,
10, 866-873.
25.Hwang, N. R., Yim, S. H., Kim, Y. M., Jeong, J.,
Song, E. J., Lee, Y., Lee, J. H., Choi, S., and Lee, K. J. (2009)
Oxidative modifications of glyceraldehyde-3-phosphate dehydrogenase
play a key role in its multiple cellular functions, Biochem. J.,
423, 253-264.
26.Mazzola, J. L., and Sirover, M. A. (2001)
Reduction of glyceraldehyde-3-phosphate dehydrogenase activity in
Alzheimer’s disease and in Huntington’s disease
fibroblasts, J. Neurochem., 76, 442-449.
27.Naletova, I., Schmalhausen, E., Kharitonov, A.,
Katrukha, A., Saso, L., Caprioli, A., and Muronetz, V. (2008)
Non-native glyceraldehyde-3-phosphate dehydrogenase can be an intrinsic
component of amyloid structures, Biochim. Biophys. Acta,
1784, 2052-2058.
28.Butterfield, D. A., Hardas, S. S., and Lange, M.
L. (2010) Oxidatively modified glyceraldehyde-3-phosphate dehydrogenase
(GAPDH) and Alzheimer’s disease: many pathways to
neurodegeneration, J. Alzheimer’s Dis., 20,
369-393.
29.Mazzola, J. L., and Sirover, M. A. (2002)
Alteration of nuclear glyceraldehyde-3-phosphate dehydrogenase
structure in Huntington’s disease fibroblasts, Brain Res. Mol.
Brain Res., 100, 95-101.
30.Bae, B.-I., Hara, M. R., Cascio, M. B.,
Wellington, C. L., Hayden, M. R., Ross, C. A., Ha, H. C., Li, X. J.,
Snyder, S. H., and Sawa, A. (2006) Mutant huntingtin: nuclear
translocation and cytotoxicity mediated by GAPDH, Proc. Natl. Acad.
Sci. USA, 103, 3405-3409.
31.Warburg, O., Posener, K., and Negelein, E. (1924)
Ueber den stoffwechsel der tumoren, Biochem. Z., 152,
319-344.
32.Warburg, O. (1956) On the origin of cancer cells,
Science, 123, 309-314.
33.Schmalhausen, E. V., and Muronetz, V. I. (1997)
An uncoupling of the processes of oxidation and phosphorylation in
glycolysis, Biosci. Rep., 17, 521-527.
34.Schmalhausen, E. V., Nagradova, N. K.,
Boschi-Muller, S., Branlant, G., and Muronetz, V. I. (1999) Mildly
oxidized GAPDH: the coupling of the dehydrogenase and acyl phosphatase
activities, FEBS Lett., 452, 219-222.
35.Weber, J. P., and Bernhard, S. A. (1982) Transfer
of 1,3-diphosphoglycerate between glyceraldehyde-3-phosphate
dehydrogenase and 3-phosphoglycerate kinase via an
enzyme–substrate–enzyme complex, Biochemistry,
21, 4189-4194.
36.Sukhodolets, M. V., Muronetz, V. I., Tsuprun, V.
L., Kaftanova, A. S., and Nagradova, N. K. (1988) Association of rabbit
muscle glyceraldehyde-3-phosphate dehydrogenase and 3-phosphoglycerate
kinase. The biochemical and electron-microscopic evidence, FEBS
Lett., 238, 161-166.
37.Tompa, P., and Batke, J. (1990)
Fructose-1,6-bisphosphate aldolase preferentially associates to
glyceraldehyde-3-phosphate dehydrogenase in a mixture of cytosolic
proteins as revealed by fluorescence energy transfer measurements,
Biochem. Int., 20, 487-494.
38.Fokina, K. V., Dainyak, M. B., Nagradova, N. K.,
and Muronetz, V. I. (1997) A study on the complexes between human
erythrocyte enzymes participating in the conversions of
1,3-diphosphoglycerate, Arch. Biochem. Biophys., 345,
185-192.
39.Clarke, F. M., and Masters, C. J. (1975) On the
association of glycolytic enzymes with structural proteins of skeletal
muscle, Biochim. Biophys. Acta, 381, 37-46.
40.Ryazanov, A. G., Ashmarina, L. I., and Muronetz,
V. I. (1988) Association of glyceraldehyde-3-phosphate dehydrogenase
with mono- and polyribosomes of rabbit reticulocytes, Eur. J.
Biochem., 171, 301-305.
41.Walsh, J. L., Keith, T. J., and Knull, H. R.
(1989) Glycolytic enzyme interactions with tubulin and microtubules,
Biochim. Biophys. Acta, 999, 64-70.
42.Sirover, M. A. (1997) Role of the glycolytic
protein, glyceraldehyde-3-phosphate dehydrogenase, in normal cell
function and in cell pathology, J. Cell. Biochem., 66,
133-140.
43.Tatton, W. G., Chalmers-Redman, R. M., Elstner,
M., Leesch, W., Jagodzinski, F. B., Stupak, D. P., Sugrue, M. M., and
Tatton, N. A. (2000) Glyceraldehyde-3-phosphate dehydrogenase in
neurodegeneration and apoptosis signaling, J. Neural Transm.
Suppl., 60, 77-100.
44.Lee, S. Y., Kim, J. H., Jung, H., Chi, S. W.,
Chung, S. J., Lee, C. K., Park, B. C., Bae, K. H., and Park, S. G.
(2012) Glyceraldehyde-3-phosphate, a glycolytic intermediate, prevents
cells from apoptosis by lowering S-nitrosylation of
glyceraldehyde-3-phosphate dehydrogenase, J. Microbiol.
Biotechnol., 22, 571-573.
45.Arutyunova, E. I., Domnina, L. V., Chudinova, A.
A., Makshakova, O. N., Arutyunov, D. Y., and Muronetz, V. I. (2013)
Localization of non-native D-glyceraldehyde-3-phosphate dehydrogenase
in growing and apoptotic HeLa cells, Biochemistry (Moscow),
78, 91-95.
46.Sevostyanova, I. A., Kulikova, K. V., Kuravsky,
M. L., Schmalhausen, E. V., and Muronetz, V. I. (2012) Sperm-specific
glyceraldehyde-3-phosphate dehydrogenase is expressed in melanoma
cells, Biochem. Biophys. Res. Commun., 427, 649-653.
47.Li, Y., Nowotny, P., Holmans, P., Smemo, S.,
Kauwe, J. S., Hinrichs, A. L., Tacey, K., Doil, L., Van Luchene, R.,
Garcia, V., Rowland, C., Schrodi, S., Leong, D., Gogic, G., Chan, J.,
Cravchik, A., Ross, D., Lau, K., Kwok, S., Chang, S. Y., Catanese, J.,
Sninsky, J., White, T. J., Hardy, J., Powell, J., Lovestone, S.,
Morris, J. C., Thal, L., Owen, M., Williams, J., Goate, A, and Grupe,
A. (2004) Association of late-onset Alzheimer’s disease with
genetic variation in multiple members of the GAPD gene family, Proc.
Natl. Acad. Sci. USA, 101, 15688-15693.
48.Welch, J. E., Schatte, E. C., O’Brien, D.
A., and Eddy, E. M. (1992) Expression of a glyceraldehyde 3-phosphate
dehydrogenase gene specific to mouse spermatogenic cells, Biol.
Reprod., 46, 869-878.
49.Welch, J. E., Brown, P. L., O’Brien, D. A.,
Magyar, P. L., Bunch, D. O., Mori, C., and Eddy, E. M. (2000) Human
glyceraldehyde 3-phosphate dehydrogenase-2 gene is expressed
specifically in spermatogenic cells, J. Androl., 21,
328-338.
50.Bunch, D. O., Welch, J. E., Magyar, P. L., Eddy,
E. M., and O’Brien, D. A. (1998) Glyceraldehyde 3-phosphate
dehydrogenase-S protein distribution during mouse spermatogenesis,
Biol. Reprod., 58, 834-841.
51.Kuravsky, M. L., and Muronetz, V. I. (2007)
Somatic and sperm-specific isoenzymes of glyceraldehyde-3-phosphate
dehydrogenase: comparative analysis of primary structures and
functional features, Biochemistry (Moscow), 72,
744-749.
52.Kuravsky, M. L., Aleshin, V. V., Frishman, D.,
and Muronetz, V. I. (2011) Testis-specific glyceraldehyde-3-phosphate
dehydrogenase: origin and evolution, BMC Evol. Biol., 11,
160.
53.Kamp, G., Busselmann, G., and Lauterwein, J.
(1996) Spermatozoa: models for studying regulatory aspects of energy
metabolism, Experientia, 52, 487-494.
54.Turner, R. M. (2003) Tales from the tail: what do
we really know about sperm motility? J. Androl., 24,
790-803.
55.Ford, W. C. (2006) Glycolysis and sperm motility:
does a spoonful of sugar help the flagellum go round? Hum. Reprod.
Update, 12, 269-274.
56.Nevo, A. C., and Rikmenspoel, R. (1970) Diffusion
of ATP in sperm flagella, J. Theor. Biol., 26, 11-18.
57.Adam, D. E., and Wei, J. (1975) Mass transport of
ATP within the motile sperm, J. Theor. Biol., 49,
125-145.
58.Gage, M. J. (1998) Mammalian sperm morphometry,
Proc. Biol. Sci., 265, 97-103.
59.Nicholls, D. G., and Ferguson, S. J. (2002)
Bioenergetics, 3rd Edn., Academic Press.
60.Tombes, R. M., and Shapiro, B. M. (1985)
Metabolite channeling: a phosphorylcreatine shuttle to mediate high
energy phosphate transport between sperm mitochondrion and tail,
Cell, 41, 325-334.
61.Tombes, R. M., and Shapiro, B. M. (1987) Enzyme
termini of a phosphocreatine shuttle. Purification and characterization
of two creatine kinase isozymes from sea urchin sperm, J. Biol.
Chem., 262, 16011-16019.
62.Steeghs, K., Oerlemans, F., and Wieringa, B.
(1995) Mice deficient in ubiquitous mitochondrial creatine kinase are
viable and fertile, Biochim. Biophys. Acta, 1230,
130-138.
63.Yeung, C. H., Majumder, G. C., Rolf, C., Behre,
H. M., and Cooper, T. G. (1996) The role of phosphocreatine kinase in
the motility of human spermatozoa supported by different metabolic
substrates, Mol. Hum. Reprod., 2, 591-596.
64.Smith, M. B., Babcock, D. F., and Lardy, H. A.
(1985) A 31P-NMR study of the epididymis and epididymal
sperm of the bull and hamster, Biol. Reprod., 33,
1029-1040.
65.Robitaille, P. M., Robitaille, P. A., Martin, P.
A., and Brown, G. G. (1987) Phosphorus-31 nuclear magnetic resonance
studies of spermatozoa from the boar, ram, goat and bull, Comp.
Biochem. Physiol. B, 87, 285-296.
66.Mita, M., and Ueta, N. (1988) Energy metabolism
of sea urchin spermatozoa, with phosphatidylcholine as the preferred
substrate, Biochim. Biophys. Acta, 959, 361-369.
67.Mukai, C., and Okuno, M. (2004) Glycolysis plays
a major role for adenosine triphosphate supplementation in mouse sperm
flagellar movement, Biol. Reprod., 71, 540-547.
68.Hiipakka, R. A., and Hammerstedt, R. H. (1978)
2-Deoxyglucose transport and phosphorylation by bovine sperm, Biol.
Reprod., 19, 368-379.
69.Hyne, R. V., and Edwards, K. P. (1985) Influence
of 2-deoxy-D-glucose and energy substrates on guinea-pig sperm
capacitation and acrosome reaction, J. Reprod. Fertil.,
73, 59-69.
70.Miki, K., Qu, W., Goulding, E. H., Willis, W. D.,
Bunch, D. O., Strader, L. F., Perreault, S. D., Eddy, E. M., and
O’Brien, D. A. (2004) Glyceraldehyde 3-phosphate dehydrogenase-S,
a sperm-specific glycolytic enzyme, is required for sperm motility and
male fertility, Proc. Natl. Acad. Sci. USA, 101,
16501-16506.
71.Elkina, Y. L., Atroshchenko, M. M., Bragina, E.
E., Muronetz, V. I., and Schmalhausen, E. V. (2011) Oxidation of
glyceraldehyde-3-phosphate dehydrogenase decreases sperm motility,
Biochemistry (Moscow), 76, 268-272.
72.Westhoff, D., and Kamp, G. (1997) Glyceraldehyde
3-phosphate dehydrogenase is bound to the fibrous sheath of mammalian
spermatozoa, J. Cell Sci., 110, 1821-1829.
73.Frayne, J., Taylor, A., Cameron, G., and
Hadfield, A. T. (2009) Structure of insoluble rat sperm
glyceraldehyde-3-phosphate dehydrogenase (GAPDH) via heterotetramer
formation with Escherichia coli GAPDH reveals target for
contraceptive design, J. Biol. Chem., 284,
22703-22712.
74.Shchutskaya, Y. Y., Elkina, Y. L., Kuravsky, M.
L., Bragina, E. E., and Schmalhausen, E. V. (2008) Investigation of
glyceraldehyde-3-phosphate dehydrogenase from human sperms,
Biochemistry (Moscow), 73, 185-191.
75.Conway, A., and Koshland, D. E. (1968) Negative
cooperativity in enzyme action. The binding of diphosphopyridine
nucleotide to glyceraldehyde 3-phosphate dehydrogenase,
Biochemistry, 7, 4011-4023.
76.De Vijlder, J. J., and Slater, E. C. (1968) The
reaction between NAD+ and rabbit-muscle glyceraldehyde
phosphate dehydrogenase, Biochim. Biophys. Acta, 167,
23-34.
77.Kuravsky, M. L., Barinova, K. V., Asryants, R.
A., Schmalhausen, E. V., and Muronetz, V. I. (2015) Structural basis
for the NAD binding cooperativity and catalytic characteristics of
sperm-specific glyceraldehyde-3-phosphate dehydrogenase,
Biochimie, 115, 28-34.
78.Elkina, Y. L., Kuravsky, M. L., El’darov,
M. A., Stogov, S. V., Muronetz, V. I., and Schmalhausen, E. V. (2010)
Recombinant human sperm-specific glyceraldehyde-3-phosphate
dehydrogenase: structural basis for enhanced stability, Biochim.
Biophys. Acta, 1804, 2207-2212.
79.Naletova, I. N., Popova, K. M., Eldarov, M. A.,
Kuravsky, M. L., Schmalhausen, E. V., Sevostyanova, I. A., and
Muronetz, V. I. (2011) Chaperonin TRiC assists the refolding of
sperm-specific glyceraldehyde-3-phosphate dehydrogenase, Arch.
Biochem. Biophys., 516, 75-83.
80.Goldberg, E. (1972) Amino acid composition and
properties of crystalline lactate dehydrogenase X from mouse testes,
J. Biol. Chem., 247, 2044-2048.
81.Bogin, O., Peretz, M., Hacham, Y., Korkhin, Y.,
Frolow, F., Kalb(Gilboa), A. J., and Burstein, Y. (1998) Enhanced
thermal stability of Clostridium beijerinckii alcohol
dehydrogenase after strategic substitution of amino acid residues with
prolines from the homologous thermophilic Thermoanaerobacter
brockii alcohol dehydrogenase, Protein Sci., 7,
1156-1163.
82.Matthews, B. W., Nicholson, H., and Becktel, W.
J. (1987) Enhanced protein thermostability from site-directed mutations
that decrease the entropy of unfolding, Proc. Natl. Acad. Sci.
USA, 84, 6663-6667.
83.Masui, A., Fujiwara, N., and Imanaka, T. (1994)
Stabilization and rational design of serine protease AprM under highly
alkaline and high-temperature conditions, Appl. Environ.
Microbiol., 60, 3579-3584.
84.Veltman, O. R., Vriend, G., and Middelhoven, P.
J. (1996) Analysis of structural determinants of the stability of
thermolysin-like proteases by molecular modelling and site-directed
mutagenesis, Protein Eng., 9, 1181-1189.
85.Watanabe, K., Masuda, T., Ohashi, H., Mihara, H.,
and Suzuki, Y. (1994) Multiple proline substitutions cumulatively
thermostabilize Bacillus cereus ATCC7064 oligo-1,6-glucosidase.
Irrefragable proof supporting the proline rule, Eur. J.
Biochem., 226, 277-283.
86.Fan, H., Liu, J., Ren, W., Zheng, Z., Zhang, Y.,
Yang, X., Li, H., Wang, X., and Zou, G. (2008) pH induces thermal
unfolding of UTI: an implication of reversible and irreversible
mechanism based on the analysis of thermal stability, thermodynamic,
conformational characterization, J. Fluoresc., 18,
305-317.
87.Kurganov, B. I., Sugrobova, N. P., and Yakovlev,
V. A. (1972) Estimation of dissociation constant of enzyme–ligand
complex from fluorometric data by “difference” method,
FEBS Lett., 19, 308-310.
88.Patra, S., Ghosh, S., Bera, S., Roy, A., Ray, S.,
and Ray, M. (2009) Molecular characterization of tumor associated
glyceraldehyde-3-phosphate dehydrogenase, Biochemistry (Moscow),
74, 717-727.
89.Kuravsky, M. L., Schmalhausen, E. V.,
Pozdnyakova, N. V., and Muronetz, V. I. (2012) Isolation of antibodies
against different protein conformations using immunoaffinity
chromatography, Anal. Biochem., 426, 47-53.
90.Mikhailova, I. N., Lukashina, M. I., Baryshnikov,
A. Yu., Morozova, L. F., Byrova, O. S., Pankina, T. N., Kozlov, A. M.,
Golubeva, V. A., Cheremushkin, E. A., Doroshenko, M. B., Demidov, L.
V., Kiselev, S. L., Larin, S. S., and Georgiev, G. P. (2005) Melanoma
cell lines as the background for creating antitumour vaccines,
Vestnik Ross. Akad. Med. Nauk, 7, 37-40.
91.Mikhaylova, I. N., Kovalevsky, D. A., Morozova,
L. F., Golubeva, V. A., Cheremushkin, E. A., Lukashina, M. I.,
Voronina, E. S., Burova, O. S., Utyashev, I. A., Kiselev, S. L.,
Demidov, L. V., Beabealashvilli, R. Sh., and Baryshnikov, A. Y. (2008)
Cancer/testis genes expression in human melanoma cell lines,
Melanoma Res., 18, 303-313.
92.Lemehov, V. G. (2001) Epidemiology, risk factors,
screening of skin melanoma, Prakt. Onkol., 2, 3-11.
93.Ferlay, J., Shin, H. R., Bray, F., Forman, D.,
Mathers, C., and Parkin, D. M. (2010) Estimates of worldwide burden of
cancer in 2008: GLOBOCAN 2008, Int. J. Cancer, 127,
2893-2917.
94.Dvoyrin, V. V., Trapeznikov, N. N., and
Mikhailovskii, A. V. (1997) Statistics of skin melanoma in Russia,
Vestnik RONTs Blokhina RAMN, 8, 3-12.
95.Evdokimov, V. V., Barinova, K. V., Turovetskii,
V. B., Muronetz, V. I., and Schmalhausen, E. V. (2015) Low
concentrations of hydrogen peroxide activate the antioxidant defense
system in human sperm cells, Biochemistry (Moscow), 80,
1178-1185.
96.Margaryan, H., Dorosh, A., Capkova, J.,
Manaskova-Postlerova, P., Philimonenko, A., Hozak, P., and Peknicova,
J. (2015) Characterization and possible function of
glyceraldehyde-3-phosphate dehydrogenase-spermatogenic protein GAPDHS
in mammalian sperm, Reprod. Biol. Endocrinol., 13,
15.
97.Chaikuad, A., Shafqat, N., Al-Mokhtar, R.,
Cameron, G., Clarke, A. R., Brady, R. L., Oppermann, U., Frayne, J.,
and Yue, W. W. (2011) Structure and kinetic characterization of human
sperm-specific glyceraldehyde-3-phosphate dehydrogenase, GAPDS,
Biochem. J., 435, 401-409.