Influence of 10-(6′-Plastoquinonyl)decyltriphenylphosphonium (SkQ1) on Oxidative Status in Rats with Protamine Sulfate-Induced Hyperglycemia
Ya. G. Voronkova1, T. N. Popova1*, A. A. Agarkov1, and M. V. Skulachev2
1Voronezh State University, Department of Medical Biochemistry and Microbiology, 394006 Voronezh, Russia; E-mail: tpopova@bio.vsu.ru2Lomonosov Moscow State University, Institute of Mitoengineering, 119991 Moscow, Russia; E-mail: info@skq-project.ru
* To whom correspondence should be addressed.
Received April 7, 2015; Revision received July 23, 2015
An influence of 10-(6′-plastoquinonyl)decyltriphenylphosphonium (SkQ1) on oxidative status and activity of some antioxidant enzymes in the liver and blood serum from rats was examined during experimental hyperglycemia developed after injecting protamine sulfate. It was found that SkQ1 lowered glycemic level in rats treated with protamine sulfate. Moreover, it was also accompanied by restoration of the normal range of biochemiluminescence parameters indicating the rate of ongoing free radical processes, magnitude of primary products of lipid peroxidation such as diene conjugates, activity of aconitate hydratase, and level of citrate in rat liver and blood. Hence, it was demonstrated that activity of superoxide dismutase and catalase, increasing during hyperglycemia, was decreased after administering SkQ1. This might be related to the ability of SkQ1 to normalize free-radical homeostasis imbalanced during hyperglycemia.
KEY WORDS: hyperglycemia, SkQ1, free radical oxidation, biochemiluminescence, superoxide dismutase, catalase, aconitate hydratase, citrateDOI: 10.1134/S0006297915120093
Abbreviations: BCL, biochemiluminescence; LPO, lipid peroxidation; ROS, reactive oxygen species; SkQ1, 10-(6′-plastoquinonyl)decyltriphenylphosphonium; SOD, superoxide dismutase.
Hyperglycemia and diabetes mellitus are among the most urgent problems
in modern medicine. It is known that patients with diabetes have
mortality rate from ischemic heart disease, cerebral stroke, and total
mortality that are significantly higher than in people without
metabolic disturbances [1]. Friedrich Theodor von
Frerichs first described development of pathological changes in the
liver of patients suffering from diabetes mellitus. Hence, the terms
“diabetic hepatosis” and “fatty hepatosis” were
introduced into clinical practice [2, 3]. Primarily, it is due to glucose toxicity, i.e. a
condition of long-standing hyperglycemia, which contributes to
desensitization of pancreatic β-cells caused by generation of
hexosamines as products of glucose turnover in the hexosamine shunt [4].
Activation of the hexosamine shunt as well as the polyol pathway in hyperglycemia can cause augmented generation of free radicals [5]. Moreover, autooxidation of glucose and its metabolic intermediates [6], nonenzymatic or autooxidative protein glycosylation [7], accumulation of triose phosphates resulting in generation of carbonyl compounds, and upregulated intensity of oxidative phosphorylation [8] can also play a role in activating free-radical oxidation during hyperglycemia. It should be noted that during chronically upregulated glucose level in blood, production of superoxide by mitochondria [8] as well as cytochrome P-450, xanthine oxidase, and protein kinase C-dependent NADH/NADPH oxidase [7] becomes enormously increased. Hence, tissue ischemia, hypoxia, and pseudohypoxia are additional factors contributing to upregulated production of reactive oxidants in various organs and tissues [9, 10].
Markedly upregulated generation of reactive oxygen species (ROS) and release of catalytically active Fe2+ from extra- and intracellular stores can contribute to degradation of Fe-S-clusters in the active site of aconitate hydratase by free radicals, thereby allowing considering this enzyme as a crucial target for ROS under developing oxidative stress [11-13]. Upon downregulation of aconitate hydratase activity, its cognate substrate – citrate – can accumulate, and it can then take part in regulating free-radical oxidation level via chelating variable-valence metal ions.
Elevated level of ROS observed in hyperglycemia initiates lipid peroxidation (LPO) within biomembranes [5], which leads to accumulation of subsequent products.
A multilayered system of oxidative defense of the organism counters development of oxidative stress [14-17], wherein superoxide dismutase (SOD) and catalase hold an important place; as such, enzymes neutralize upstream primary ROS such as O2•– and H2O2 that can participate in reactions resulting in generation of other more reactive free radicals [18]. However, the intrinsic reserve of compounds with antioxidant activity often turns out to be insufficient for neutralizing large amounts of free radicals generated during hyperglycemia. In connection with this, examination of agents with antioxidant effect under pathological conditions coupled with oxidative stress is currently considered a high priority.
10-(6′-Plastoquinonyl)decyltriphenylphosphonium (SkQ1) is a highly efficacious mitochondria-targeted antioxidant consisting of plastoquinone (Q) providing antioxidant defense, a penetrating cation (Sk) responsible for delivering the antioxidant into mitochondria, and a decane linker. Compounds of SkQ type are regenerable antioxidants with reiterative action, which can be regenerated via complex III of the mitochondrial respiratory chain, providing repeated antioxidant activity [19].
The goal of this study was to evaluate the influence of SkQ1 on biochemiluminescence (BCL) parameters responsible for the rate of ongoing free radical processes, level of primary LPO products such as diene conjugates, activity of SOD, catalase, and aconitate hydratase, and content of citrate in liver and blood of rats with protamine sulfate-induced hyperglycemia.
MATERIALS AND METHODS
During the study, male white laboratory rats (Rattus rattus L.) of body weight 150-200 g were used. All experimental procedures done were in accordance with the principles of international guidelines on humane treatment of animals stated in sanitary regulations on selection and management of experimental and biological clinics (animal facilities).
Hyperglycemia was induced by administering protamine sulfate intramuscularly for three weeks at dose 10 mg/kg body weight, dissolved in 0.5 ml of 0.9% saline (three times per day) [20]. It is known that insulin together with heparin exhibits a stronger hypoglycemic effect [21, 22]. In contrast, binding of endogenous heparin by protamine sulfate enhances the diabetogenic effect, e.g. of alloxan and blood serum-derived diabetogenic factor [23], whereas resistance to hypoglycemic action both of endogenous and exogenous insulin develops in healthy rats upon binding of the entire endogenous heparin in the blood flow by protamine sulfate [24]. Under these circumstances, insulin resistance can be effectively abolished by administering heparin [25].
Three weeks after the onset of inducing hyperglycemia, anesthetized animals were sacrificed for further examination. The experimental rats were divided into three groups: group 1 (n = 12) – the animals were housed under standard conditions; group 2 (n = 12) – rats with protamine sulfate-induced hyperglycemia; group 3 (n = 12) – animals with hyperglycemia administered with SkQ1 solution intraperitoneally at dose 1250 nmol/kg, once per day, starting from the second week.
Tissue homogenate was prepared by taking a sample of liver tissue to be homogenized in a 4× volume of cooled isolation medium (0.1 M Tris-HCl buffer, pH 7.8, containing 1 mM EDTA, 1% β-mercaptoethanol) and centrifuged at 10,000g for 15 min. Serum was obtained from venous blood.
Development of hyperglycemia was verified by measuring the amount of glucose in the blood serum by using reagents purchased from Vital Diagnosticum (Russia).
Intensity of free-radical oxidation and total antioxidant activity were measured by applying a Fe2+-induced biochemiluminescence based on catalytic degradation of peroxide by metal ions with variable valence (Fe2+) according to the Fenton reaction. Emerging free radicals then initiate free-radical oxidation in the examined biological substrate. Recombination of RO2 radicals results in generation of unstable tetroxide, which then degrades and emits photons. A BCL kinetic curve was recorded for 30 s on a BCL-07 instrument (Medozons Ltd., Russia) supplied with software for measuring the following parameters: chemiluminescence light sum (S) and flash intensity (Imax) (characterizing intensity of free-radical oxidation) and tangent of slope of BCL time-dependence (tanα2) (depicting total antioxidant activity).
Medium for BCL measurements contained 0.4 ml 0.02 M potassium-phosphate buffer (pH 7.5), 0.4 ml 0.01 M FeSO4, and 0.2 ml 2% H2O2 solution (added immediately before measurements). Before the procedure, 0.1 ml of the examined sample was added.
The amount of diene conjugate was measured spectrophotometrically implying that during the LPO at the stage of free radical generation a system of conjugated double bonds is formed in molecules of polyunsaturated fatty acids, which is accompanied by appearance of a maximum in the absorption spectrum at 233 nm [26].
Activity of aconitate hydratase was measured at 233 nm using a Hitachi U-1900 spectrophotometer (Hitachi High-Technologies, Japan) suppled with installed software in medium containing 0.05 M Tris-HCl, pH 7.8, and 4 mM citrate [27]. The enzyme unit (U) was defined as the amount of the enzyme necessary for converting 1 µmol of substrate per min at 25°C. The concentration of citrate was measured according to Natelson’s method [28].
Activity of SOD was determined by inhibition of the rate of nitroblue tetrazolium (NBT) reduction in a nonenzymatic system consisting of phenazine methosulfate (PMS) and NADH. The incubation medium (3 ml) contained 0.1 M phosphate buffer, pH 7.8, 0.33 mM EDTA, 0.41 mM NBT, 0.01 mM PMS, and 0.8 mM NADH. Enzymatic activity was measured spectrophotometrically by the increase in optical density at 540 nm during 5 min using the Hitachi U-1900 spectrophotometer [29].
Activity of catalase was measured at 410 nm by the method based on formation of stable stained complex of hydrogen peroxide with ammonium molybdate [30].
During the study, the following chemicals were applied: protamine sulfate, Tris-HCl, citrate, NBT, PMS, and NADH from Sigma (USA); EDTA from Reanal (Hungary); all other chemicals of “chemically pure” or “analytically pure” grade were purchased from Russian manufacturers. SkQ1 was synthesized according to the previous protocol [19].
Experiments were done at least in 12 biological and two analytical replicates. Experimental results were compared with control group. The data were processed by applying Student’s t-test and calculating means and standard deviation. Significance level was set at p ≤ 0.05 [31].
RESULTS AND DISCUSSION
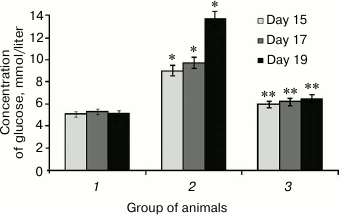
It was found that SkQ1 administered to rats with hyperglycemia lowered glycemic level, which was initially upregulated 2.7-fold (Fig. 1). For instance, SkQ1 administered on day 19 elicited decrease in blood glucose level by 2-fold (Fig. 1). There are the data showing that upregulated production of free radicals during elevated glycemic level activates the major stress-sensing factors (protein kinase C (PKC), nuclear transcription factor κB (NF-κB), mitogen-activated protein kinase (p38MAPK)) aggravating insulin resistance and lowering compensatory abilities of pancreatic islets [32]. Perhaps, a decrease in glucose concentration in rats after administering SkQ1 with induced hyperglycemia may is related to its antioxidant properties, which removes free radicals and subsequently decreases activity of PKC, NF-κB [33], and expression of inducible NO-synthase, eventually avoiding upregulated NO production and subsequent production of peroxynitrite, a powerful prooxidant that triggers apoptosis in pancreatic β-cells [18].
Fig. 1. Concentration of glucose in rats from control group (1), animals with protamine sulfate-induced hyperglycemia, untreated (2) and SkQ1-treated (3), measured on day 15, 17, and 19 from the beginning of the experiment (significant differences are shown at p ≤ 0.05; * compared to control group, ** compared to group with experimental hyperglycemia; hereinafter for Figs. 1-6).
Moreover, it was demonstrated that during development of protamine sulfate-induced hyperglycemia the magnitude of BCL light sum (S) in rat liver was increased 2.6-fold, in blood serum by 2.4-fold, with maximum flash intensity (Imax) by 2.1- and 2.0-fold, respectively, compared to the control group (table), evidencing increased intensity of free-radical oxidation. Hence, oxidative stress can result from various mechanisms: enhanced production of reactive oxidants generated under oxidation of both carbohydrates per se and those making complexes with different proteins, as well as due to autooxidation of fatty acids within triglycerides, phospholipids, and cholesterol esters; disturbed activity of enzymes from polyol glucose metabolism, mitochondrial oxidation, turnover of prostaglandins and leukotrienes, reduced activity of glyoxalase, etc. [4].
Biochemiluminescence parameters measured in the liver and blood serum
from rats of control group, untreated and SkQ1-injected animals with
protamine sulfate-induced hyperglycemia
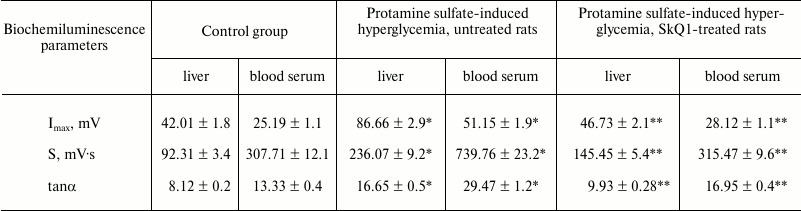
Note: Significant differences are shown at p ≤ 0.05; *
compared to control group; ** compared to group with experimental
hyperglycemia.
During pathological conditions, enhancement of compensatory mechanisms aimed at lowering in vivo level of free-radical oxidation can be judged by evaluating the magnitude of tanα2 characterizing total antioxidant activity. It was found that tanα2 increased by 2.1- and 2.2-fold in the liver and blood serum from rats with hyperglycemia, respectively, compared to the control group (table).
When SkQ1 was administered to rats with hyperglycemia, it was observed to lower the magnitude of S-value by 1.6- and 2.3-fold in the liver and blood serum, respectively, compared to its magnitude in type 2 diabetes mellitus. In addition, it was accompanied by decrease in magnitude of Imax by 1.9- and 1.8-fold in the liver and blood serum, respectively (table). These data can be interpreted from the viewpoint of antioxidant properties exhibited by SkQ1 [34]. Apart from that, use of SkQ1 was demonstrated to decrease the magnitude of tanα2 by 1.6- and 1.7-fold in the liver and blood serum, respectively, in treated vs. untreated animals with hyperglycemia (table). It is known that SkQ1 influences LPO by lowering the intensity of free radical processes, thereby normalizing or partially compensating downregulated antioxidant enzymes [34].
It was found that upon protamine sulfate-induced hyperglycemia, level of diene conjugates referred to toxic metabolites damaging lipoproteins, proteins, enzymes, and nucleic acids was elevated. In particular, the level of diene conjugates increased by 4.1- and 1.6-fold in the liver (Fig. 2a) and blood serum (Fig. 2b), respectively, compared to the control group. It cannot be ruled out that increased level of diene conjugates is one of the factors involved in the pathogenesis of microangiopathies [4, 14].
Fig. 2. Concentration of diene conjugates in liver (a) and blood serum (b) in rats from control group (1), animals with protamine sulfate-induced hyperglycemia untreated (2) and SkQ1-treated (3).
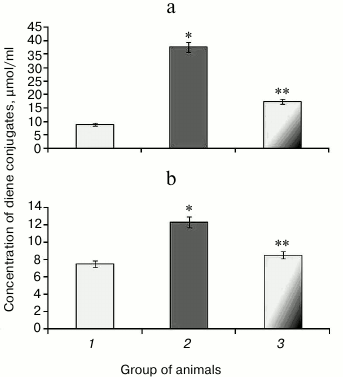
After administration of SkQ1, the level of detectable primary products of LPO was decreased 2.2- (Fig. 2a) and 1.4-fold (Fig. 2b) in the liver and blood serum, respectively, compared to untreated animals with hyperglycemia, thereby showing the suppressive effect of SkQ1 on LPO processes. The activity of respiratory chain complexes stayed at a rather high level in the presence of SkQ1, which might be due to its ability to normalize ROS production inhibiting activity of the key enzymes in the Krebs cycle [34]. The intensity of LPO largely depends on the level of metals with variable valence, particularly, Fe2+, which can increase during degradation of Fe-S cluster in aconitate hydratase by free radicals [11-13].
It was found that during protamine sulfate-induced hyperglycemia, specific activity (U/mg protein) of aconitate hydratase, a marker of oxidative stress, decreased by 1.9- and 2.8-fold in rat liver and blood serum (Figs. 3a and 3b), respectively. Alternatively, the value of the liver enzyme activity, expressed in U/g liver wet weight, decreased 1.5-fold (Fig. 3c), and the value of the serum enzyme activity, expressed in U/ml blood serum, decreased 2.1-fold (Fig. 3d). Accumulation of free radicals under hyperglycemia contributed to destruction of the active site in the enzyme and loss of its activity [35, 36]. Administration of SkQ1 to rats with hyperglycemia revealed that specific activity of aconitate hydratase was upregulated by 1.4- and 1.9-fold in the liver and blood serum, respectively (Figs. 3a and 3b). The value of the liver enzyme activity, expressed in U/g liver wet weight, increased 1.1-fold (Fig. 3c), and the value of the blood serum enzyme activity, expressed in U/ml, increased 2.0-fold (Fig. 3d). It seems that degree of free radical injury in molecules of aconitate hydratase was due to antioxidant properties exhibited by SkQ1, which, subsequently, influenced its activity.
Fig. 3. Activity of aconitate hydratase in rats from control group (1) and animals with protamine sulfate-induced hyperglycemia either untreated (2) or SkQ1-treated (3): a) specific enzymatic activity in liver; b) specific enzymatic activity in blood serum; c) activity of aconitate hydratase expressed in U/g liver wet weight; d) activity of aconitate hydratase expressed in U/ml blood serum.
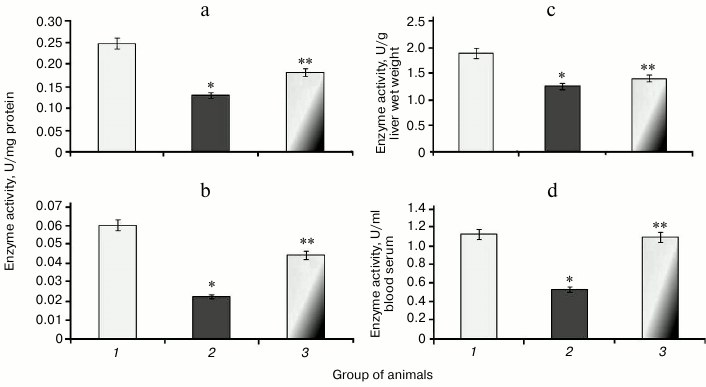
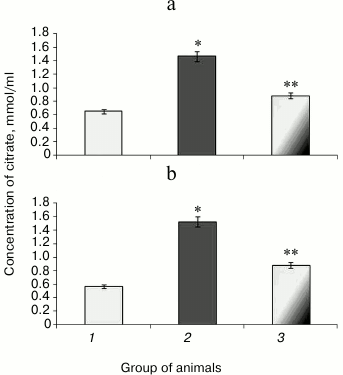
Along with decreased aconitate hydratase activity observed during hyperglycemia, the amount of the low molecular weight antioxidant citrate, its cognate substrate, was upregulated, apparently due to its impaired utilization [37]. Animals with experimental protamine sulfate-induced hyperglycemia were found to have citrate upregulated by 2.3- (Fig. 4a) and 2.7-fold in liver and blood serum (Fig. 4b), respectively. Administration of SkQ1 at the examined dose resulted in 2-fold decrease in amount of citrate in liver (Fig. 4a) and 1.7-fold decrease in blood serum (Fig. 4b) compared to untreated animals. Injection of SkQ1 probably led to enhanced antioxidant potential, and the amount of citrate tended to normalize.
Fig. 4. Content of citrate in liver (a) and blood serum (b) in rats from control group (1) and animals with protamine sulfate-induced hyperglycemia either untreated (2) or SkQ1-treated (3).
During the study, it was found that animals with protamine sulfate-induced hyperglycemia had upregulated SOD specific activity in liver and blood serum by 2.6- and 1.9-fold, respectively (Figs. 5a and 5b) compared to the control group. Enzyme activity in liver and blood serum expressed in U/g liver wet weight and U/ml blood serum increased 2- (Fig. 5c) and 1.5-fold (Fig. 5d), respectively. It seems that upregulated production of superoxide anion by mitochondria observed under chronic hyperglycemia [7, 8] results in compensatory enhancement of SOD activity. There are cytoplasmic, extracellular, and mitochondrial isoforms of superoxide dismutase containing heme metal ions such as Zn2+ and Cu2+ (cytoplasmic and extracellular SOD), whereas the latter contains Mn2+ [38]. It is supposed that extracellular SOD functions to defend endothelial cells in the entire body. The protective action of cytoplasmic SOD is related to detoxication of not only superoxide anion radical, but hydroxyl radical as well. In particular, it was shown that after administration of agents containing SOD and catalase, extracellular SOD displayed a more pronounced effect in lowering ROS than the intracellular isoform [39]. Apparently, apart from the extracellular SOD, the serum pool of the enzyme can consist of other isoforms that are released from cells after their death. It is known that during development of diabetes mellitus, apoptotic processes become activated [40].
Fig. 5. Activity of SOD in rats from control group (1) and animals with protamine sulfate-induced hyperglycemia either untreated (2) or SkQ1-treated (3): a) specific SOD activity in the liver; b) specific SOD activity in the blood serum; c) SOD activity expressed in U/g liver wet weight; d) SOD activity expressed in U/ml blood serum.
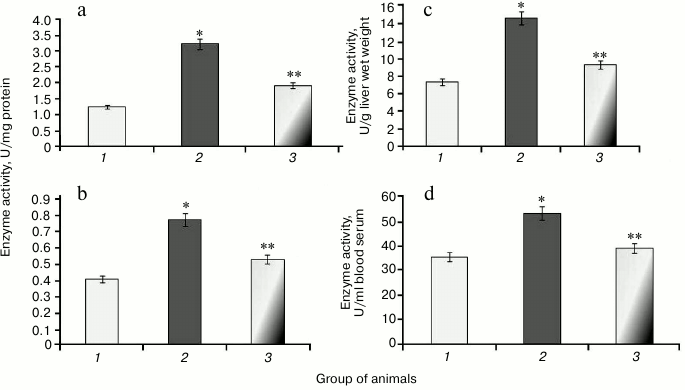
After administration of SkQ1 to animals with hyperglycemia, it was noted that specific activity of SOD decreased 1.7- and 1.5-fold, respectively (Figs. 5a and 5b). Enzyme activity expressed in U/g liver wet weight increased 1.6-fold (Fig. 5c), and in U/ml blood serum by 1.4-fold (Fig. 5d). It is known that SkQ1 can exhibit antioxidant properties due to containing plastoquinone. Moreover, SkQ1 is a lipophilic molecule capable of penetrating into cells [32]. Probably, it contributed to lowering burden on SOD, which was accompanied by return of its activity to the normal range.
It was found that development of protamine sulfate-induced hyperglycemia in rats was accompanied by increased specific activity of catalase in both liver and blood serum by 2.7-fold (Figs. 6a and 6b) compared to the control group. Enzyme activity expressed in U/g liver wet weight and U/ml blood serum was increased 2.3- and 2.1-fold, respectively (Figs. 6c and 6d).
Fig. 6. Activity of catalase in rats from control group (1) and in animals with protamine sulfate-induced hyperglycemia either untreated (2) or SkQ1-treated (3): a) specific activity in liver; b) specific activity in blood serum; c) activity of catalase expressed in U/g liver wet weight; d) activity of catalase expressed in U/ml blood serum.
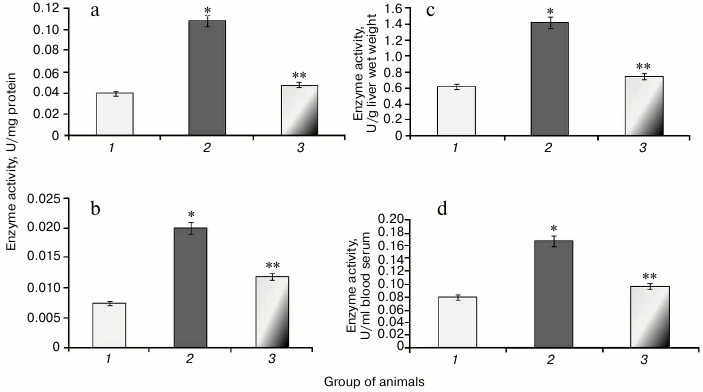
Administration of SkQ1 was noted to decrease specific activity of catalase in liver and blood serum by 2.2- and 1.6-fold, respectively (Figs. 6a and 6b). In addition, enzyme activity expressed in U/g liver wet weight and U/ml blood serum was reduced 1.9- and 1.7-fold, respectively (Figs. 6c and 6d).
The data suggest that the antioxidant potential exhibited in SkQ1-treated animals positively impacted free radical homeostasis in the body thus resulting in decreased stimulation of the antioxidant system in the cells and, in particular, catalase.
Thus, administration of SkQ1 to rats with protamine sulfate-induced hyperglycemia lowers the level of free-radical oxidation, which is reflected by downregulated BCL parameters such as S and Imax denoting the rate of free radical processes, concentration of the primary LPO products – diene conjugates, activity of aconitate hydratase – a target for ROS, and concentration of citrate as well as restored activity of antioxidant enzymes towards the normal range. Apparently, it can be related to the antioxidant effect exhibited by SkQ1. Decreased concentration of glucose observed after administering SkQ1 to animals with hyperglycemia might be coupled with its influence on activity of some factors contributing to enhanced free radical processes and aggravated insulin resistance.
The authors are grateful to Prof. Skulachev Vladimir Petrovich, Director of the Belozersky Institute of Physico-Chemical Biology, for interest in our study and valuable comments during preparation of the manuscript.
REFERENCES
1.Chazova, V. B., and Mychka, V. B. (2003) Metabolic
syndrome, type 2 diabetes mellitus and arterial hypertension,
Serdtse, 3, 32-38.
2.Bayzhanova, Zh. Zh., Ignatova, T. M., Severova, M.
M., and Burnevich, E. Z. (2012) Hepatic steatosis and insulin
resistance during chronic hepatitis C virus infection,
Farmateka, 7, 26-29.
3.Von Frerichs, Fr. Th. (1884) Ueber den
Diabetes, Berlin, 272.
4.Balabolkin, M. I., Kreminskaya, V. M., and
Klebanova, E. M. (2005) A role of oxidative stress in pathogenesis of
diabetic neuropathy and opportunity for its correction by
α-lipoic acid-containing drugs, Probl. Endokrinol.,
51, 22-33.
5.De Haan, J. B., Stefanovic, N., and
Nikolic-Paterson, D. (2005) Kidney expression of glutathione
peroxidase-1 is not protective against streptozotocin-induced diabetic
nephropathy, Am. J. Physiol. Renal. Physiol., 289,
544-551.
6.Martinov, M. V. (2010) The logic of the hepatic
methionine metabolic cycle, Biochim. Biophys. Acta, 1,
89-96.
7.Kondrat’eva, E. I., and Kosyankova, T. V.
(2002) Nitric oxide synthase genes in pathogenesis of diabetes mellitus
and its complications, Probl. Endokrinol., 2,
33-37.
8.Voskresensky, O. N. (2002) Antioxidant system,
ontogenesis, and aging, Vopr. Med. Khim., 1, 14-27.
9.Alonso-Magdalena, P., Ropero, A. B., Soriano, S.,
Quesada, I., and Nadal, A. (2010) Bisphenol-A: a new diabetogenic
factor? Hormones (Athens), 9, 118-126.
10.Krotz, F. (2009) NAD(P)H oxidase-dependent
platelet superoxide anion release increases platelet recruitment,
Blood, 100, 917-924.
11.Skulachev, V. P. (2000) Oxygen and Events of
Programmed Death [in Russian], IBKh RAMN, Moscow.
12.Beinert, H. (1986) Iron-sulfur clusters: agents
of electron transfer and storage, and direct participants in enzymic
reactions, Biochem. Soc. Trans., 14, 527-533.
13.Eberly, D., Clanke, R., and Kaplowitz, N. (1981)
Rapid oxidation in vitro of endogenous and exogenous glutathione
in bile of rats, J. Biol. Chem., 256, 2115-2117.
14.Severin, E. S. (2008) Biochemistry [in
Russian], GEOTAR-Media, Moscow.
15.Zanozina, O. V., Borovkov, N. N., and
Shcherbatyuk, T. G. (2010) Free radical oxidation during type 2
diabetes mellitus: source of generation comprising pathogenetic
mechanisms of toxicity, Sovr. Tekhnol. Med., 3,
104-112.
16.Kazimirko, V. K., Mal’tsev, V. I., Butylin,
V. Yu., and Gorobets, N. I. (2004) Free Radical Oxidation and
Antioxidant System [in Russian], Morion, Kiev.
17.Evans, J. L., Goldfine, I. D., Maddux, B. A., and
Grodsky, G. M. (2009) Oxidative stress and stress-activated signaling
pathways: a unifying hypothesis of type 2 diabetes, Endocrin.
Rev., 23, 599-622.
18.Lin, K. T., Xue, J. Y., and Nomen, M. J. (1995)
Peroxynitrite-induced apoptosis in HL-60 cells, Biol. Chem.,
270, 16487-16490.
19.Antonenko, Y. N., Avetisyan, A. V., Bakeeva, L.
E., Chernyak, B. V., Chertkov, V. A., Domnina, L. V., Ivanova, O. Y.,
Izyumov, D. S., Khailova, L. S., Klishin, S. S., Korshunova, G. A.,
Lyamzaev, K. G., Muntyan, M. S., Nepryakhina, O. K., Pashkovskaya, A.
A., Pletjushkina, O. Y., Pustovidko, A. V., Roginsky, V. A.,
Rokitskaya, T. I., Ruuge, E. K., Saprunova, V. B., Severina, I. I.,
Simonyan, R. A., Skulachev, I. V., Skulachev, M. V., Sumbatyan, N. V.,
Sviryaeva, I. V., Tashlitsky, V. N., Vassiliev, J. M., Vyssokikh, M.
Y., Yaguzhinsky, L. S., Zamyatnin, A. A., Jr., and Skulachev, V. P.
(2008) Mitochondria-targeted plastoquinone derivatives as tools to
interrupt execution of the aging program. 1. Cationic plastoquinone
derivatives: synthesis and in vitro studies, Biochemistry
(Moscow), 73, 1273-1287.
20.Ul’yanov, A. M., and Tarasov, Yu. A. (2000)
Pancreatic insular apparatus in animals under chronic heparin
deficiency, Vopr. Med. Khim., 2, 149-154.
21.Kudryashov, B. A., Pytel’, Yu. A., Lyapina,
L. A., and Baskakova, G. M. (1981) Insulin–heparin complex and
its physiological properties, Vopr. Med. Khim., 27,
547-552.
22.Ul’yanov, A. M., Shapiro, F. B., and
Lyapina, L. A. (1989) Hypoglycemic activity of insulin–heparin
complex and conditions for its manifestation, Patol. Fiziol. Eksp.
Ter., 1, 54-56.
23.Kudryashov, B. A., Ul’yanov, A. M., and
Tarasov, Yu. A. (1989) Protamine sulfate enhances diabetogenic effect
of alloxan, Vopr. Med. Khim., 35, 128-131.
24.Kudrjashov, B. A., Shapiro, F. B., and Ulyanov,
A. M. (1987) Role of heparin of realization of hypoglycaemic action of
insulin, Acta Physiol. Hung., 69, 197-202.
25.Ul’yanov, A. M., Shapiro, F. B., and
Bazaz’yan, G. G. (1987) Lowered sensitivity to hypoglycemic
effects of insulin in animals with reduced concentration of blood
heparin, Byul. Eksp. Biol. Med., 103, 522-524.
26.Stal’naya, I. D. (1977) A Method for
Detecting Diene Conjugates of Unsaturated Higher Fatty Acids. Modern
Methods in Biochemistry [in Russian], Meditsina, Moscow, pp.
63-64.
27.Buzlama, B. C., Retskiy, M. I., Meshcheryakov, N.
P., and Rogacheva, T. E. (1997) A Textbook on Investigating Lipid
Peroxidation Processes and Body Antioxidant Defense System in
Animals [in Russian], Voronezh.
28.Afanas’ev, V. G., Zaytsev, V. S., and
Vol’fson, T. I. (1973) To a micromethod of detecting citric acid
in the blood serum by photoelectrocolorimeter, Lab. Delo,
4, 115-116.
29.Matyushina, B. N., Loginov, A. S., and Tkachev,
V. D. (1991) Detection of superoxide dismutase activity in samples of
needle liver biopsy during chronic hepatic injuries, Lab. Delo,
7, 16-19.
30.Korolyuk, M. A., Ivanova, L. I., and Mayorova, I.
T. (1988) A method for detecting catalase activity, Lab. Delo,
1, 16-19.
31.Lloyd, E., and Lederman, U. (1990) Guidelines
on Applied Statistics [in Russian], Finansy i Statistika,
Moscow.
32.Chistyakov, V. A., Prazdnova, E. V., Gutnikova,
L. V., Sazykina, M. A., and Sazykin, I. S. (2012) Superoxide scavenging
activity of plastoquinone derivative
10-(6′-plastoquinonyl)decyltriphenylphosphonium (SkQ1),
Biochemistry (Moscow), 77, 776-778.
33.Zinovkin, R. A., Romaschenko, V. P., Galkin, I.
I., Zakharova, V. V., Pletjushkina, O. Yu., Chernyak, B. V., and
Popova, E. N. (2014) Role of mitochondrial reactive oxygen species in
age‐related inflammatory activation of endothelium,
Aging, 6, 661-674.
34.Antonenko, Y. N., Roginsky, V. A., Pashkovskaya,
A. A., Rokitskaya, T. I., Kotova, E. A., Zaspa, A. A., Chernyak, B. V.,
and Skulachev, V. P. (2008) Protective effects of mitochondria-targeted
antioxidant SkQ in aqueous and lipid membrane environments, J.
Membr. Biol., 222, 141-149.
35.Evans, J. L., Goldfine, I. D., Maddux, B. A., and
Grodsky, G. M. (2009) Oxidative stress and stress-activated signaling
pathways: a unifying hypothesis of type 2 diabetes, Endocrin.
Rev., 23, 599-622.
36.Murakami, K., and Yoshino, M. (1997) Inactivation
of aconitase in yeast exposed to oxidative stress, Biochem. Mol.
Biol. Int., 41, 481-486.
37.Gardner, P. R., Nguyen, D. M., and White, C. W.
(1994) Aconitase is a sensitive and critical target of oxygen poisoning
in cultured mammalian cells and in rat lungs, Proc. Natl. Acad. Sci.
USA, 91, 12248-12252.
38.Turpaev, K. T. (2002) Reactive oxygen
intermediates and gene expression, Biochemistry (Moscow),
67, 281-292.
39.Dubinina, E. E. (1995) Characteristics of
extracellular superoxide dismutase, Vopr. Med. Khim., 41,
8-12.
40.Popova, T. N., Agarkov, A. A., and Verevkin, A.
N. (2013) Intensity of free radical processes in rat liver during type
2 diabetes mellitus and in response to administered epifamine, Acta
Naturae, 5, 129-134.