MINI-REVIEW: DNA Methylation, Mitochondria, and Programmed Aging
L. A. Zinovkina1 and R. A. Zinovkin2*
1Lomonosov Moscow State University, Faculty of Bioengineering and Bioinformatics, 119991 Moscow, Russia2Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; fax: +7 (495) 939-0338; E-mail: roman.zinovkin@gmail.com
* To whom correspondence should be addressed.
Received July 22, 2015; Revision received August 28, 2015
DNA methylation is a key epigenetic process involved in the regulation of nuclear gene expression. Progress in the study of genomic DNA methylation led to the precise identification of methylation sites reflecting biological age of cells and tissues. However, the functional significance of mitochondrial DNA (mtDNA) methylation remains unknown. Growing evidence suggests that mtDNA methylation is linked to aging and oxidative stress. This mini-review summarizes information about the methylation of nuclear and mtDNA in mammals, indicating the connection of these processes to programmed aging.
KEY WORDS: epigenetics, DNA methylation, mitochondria, agingDOI: 10.1134/S0006297915120044
Abbreviations: DNMT, DNA-methyltransferase; 5hmC, 5-hydroxymethylcytosine; 5mC, 5-methylcytosine; mtDNA, mitochondrial DNA; mtROS, mitochondrial reactive oxygen species; nDNA, nuclear DNA; TET, ten-eleven-translocation oxygenase.
NUCLEAR DNA METHYLATION
DNA methylation is one of the key epigenetic modifications regulating gene expression. DNA methylation involves the transfer of a methyl group to the carbon atom in the fifth position of cytosine using S-adenosyl-L-methionine (SAM) as the methyl group donor. This process results in the formation of 5-methylcytosine (5mC) and S-adenosyl-L-homocysteine. In humans, there are several cytosine DNA methyltransferases – DNMT1, DNMT3a, DNMT3b, and DNMT3L. All these enzymes contain a highly conservative C-terminal catalytic domain and N-terminal regulatory variable regions, interacting with other proteins and chromatin [1]. Enzymes of the TET group (ten eleven translocation) cause 5mC oxidation, which results in the formation of 5-hydroxymethylcytosine (5hmC) [2].
DNA methylation primarily takes place in CpG sequences, located within CpG-islands found in the promoter areas of transcriptionally active genes. According to recent findings, cytosine methylation can occur also outside of CpG dinucleotides [3, 4]. Initial methylation patterns are formed by methyltransferases DNMT3a and DNMT3b, which are active in embryogenesis. DNMT3L apparently functions as an adapter protein required for DNA methylation in gametogenesis [5]. DNMT3a and DNMT3b and to a lesser extent, DNMT1, possess de novo methylating activity. Hemimethylated DNA molecules are formed during replication, and DNMT1 is responsible for the restoration and maintenance of the methylation patterns in mammals. A recently proposed stochastic methylation model has gained popularity. According to this model, methylation in each site results from two opposite processes – methylation and demethylation dependent on the activity of DNA methyltransferases, enzymes of the TET family, and on the chromatin state [6].
METHYLATION OF mtDNA
Cytosine methylation in mitochondrial DNA (mtDNA) was discovered in the 1970s [7-9]. It is important to note that specificity of cytosine DNA-methyltransferase isolated from animal mitochondria was different from that of nuclear DNA-methyltransferase [10]. The methylation of mtDNA was confirmed in subsequent studies [11, 12], but its functional significance has not been determined.
Research in this area has intensified in the last decade. The presence of 5hmC was reported for mtDNA [13], and methylation patterns have been partially determined. These patterns are nearly identical for the entire mitochondrial genome in various tissues and cell types, except for several areas where methylation differed depending on tissue specificity [14]. Regulatory regions of mtDNA (promoter regions, especially in the conserved sequence blocks CSB II and CSB III) were enriched in the methylated cytosine residues [15]. These areas play an important role in binding with TEFM protein responsible for switching between synthesis of short RNA primers for replication and mtDNA transcription [16].
Some regulatory regions of mtDNA include several CpG sites that are protected from methylation by exogenous bacterial enzymes in vitro [17]. The presence of these sites has been demonstrated at the origin of light strand synthesis OL and TERM sequence, where transcription from LSP and HSP1 promoters is terminated. It is assumed that methylation of these areas can affect replication and transcription in mitochondria [18].
In addition, many methylated cytosines are located in the termination-associated sequence (TAS), where 7S DNA synthesis is terminated (7S DNA is required for the formation of the D-loop in mtDNA). The D-loop is not always present in mtDNA molecules, and its functions remain unknown. It is assumed that D-loop might be involved in regulation of replication and in mtDNA recombination; it might provide open mtDNA conformation for free access to the enzyme, as well as anchoring of mtDNA to the inner membrane. The regulation mechanism of D-loop synthesis has not been studied, but it likely takes place in the TAS area [19].
Cytosine methylation in mitochondria is probably carried out by the mitochondrial isoform of DNMT1 (mtDNMT1). This isoform is synthesized from the same gene and its elongated transcript contains a mitochondrial targeting sequence [13]. Some studies have shown the presence of not only DNMT1, but also DNMT3a/3b in mitochondria [15, 20].
Interestingly, mitochondrial methylation occurs not only at CpG sites: dinucleotides CpA, CpC, and CpT were predominantly methylated in the D-loop area [15]. The functional significance and corresponding methylation enzymes have not yet been determined.
Only in the last few years, changes in mtDNA methylation pattern were shown to be influenced by various factors [18, 21, 22].
Hypermethylation of 12S rRNA and Phe-tRNA genes and the D-loop area was observed in workers whose professional activity was associated with long-term exposure to airborne pollutants [23]. The quantity of 12S rRNA limits mitochondrial translation, one of the promoters is located in the Phe-tRNA gene, and many mtDNA regulatory elements are concentrated in the D-loop. It seems likely that environmental conditions can affect mtDNA methylation by altering the expression of mitochondrial genes.
Various oncological diseases can be accompanied by mtDNA hypermethylation [24]. It is possible that this process changes the expression of mitochondrial genes leading to reduced activity of the mitochondrial respiratory chain and development of the Warburg effect. This in turn can result in changes in the expression profile of nuclear genes, thereby promoting oncogenesis.
It was shown that expression of certain mitochondrial genes was altered in cells overexpressing mtDNMT1: the amount of ND6 encoded by the L-chain was reduced, while the amount of ND1 encoded by the H-chain increased [13]. Interestingly, transcription factors NRF1 and PGC1-α, known to be activated by oxidative stress, also increased DNMT1 expression [13]. Perhaps mitochondrial genes’ expression is regulated via methylation change caused by the oxidative stress.
Involvement of DNMT1 in the proper functioning of mitochondria is indirectly confirmed by the fact that certain hereditary diseases are associated with mutations in this gene and phenotypically manifested as mitochondrial diseases [1].
DNA METHYLATION AND AGING
A connection between aging and reduced DNA methylation level in mammals was established over 40 years ago [25]. Then, it was discovered that the decrease in DNA methylation in mammalian fibroblast culture positively correlated with the number of divisions of these cells, whereas methylation level in immortalized cells remained unchanged [26]. Based on these data, a hypothesis was proposed explaining age-related dysfunctions in cells and tissues by impaired DNA methylation [27]. Development of modern methods of analysis of genome epigenetic markers provided refined results. It was found that the level of genomic DNA methylation varies with age, and these changes are multidirectional in different CpG areas (see review [28]). It is difficult to explain this multidirectional change by purely stochastic processes accompanying DNA methylation and demethylation, which is in good agreement with the concept of programmed aging that could be caused by changes in DNA methylation [29].
The discovery of biomarkers of aging – epigenetic clocks that enable determination of biological age of cells and tissues – became the most important event for gerontology and epigenetics [30]. Several hundreds of CpG sites with methylation level correlating with biological age were precisely mapped. It is important to note another observation from the same study: it was shown that “manipulations used to obtain pluripotent cells move the epigenetic clock to the zero mark” [30].
Chronic inflammation is one of the most thoroughly studied factors causing changes in methylation level [31, 32], but the reasons of this change remain unknown. According to the “inflammaging theory”, prolonged inflammation causes an imbalance of the immune system and facilitates the development of senile phenotype [33]. The role of mitochondria in this process was first described in 2006 [34]: when cellular mtDNA was replaced for another haplotype, cytokine expression was affected both at basal conditions and when stimulated by IL-6. This indicates the presence of a retrograde signaling pathway between the mitochondrial and nuclear genomes that can guide inflammatory processes accompanying aging.
Functioning of the mitochondrial respiratory chain is coupled to the production of toxic mtROS (mitochondrial reactive oxygen species) causing oxidative stress in cells, tissues, and organs. The mtROS act as messengers of inflammatory response [35] and accompany virtually any type of oxidative stress caused by exogenous or endogenous factors [36]. It has been shown that oxidative stress induced by hydrogen peroxide leads to the binding of DNMT1 with chromatin; it also changes the level of nuclear DNA (nDNA) methylation [37]. The critical role of mtROS in the process of aging has been discussed in detail in the works of Skulachev [38, 39] and is not the purpose of this review.
A previously popular hypothesis stated that aging in mammals is caused by mutations accumulated in mtDNA. It suggested a scheme of a so-called “vicious cycle”: mtDNA mutations → respiratory chain dysfunction → increased mtROS production → mtDNA mutations [40, 41]. This hypothesis was supported by the following facts: (i) the level of somatic mutations is higher in mtDNA than in nDNA [42]; (ii) mtDNA repair pathways are limited compared to nDNA [43]. ROS cause the formation of 8-oxo-dG resulting in G:C → T:A transversions after mtDNA replication [44, 45]; (iii) transgenic “mutator” mice with impaired proofreading activity of polymerase γ responsible for mtDNA replication quickly accumulated a large number of mtDNA mutations, aged prematurely, and died [46].
The overall level of mtDNA mutations was found to be low in most tissues, and accumulation of mtDNA mutations leads to clonal spread of mtDNA versions and dysfunction of tissues and organs [47]. It is worth noting that reparative processes in mitochondria were also underestimated. It was shown that the base excision repair (BER) system effectively works in mitochondria; components of the system mismatch repair (MMR) are also present [48]. It is believed that mitochondria lack the nucleotide excision repair (NER) system, although the nuclear components of this system, proteins CSA and CSB, are imported into mitochondria under oxidative stress. It is assumed that CSB is involved in the regulation of transcription and BER in mitochondria [49]. The possibility of repair of double-stranded mtDNA damages has not been studied, but it was shown, that the key enzyme of repair based on homologous recombination, Rad51, was imported into mitochondria under oxidative stress and was involved in replication [50].
The leading role of 8-oxo-dG in mtDNA mutagenesis has also been argued. The frequency of transversions, including G:C → T:A, did not vary with age in mitochondria, but the frequency of transitions increased. Apparently, the mitochondrial repair systems effectively eliminate damage caused by 8-oxo-dG [51].
Heterozygous mutator mice had normal lifespan, although they accumulated hundreds of times more mutations in mtDNA than aging wild-type mice [52]. Thus, the mitochondrial stochastic theory of aging was rejected.
So far, very few studies have been performed concerning aging and mtDNA methylation. 5mC level in the 12S mtDNA gene decreased with age [22]. In mtDNA of mice cerebral cortex, the level of 5hmC, but not 5mC, increased with age [53]. The amount of the mitochondrial transcripts ND2, ND4, ND4L, ND5, and ND6 increase with age in the cortex, but not in the cerebellum. It remains unclear if there is any relationship between 5hmC increase and transcriptional upregulation of the mitochondrial complex I genes.
The expression of mitochondrial methylation and hydroxymethylation enzymes was shown to be age-dependent. The level of mtDNMT1 mRNA decreased and the level of TET1-TET3 mRNA remained unaffected in the cerebral cortex of mice. In the cerebellum, the level of TET2 and TET3 mRNA increased, and the level of mtDNMT1 mRNA did not change with age [53]. Methylation of sites located before mitochondrial genes ND6, ATP6, and COX1 decreased during embryonic development of the human brain [14].
EVIDENCE OF EPIGENETIC MECHANISMS IN AGING
Methylation of CpG islands in the nuclear genome of rho0 cells lacking mtDNA was changed compared to the original cell lines. Introduction of mtDNA into rho0 cells resulted in partial restoration of the methylation profile [21]. The degree of methylation of nuclear genes encoding mitochondrial proteins was shown to be tissue-specific; it correlated with the level of gene expression and mitochondrial activity [54].
Data obtained by the group of Prof. J. Hayashi [55] provide the most striking evidence of the functioning of epigenetic mechanisms in aging. The respiratory chain does not work so effectively in old people leading to decrease in oxygen consumption rate and formation of “senile” cell phenotype. They restored the normal functioning of the respiratory chain in cells with “senile” phenotype by cell reprogramming. Analysis of gene expression showed the epigenetic reduction of the expression of nuclear gene GCAT that caused changes in the functioning of the respiratory chain. (Gene GCAT encodes glycine C-acetyltransferase involved in glycine biosynthesis in mitochondria.) Addition of glycine to the medium with fibroblasts with “senile” phenotype partially restored respiratory functions. Since the product of the gene SHMT2, serine hydroxymethyltransferase, is also involved in mitochondrial glycine biosynthesis, an additional study revealed that SHMT2 mRNA level is significantly reduced in the fibroblasts of old people compared to the young individuals.
Decreased GCAT and SHMT2 gene expression by shRNA and siRNA, respectively, in the fibroblasts of young patients led to the respiratory chain dysfunction typical for the “senile phenotype”. Thus, epigenetic processes led to age-related mitochondrial dysfunctions by modifying expression of genes responsible for the mitochondrial metabolism, in particular, in the formation of glycine from serine (SHMT2) and L-threonine (GCAT). The lack of glycine in mitochondria leads to impaired mitochondrial translation causing respiratory chain dysfunctions leading to “senile phenotype” [55]. Furthermore, SHMT-dependent mitochondrial and cytoplasmic folate cycles are coupled to the methionine cycle converting methionine to SAM – the donor of methyl groups for the methylation reactions of mitochondrial and cytoplasmic DNA. Therefore, the reduction in the activity of enzymes involved in glycine synthesis can significantly affect not only translation, but also methylation, providing epigenetic changes in gene expression leading to metabolic “senile” changes.
It would be logical to suggest that the scheme “age-related epigenetic changes in mitochondria and nucleus → changes in the expression of nuclear and/or mitochondrial genes → altered mitochondrial metabolism → formation of “senile phenotype” can explain functioning of the “aging program” involving many metabolic pathways and genes (figure). The following factors may play the key role in this program. (1) Epigenetic changes in the nuclear encoded mitochondrial enzymes (primarily DNA-polymerase γ, RNA-polymerase POLRMT, other replication proteins, and transcription factors) may drastically affect mitochondrial metabolism. (2) The variety of ways to form mitochondrial isoforms of enzymes active in both nucleus and mitochondria suggests many opportunities for epigenetic and other types of regulation. Mitochondrial isoforms are synthesized by: (i) alternative splicing (glycosylase OGG1) [56]; (ii) transcription from different promoters of a single gene (DNMT1 and uracil glycosylase UNG) [13, 56]; (iii) alternative translation initiation from a common transcript (topoisomerase TOP3Amt) [57]; (iv) limited proteolysis of the nuclear isoform (topoisomerase TOP2Bmt) [58]; (v) carrying both nuclear and mitochondrial localization sequences (thymine glycol DNA glycosylase hNTHL1) [56].
Hypothetical scheme of an aging program involving nuclear and mitochondrial epigenetic mechanisms. Explanations are provided in the text
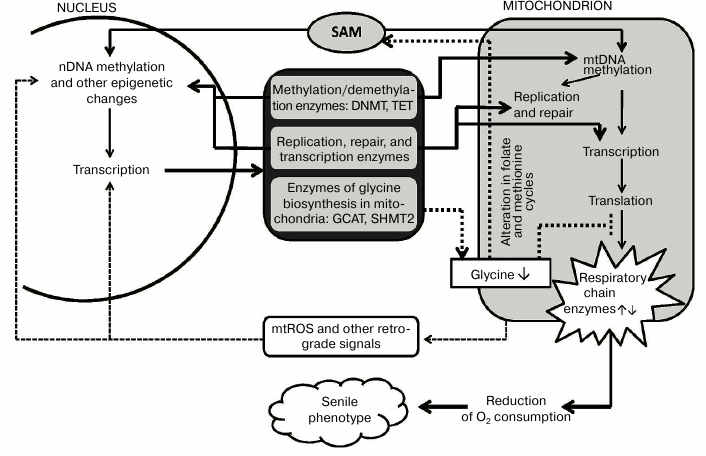
In addition, some nuclear repair enzymes are targeted to mitochondria under oxidative stress. Interestingly, these enzymes apparently perform somewhat different functions in mitochondria than in the nucleus. For example, Rad51, the key factor in homologous recombination of DNA during double strand break, is also involved in mitochondrial replication and targeted to the mitochondria only under oxidative stress [50]. Oxidative stress greatly increases the levels of AP-endonuclease APEX1 as well as the NER components CSA and CSB. It was found that in mitochondria CSB is probably involved in transcription but not repair enhancing processivity of POLRMT [49]. It seems possible that oxidative stress affects mitochondrial replication and transcription.
The results suggest a close relationship in the regulation of mitochondrial and nuclear gene expression. Methylation might be one of these regulation methods, also functioning in aging. Both SAM and methylation/demethylation enzymes DNMT and TET are active in the nucleus and in mitochondria. Age-related changes in the levels of mitochondrial DNMT and TET [53] indicate the involvement of the mitochondrial methylation system in aging processes. Aging is often accompanied by oxidative stress that activates transcription factors NRF1 and PGC1-α, leading to increased expression of mitochondrial DNMT1 [13]. Although we know that methylation in the nucleus can alter gene expression, including those of mitochondrial proteins, the effect of changes in methylation and expression of the mitochondrial genome on the expression of nuclear genes remains unexplored. Nevertheless, these retrograde effects are also possible at both epigenetic and transcriptional regulation levels.
Recent advances in the determination of the nDNA methylation sites reflecting biological age [30], as well as discovery of the key role of epigenetic modifications during cell senescence [55], do support the earlier hypothesis by Prof. B. F. Vanyushin [29] about nDNA methylation as a mechanism of programmed aging (phenoptosis) proposed by Prof. V. P. Skulachev [59]. The methylation of mtDNA and retrograde signaling might also play an important role in the realization of this program.
Authors would like to express their deep gratitude to Prof. V. P. Skulachev.
This work was supported by the Russian Science Foundation (project No. 14-24-00107).
REFERENCES
1.Maresca, A., Zaffagnini, M., Caporali, L., Carelli,
V., and Zanna, C. (2015) DNA methyltransferase 1 mutations and
mitochondrial pathology: is mtDNA methylated? Front. Genet.,
6, 90.
2.Kohli, R. M., and Zhang, Y. (2013) TET enzymes, TDG
and the dynamics of DNA demethylation, Nature, 502,
472-479.
3.Arand, J., Spieler, D., Karius, T., Branco, M. R.,
Meilinger, D., Meissner, A., Jenuwein, T., Xu, G., Leonhardt, H., Wolf,
V., and Walter, J. (2012) In vivo control of CpG and non-CpG DNA
methylation by DNA methyltransferases, PLoS Genet., 8,
e1002750.
4.Guo, J. U., Su, Y., Shin, J. H., Shin, J., Li, H.,
Xie, B., Zhong, C., Hu, S., Le, T., Fan, G., Zhu, H., Chang, Q., Gao,
Y., Ming, G. L., and Song, H. (2014) Distribution, recognition, and
regulation of non-CpG methylation in the adult mammalian brain, Nat.
Neurosci., 17, 215-222.
5.Bourc’his, D., Xu, G. L., Lin, C. S.,
Bollman, B., and Bestor, T. H. (2001) Dnmt3L and the establishment of
maternal genomic imprints, Science, 294, 2536-2539.
6.Jeltsch, A., and Jurkowska, R. Z. (2014) New
concepts in DNA methylation, Trends Biochem. Sci., 39,
310-318.
7.Vanyushin, B. F., Kiryanov, G. I., Kudryashova, I.
B., and Belozersky, A. N. (1971) DNA-methylase in loach embryos
(Misgurnus fossilis), FEBS Lett., 15, 313-316.
8.Vanyushin, B. F., and Kirnos, M. D. (1974) The
nucleotide composition and pyrimidine clusters in DNA from beef heart
mitochondria, FEBS Lett., 39, 195-199.
9.Vanyushin, B. F., and Kirnos, M. D. (1977) The
structure of animal mitochondrial DNA (base composition, pyrimidine
clusters, character of methylation), Mol. Cell. Biochem.,
14, 31-36.
10.Kudriashova, I. B., Kirnos, M. D., and Vaniushin,
B. F. (1976) DNA-methylase activities from animal mitochondria and
nuclei: different specificity of DNA methylation, Biokhimiya,
41, 1968-1977.
11.Shmookler Reis, R. J., and Goldstein, S. (1983)
Mitochondrial DNA in mortal and immortal human cells. Genome number,
integrity, and methylation, J. Biol. Chem., 258,
9078-9085.
12.Pollack, Y., Kasir, J., Shemer, R., Metzger, S.,
and Szyf, M. (1984) Methylation pattern of mouse mitochondrial DNA,
Nucleic Acids Res., 12, 4811-4824.
13.Shock, L. S., Thakkar, P. V., Peterson, E. J.,
Moran, R. G., and Taylor, S. M. (2011) DNA methyltransferase 1,
cytosine methylation, and cytosine hydroxymethylation in mammalian
mitochondria, Proc. Natl. Acad. Sci. USA, 108,
3630-3635.
14.Ghosh, S., Sengupta, S., and Scaria, V. (2014)
Comparative analysis of human mitochondrial methylomes shows distinct
patterns of epigenetic regulation in mitochondria,
Mitochondrion, 18, 58-62.
15.Bellizzi, D., D’Aquila, P., Scafone, T.,
Giordano, M., Riso, V., Riccio, A., and Passarino, G. (2013) The
control region of mitochondrial DNA shows an unusual CpG and non-CpG
methylation pattern, DNA Res., 20, 537-547.
16.Agaronyan, K., Morozov, Y. I., Anikin, M., and
Temiakov, D. (2015) Mitochondrial biology. Replication-transcription
switch in human mitochondria, Science, 347, 548-551.
17.Rebelo, A. P., Williams, S. L., and Moraes, C. T.
(2009) In vivo methylation of mtDNA reveals the dynamics of
protein–mtDNA interactions, Nucleic Acids Res., 37,
6701-6715.
18.Ferreira, A., Serafim, T. L., Sardao, V. A., and
Cunha-Oliveira, T. (2015) Role of mtDNA-related mitoepigenetic
phenomena in cancer, Eur. J. Clin. Invest., 45 (Suppl.
1), 44-49.
19.Nicholls, T. J., and Minczuk, M. (2014) In
D-loop: 40 years of mitochondrial 7S DNA, Exp. Gerontol.,
56, 175-181.
20.Chestnut, B. A., Chang, Q., Price, A., Lesuisse,
C., Wong, M., and Martin, L. J. (2011) Epigenetic regulation of motor
neuron cell death through DNA methylation, J. Neurosci.,
31, 16619-16636.
21.Minocherhomji, S., Tollefsbol, T. O., and Singh,
K. K. (2012) Mitochondrial regulation of epigenetics and its role in
human diseases, Epigenetics, 7, 326-334.
22.Iacobazzi, V., Castegna, A., Infantino, V., and
Andria, G. (2013) Mitochondrial DNA methylation as a next-generation
biomarker and diagnostic tool, Mol. Genet. Metab., 110,
25-34.
23.Byun, H.-M., Panni, T., Motta, V., Hou, L.,
Nordio, F., Apostoli, P., Bertazzi, P. A., and Baccarelli, A. A. (2013)
Effects of airborne pollutants on mitochondrial DNA methylation,
Part. Fibre Toxicol., 10, 18.
24.Sun, C., Reimers, L. L., and Burk, R. D. (2011)
Methylation of HPV16 genome CpG sites is associated with cervix
precancer and cancer, Gynecol. Oncol., 121, 59-63.
25.Vanyushin, B. F., Nemirovsky, L. E., Klimenko, V.
V., Vasiliev, V. K., and Belozersky, A. N. (1973) The 5-methylcytosine
in DNA of rats. Tissue and age specificity and the changes induced by
hydrocortisone and other agents, Gerontologia, 19,
138-152.
26.Wilson, V. L., and Jones, P. A. (1983) DNA
methylation decreases in aging but not in immortal cells,
Science, 220, 1055-1057.
27.Holliday, R. (1985) The significance of DNA
methylation in cellular aging, Basic Life Sci., 35,
269-283.
28.Richardson, B. (2003) Impact of aging on DNA
methylation, Ageing Res. Rev., 2, 245-261.
29.Vanyushin, B. F. (2013) Epigenetics today and
tomorrow, Vavilov Zh. Genet. Selekt., 17, 805-831.
30.Horvath, S. (2013) DNA methylation age of human
tissues and cell types, Genome Biol., 14, 115.
31.Issa, J.-P. J., Ahuja, N., Toyota, M., Bronner,
M. P., and Brentnall, T. A. (2001) Accelerated age-related CpG island
methylation in ulcerative colitis, Cancer Res., 61,
3573-3577.
32.Kang, G. H., Lee, H. J., Hwang, K. S., Lee, S.,
Kim, J.-H., and Kim, J.-S. (2003) Aberrant CpG island hypermethylation
of chronic gastritis, in relation to aging, gender, intestinal
metaplasia, and chronic inflammation, Am. J. Pathol.,
163, 1551-1556.
33.Franceschi, C., Bonafe, M., Valensin, S.,
Olivieri, F., De Luca, M., Ottaviani, E., and De Benedictis, G. (2000)
Inflamm-aging: an evolutionary perspective on immunosenescence, Ann.
N.Y. Acad. Sci., 908, 244-254.
34.Bellizzi, D., Cavalcante, P., Taverna, D., Rose,
G., Passarino, G., Salvioli, S., Franceschi, C., and De Benedictis, G.
(2006) Gene expression of cytokines and cytokine receptors is modulated
by the common variability of the mitochondrial DNA in cybrid cell
lines, Genes Cells, 11, 883-891.
35.Zinovkin, R. A., Romaschenko, V. P., Galkin, I.
I., Zakharova, V. V., Pletjushkina, O. Y., Chernyak, B. V., and Popova,
E. N. (2014) Role of mitochondrial reactive oxygen species in
age-related inflammatory activation of endothelium, Aging (Albany
N.Y.), 6, 661.
36.Zorov, D. B., Filburn, C. R., Klotz, L.-O.,
Zweier, J. L., and Sollott, S. J. (2000) Reactive oxygen species
(ROS-induced) ROS release a new phenomenon accompanying induction of
the mitochondrial permeability transition in cardiac myocytes, J.
Exp. Med., 192, 1001-1014.
37.O’Hagan, H. M., Wang, W., Sen, S., Shields,
C. D., Lee, S. S., Zhang, Y. W., Clements, E. G., Cai, Y., Van Neste,
L., and Easwaran, H. (2011) Oxidative damage targets complexes
containing DNA methyltransferases, SIRT1, and polycomb members to
promoter CpG Islands, Cancer Cell, 20, 606-619.
38.Skulachev, V. P., and Longo, V. D. (2005) Aging
as a mitochondria-mediated atavistic program: can aging be switched
off? Ann. N.Y. Acad. Sci., 1057, 145-164.
39.Skulachev, V. P., Anisimov, V. N., Antonenko, Y.
N., Bakeeva, L. E., Chernyak, B. V., Erichev, V. P., Filenko, O. F.,
Kalinina, N. I., Kapelko, V. I., and Kolosova, N. G., Kopnin, B. P.,
Korshunova, G. A., Lichinitser, M. R., Obukhova, L. A., Pasyukova, E.
G., Pisarenko, O. I., Roginsky, V. A., Ruuge, E. K., Senin, I. I.,
Severina, I. I., Skulachev, M. V., Spivak, I. M., Tashlitsky, V. N.,
Tkachuk, V. A., Vyssokikh, M. Y., Yaguzhinsky, L. S., and Zorov, D. B.
(2009) An attempt to prevent senescence: a mitochondrial approach,
Biochim. Biophys. Acta, 1787, 437-461.
40.Harman, D. (1972) The biologic clock: the
mitochondria? J. Am. Geriatr. Soc., 20, 145-147.
41.Wallace, D. C. (2005) A mitochondrial paradigm of
metabolic and degenerative diseases, aging, and cancer: a dawn for
evolutionary medicine, Annu. Rev. Genet., 39,
359-407.
42.Khrapko, K., Coller, H. A., Andre, P. C., Li, X.
C., Hanekamp, J. S., and Thilly, W. G. (1997) Mitochondrial mutational
spectra in human cells and tissues, Proc. Natl. Acad. Sci. USA,
94, 13798-13803.
43.Brierley, E. J., Johnson, M. A., Lightowlers, R.
N., James, O. F., and Turnbull, D. M. (1998) Role of mitochondrial DNA
mutations in human aging: implications for the central nervous system
and muscle, Ann. Neurol., 43, 217-223.
44.Richter, C., Park, J. W., and Ames, B. N. (1988)
Normal oxidative damage to mitochondrial and nuclear DNA is extensive,
Proc. Natl. Acad. Sci. USA, 85, 6465-6467.
45.De Bont, R., and Van Larebeke, N. (2004)
Endogenous DNA damage in humans: a review of quantitative data,
Mutagenesis, 19, 169-185.
46.Kujoth, G. C., Hiona, A., Pugh, T. D., Someya,
S., Panzer, K., Wohlgemuth, S. E., Hofer, T., Seo, A. Y., Sullivan, R.,
Jobling, W. A., Morrow, J. D., Van Remmen, H., Sedivy, J. M., Yamasoba,
T., Tanokura, M., Weindruch, R., Leeuwenburgh, C., and Prolla, T. A.
(2005) Mitochondrial DNA mutations, oxidative stress, and apoptosis in
mammalian aging, Science, 309, 481-484.
47.Khrapko, K., and Turnbull, D. (2014)
Mitochondrial DNA mutations in aging, Prog. Mol. Biol. Transl.
Sci., 127, 29-62.
48.Kazak, L., Reyes, A., and Holt, I. J. (2012)
Minimizing the damage: repair pathways keep mitochondrial DNA intact,
Nat. Rev. Mol. Cell Biol., 13, 659-671.
49.Kamenisch, Y., and Berneburg, M. (2013)
Mitochondrial CSA and CSB: protein interactions and protection from
ageing associated DNA mutations, Mech. Ageing Dev., 134,
270-274.
50.Sage, J. M., and Knight, K. L. (2013) Human Rad51
promotes mitochondrial DNA synthesis under conditions of increased
replication stress, Mitochondrion, 13, 350-356.
51.Kennedy, S. R., Salk, J. J., Schmitt, M. W., and
Loeb, L. A. (2013) Ultra-sensitive sequencing reveals an age-related
increase in somatic mitochondrial mutations that are inconsistent with
oxidative damage, PLoS Genet., 9, e1003794.
52.Vermulst, M., Bielas, J. H., Kujoth, G. C.,
Ladiges, W. C., Rabinovitch, P. S., Prolla, T. A., and Loeb, L. A.
(2007) Mitochondrial point mutations do not limit the natural lifespan
of mice, Nat. Genet., 39, 540-543.
53.Dzitoyeva, S., Chen, H., and Manev, H. (2012)
Effect of aging on 5-hydroxymethylcytosine in brain mitochondria,
Neurobiol. Aging, 33, 2881-2891.
54.Takasugi, M., Yagi, S., Hirabayashi, K., and
Shiota, K. (2010) DNA methylation status of nuclear-encoded
mitochondrial genes underlies the tissue-dependent mitochondrial
functions, BMC Genomics, 11, 481.
55.Hashizume, O., Ohnishi, S., Mito, T., Shimizu,
A., Iashikawa, K., Nakada, K., Soda, M., Mano, H., Togayachi, S.,
Miyoshi, H., Okita, K., and Hayashi, J. (2015) Epigenetic regulation of
the nuclear-coded GCAT and SHMT2 genes confers human age-associated
mitochondrial respiration defects, Sci. Rep., 5,
10434.
56.Boesch, P., Weber-Lotfi, F., Ibrahim, N.,
Tarasenko, V., Cosset, A., Paulus, F., Lightowlers, R. N., and
Dietrich, A. (2011) DNA repair in organelles: pathways, organization,
regulation, relevance in disease and aging, Biochim. Biophys.
Acta, 1813, 186-200.
57.Wang, Y., Lyu, Y. L., and Wang, J. C. (2002) Dual
localization of human DNA topoisomerase 3α to mitochondria and
nucleus, Proc. Natl. Acad. Sci. USA, 99, 12114-12119.
58.Low, R. L., Orton, S., and Friedman, D. B. (2003)
A truncated form of DNA topoisomerase 2β associates with the mtDNA
genome in mammalian mitochondria, Eur. J. Biochem., 270,
4173-4186.
59.Skulachev, V. P. (1999) Phenoptosis: programmed
death of an organism, Biochemistry (Moscow), 64,
1418-1426.