A Rapid and Cost-Effective Method for DNA Extraction from Archival Herbarium Specimens
A. A. Krinitsina1*, T. V. Sizova2, M. A. Zaika1, A. S. Speranskaya1,3, and A. P. Sukhorukov1
1Lomonosov Moscow State University, Faculty of Biology, 119991 Moscow, Russia; fax: +7 (495) 939-4309; E-mail: krinitsina@mail.ru2Vavilov Institute of General Genetics, Russian Academy of Sciences, 119333 Moscow, Russia; fax: +7 (499) 132-8962
3Central Research Institute of Epidemiology, Federal Service on Customers Rights Protection and Human Well-being Surveillance, 111123 Moscow, Russia; fax: +7 (495) 304-2209
* To whom correspondence should be addressed.
Received July 6, 2015; Revision received July 24, 2015
Here we report a rapid and cost-effective method for the extraction of total DNA from herbarium specimens up to 50-90-year-old. The method takes about 2 h, uses AMPure XP magnetic beads diluted by PEG-8000-containing buffer, and does not require use of traditional volatile components like chloroform, phenol, and liquid nitrogen. It yields up to 4 µg of total nucleic acid with high purity from about 30 mg of dry material. The quality of the extracted DNA was tested by PCR amplification of 5S rRNA and rbcL genes (nuclear and chloroplast DNA markers) and compared against the traditional chloroform/isoamyl alcohol method. Our results demonstrate that the use of the magnetic beads is crucial for extraction of DNA suitable for subsequent PCR from herbarium samples due to the decreasing inhibitor concentrations, reducing short fragments of degraded DNA, and increasing median DNA fragment sizes.
KEY WORDS: DNA extraction, herbarium, PCR, 5S rRNA, rbcL, genomic markers, sequenceDOI: 10.1134/S0006297915110097
Abbreviations: CTAB, cetyltrimethylammonium bromide; MHA, Main Botanical Garden of the Russian Academy of Sciences; MW, Herbarium of the Biological Faculty of Moscow State University; PCR, polymerase chain reaction; PVP40, polyvinylpyrrolidone, 40 kDa.
Nucleotide sequencing of chloroplast DNA markers or nuclear ribosomal
DNA markers is now routinely used to identify systematic position and
infer evolutionary relationships between different groups of plants in
combination with analysis of their morphological and anatomical traits
[1]. Besides taxonomic and phylogenetic studies,
the analysis of marker sequences can also be applied as tools for DNA
barcoding of wild species and characterization of cultivars [2-4]. Herbaria collections are one
of the main sources of preserved material for plant diversity studies.
It has been estimated that worldwide there are at least 3400 officially
registered herbaria, together harboring ~350 million specimens
collected mostly during the last two hundred years (New York Botanical
Garden’s Virtual Herbarium, http://sweetgum.nybg.org/ih/).
Herbaria have not been extensively sampled for DNA-based research,
mainly due to poor usability of herbarium specimens for successful
extraction and PCR amplification of DNA caused by various gathering and
drying methods used followed by different subsequent preservation
history and age of specimens [5]. Besides, both
nuclear and plastid DNA degrades through time [6].
The great decrease in fragment length over time greatly complicates PCR
and DNA sequencing from older herbarium specimens. The quality of the
extracted DNA also depends on taxon-specific factors of herbarium DNA
since different plant species contain a wide spectrum of secondary
compounds present in plant cells, like polyphenols polysaccharides and
other substances interfering with subsequent PCR and sequencing
procedures [6].
Numerous protocols for total DNA extraction from plants have been published. One of the most commonly used methods to extract DNA from plants uses the ionic detergent cetyltrimethylammonium bromide (CTAB) [7]. This method and it various modifications are especially effective for plants with high polyphenolics or polysaccharide content [8, 9]. Therefore, methods for obtaining genomic DNA of quality suitable for PCR from old, dry herbarium specimens and development of new rapid and cost-effective extraction methods yielding high-quality DNA and not requiring specialized equipment and undesirable volatile materials are still required [10].
Here we describe a DNA extraction method that is a combination of modified CTAB and methods where magnetic beads are used. The results show that combination of lysis by CTAB followed by magnetic beads purification strongly affects DNA extraction and PCR success, especially the availability of fragments longer than 350 bp from specimens containing high amounts of secondary metabolites and collected before the 1960s.
MATERIALS AND METHODS
Total DNA was extracted from dried leaf fragments (about 30 mg) of plant herbarium specimens related to genera Scorzonera (Asteraceae). Plant specimens (total 12) were collected during 1920-1960 (Table 1) and deposited in the Herbarium of the Biological Faculty of Moscow State University (acronym MW) and the N. V. Tsitsin Main Botanical Garden of the Russian Academy of Sciences (acronym MHA). For all 12 specimens, total DNA was extracted using three methods listed below. Method 1: DNA extraction using CTAB lysis buffer and the traditional chloroform–isoamyl alcohol purification step (Stewart and Via [8]). Method 2: DNA extraction by Method 1 followed by purification of samples with Agencourt AMPure XP magnetic beads (Beckman Coulter, USA) diluted fourfold by buffer A. Method 3: DNA extraction using CTAB lysis buffer and Agencourt AMPure XP magnetic beads diluted fourfold by buffer A (without chloroform–isoamyl alcohol purification step). Lysis buffer composition: 2% (w/v) CTAB, 100 mM Tris-HCl (pH 8.0), 20 mM EDTA, 1.4 M NaCl, 1% (w/v) PVP40. Buffer A composition: 18% (w/v) PEG-8000, 1 M NaCl, 10 mM Tris-HCl (pH 8.0), 1 mM EDTA.
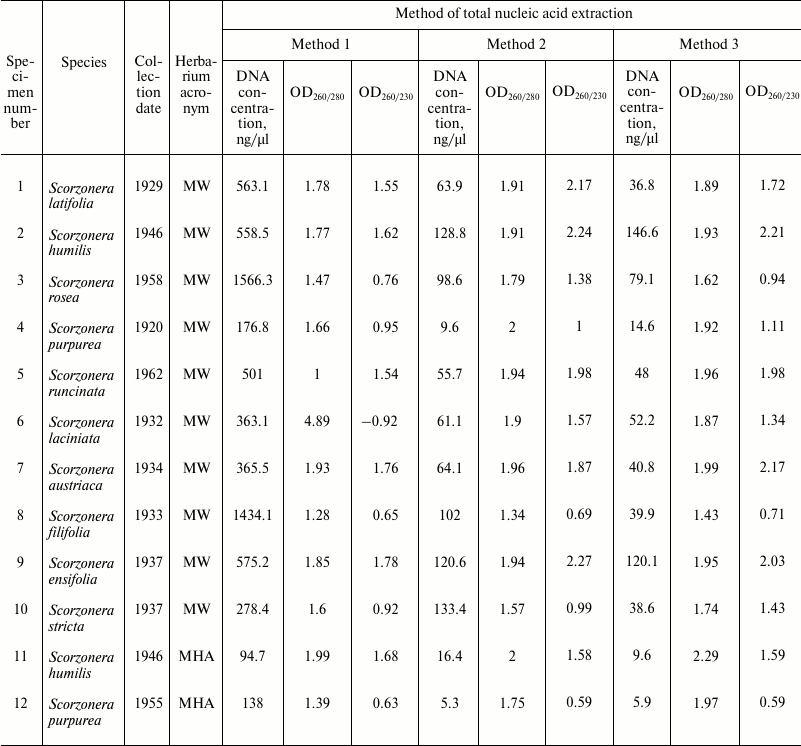
Table 1. DNA concentrations, 260/280 and
260/230 absorbance ratios of samples obtained using different methods
of total nucleic acid extraction from herbarium specimens of the genus
Scorzonera
Method 1. Reagents and solutions: lysis buffer, chloroform–isoamyl alcohol 24 : 1, isopropanol (isopropyl alcohol), 80% ethanol, deionized water. Steps: (1) grind about 30 mg of dry plant sample (leaf) to powder by hand using a paper envelope or medical powder-free gloves, put the powdered sample into a 1.5 ml microcentrifuge tube; (2) add 500 µl of lysis buffer to the powdered sample and resuspend it by pipetting; (3) incubate the sample at 60°C for 60 min; during incubation, invert the tube every 10-15 min; (4) add an equal volume of chloroform–isoamyl alcohol 24 : 1 (500 µl) to lysate and mix carefully by inverting tube during 15-30 s; (5) centrifuge the mixture for 5 min at 13,400 rpm (12,100g); (6) transfer the aqueous phase into a new 1.5-ml tube and repeat steps 4-6; (7) transfer the aqueous phase into a fresh 1.5-ml tube and add equal volume of isopropanol; thoroughly mix and incubate at room temperature for 10 min; (8) centrifuge the mixture for 10 min at 13,400 rpm (12,100g); (9) discard the supernatant and wash the pellet with 500 µl of 80% ethanol; centrifuge the mixture for 2 min at 13,400 rpm (12,100g); remove the supernatant and repeat the washing step twice; (10) dry the pellet during 3-5 min at 60°C; (11) dissolve pellet in 30 µl of deionized water and incubate at 60°C for 5 min.
Method 2. Reagents and solutions: lysis buffer, chloroform–isoamyl alcohol 24 : 1; isopropanol (isopropyl alcohol), 80% ethanol, deionized water, Agencourt AMPure XP magnetic beads, buffer A. Steps: extract DNA from 30 mg of dry sample using Method 1 protocol (steps 1-11); (12) gently shake the Agencourt AMPure XP magnetic beads to achieve a homogeneous suspension and collect an aliquot of the suspension into a fresh microcentrifuge tube, then add triple volume of buffer A and resuspend to homogeneity; (13) add diluted AMPure XP to a sample of isolated nucleic acids at the ratio 1 : 1 v/v; (14) carefully mix by pipetting and incubate for 5 min at room temperature; (15) place tube on magnet stand, incubate on stand for 2 min, remove and discard supernatant without disturbing beads; (16) add 200 µl 80% ethanol for washing, incubate tube on stand for 1 min turning the tube around it axis, remove and discard supernatant, repeat washing once; (17) carefully remove and discard all of the supernatant without disturbing beads, let bead pellet sit open at room temperature to air-dry completely (5-10 min); (18) remove tube from magnetic stand, add 30 µl of deionized water to dry bead pellet, carefully resuspend by repeated pipetting, and incubate at room temperature for 5 min; (19) place tube on magnet stand, incubate on stand for about 2 min (until the solution appears clear), and transfer 28 µl of supernatant to a fresh tube without touching beads.
Method 3. Reagents and solutions: lysis buffer, isopropanol (isopropyl alcohol), 80% ethanol, deionized water, Agencourt AMPure XP magnetic beads, buffer A. Steps: (1) grind about 30 mg of dry plant sample (leaf) to powder by hand using a paper envelope or medical powder-free gloves, put the powdered sample into a 1.5-ml microcentrifuge tube; (2) add 500 µl of lysis buffer to the powdered sample and resuspend it by pipetting, incubate the sample at 60°C for 60 min; during incubation, invert the tube every 10-15 min; (3) centrifuge the sample for 5 min at 13,400 rpm (12,100g) to pellet debris; (4) transfer the supernatant into a fresh 1.5-ml tube and add equal volume of isopropanol; (5) thoroughly mix and incubate at room temperature for 10 min; (6) centrifuge the mixture for 10 min at 13,400 rpm (12,100g); (7) discard the supernatant and wash the pellet with 500 µl of 80% ethanol, centrifuge the mixture for 2 min at 13,400 rpm (12,100g), remove the supernatant and repeat the washing step two times; (8) dry the pellet during 3-5 min; (9) dissolve pellet in 30 µl of deionized water and incubate at 60°C for 5 min; (10) gently shake the Agencourt AMPure XP magnetic beads to achieve a homogeneous suspension and collect an aliquot of the suspension into a fresh microcentrifuge tube, then add triple volume of buffer A and resuspend to homogeneity; (11) add diluted AMPure XP to a sample of isolated nuclei acids at the ratio 1 : 1 v/v; (12) carefully mix by pipetting, and incubate for 5 min at room temperature; (13) place tube on magnet stand, incubate on stand for 2 min, remove and discard supernatant without disturbing beads; (14) add 200 µl of 80% ethanol for washing, incubate tube on stand for 1 min turning the tube around it axis, remove and discard supernatant, repeat washing once; (15) carefully remove and discard all of the supernatant without disturbing beads, let bead pellet sit open at room temperature to air-dry completely (5-10 min); (16) remove tube from magnetic stand, add 30 µl of deionized water to dry bead pellet, carefully resuspend by repeated pipetting, and incubate at room temperature for 5 min; (17) place tube on magnet stand, incubate on stand for about 2 min (until the solution appears clear), and transfer 28 µl of supernatant to new tube without touching beads.
The DNA extraction procedures were carried out using the following equipment: dry block thermostat CH-100 (BioSan, Latvia), minicentrifuge MiniSpin Plus and rotor F-45-12-11 (Eppendorf, Germany), magnetic stand (IsoGel, Russia).
The concentration of total nucleic acids obtained and their quality was estimated using a NanoDrop 2000c spectrophotometer (Thermo Scientific, USA) and Qubit 2.0 fluorometer (Invitrogen, USA). The average DNA fragment size was analyzed using 0.8% agarose gel electrophoresis stained with ethidium bromide. The distribution of the DNA fragment lengths was estimated using an Agilent Bioanalyzer 2100 (Agilent Technologies, USA).
All DNA extracted samples were tested for PCR amplification yield of marker sequences. Two sets of degenerate oligonucleotide primers were used for PCR amplification of chloroplast genome regions: rbcLa-F/rbcLa-R for short fragments of rbcL gene and UrbcL1/UrbcL2 for entire rbcL gene amplification. One set of degenerate oligonucleotide primers – 5SluA-F/5SluA-R – was used for nucleus genome 5S rRNA gene fragment amplification (Table 2).
Table 2. Primer sequences for
PCR-amplification of DNA regions coding 5S rRNA and
rbcL
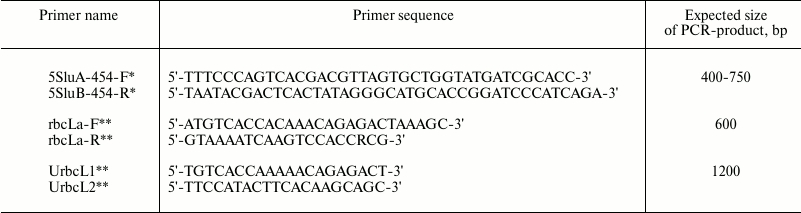
* Oligonucleotides 5SluA-F and 5SluA-R are designed for the 454
sequencing platform (Roche, Switzerland) and are courtesy of N. A.
Mel’nikova.
** rbcLa-F, rbcLa-R, UrbcL1, and UrbcL2 were designed according to the
data from W. J. Kress et al. [3] and R. A. Levin et
al. [10] and synthesized by Syntol (Russia).
PCR was carried out using Taq DNA polymerase (New England Biolabs, USA) according to manufacturer’s instructions. A 50-ng portion of template DNA was added to the reaction mixture (all samples were previously normalized to 10 ng/µl). The PCR protocol used in this study for amplification of 5S rRNA and short fragments of rbcL genes was as follows: 1 cycle of denaturation at 95°C for 10 min, 30 cycles of amplification (5 s denaturation at 95°C, 30 s annealing at 57°C, and 30 s extension at 72°C), and a final step of 72°C for 5 min. The PCR protocol used for amplification of entire rbcL genes was as follows: 1 cycle of denaturation at 95°C for 10 min, 30 cycles of amplification (5 s denaturation at 95°C, 30 s annealing at 50°C, and 35 s extension at 72°C), and a final step of 72°C for 5 min. PCR amplification was done with a PCR system T-100 Thermal Cycler (Bio-Rad, USA).
RESULTS AND DISCUSSION
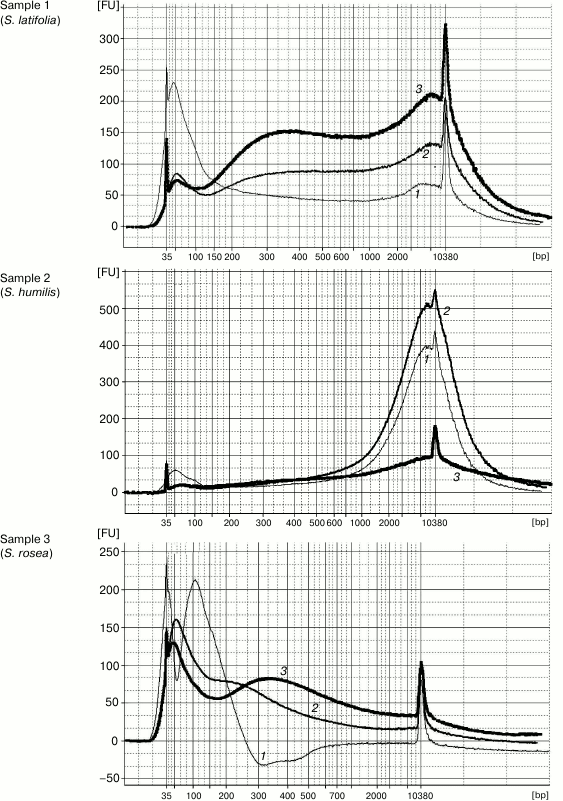
DNA extraction by Method 1 resulted in 100-12,000-bp-long genomic DNA fragments contaminated by RNA. The high level of DNA degradation can be explained by the process of post-collection DNA degradation over time (the youngest specimens were more than 50-year-old). The protocol of Method 2 was the same but was followed by a magnetic beads purification stage to remove unwanted short DNA fragments (<200 bp). Method 3 was a combination of first two stages (CTAB buffer lysis and isopropanol precipitation) from Method 1 and the magnetic beads purification. The original Agencourt AMPure XP magnetic beads reagent was diluted fourfold for decreasing the cost of experiments. The results demonstrated that both methods containing the magnetic beads purification stage yielded DNA samples with high purity, unlike that obtained by Method 1. The Agilent 2100 bioanalyzer provides evidence that purifying DNA samples using magnetic beads increases average DNA fragment size (longer than 300 bp) (Fig. 1). The cleaning of template from short disrupted DNA fragments and polyphenolic compounds giving brown color reduces useless template for molecular studies and improves the probability of generating a PCR target. Therefore, the magnetic beads purification stage is significant for old plant specimens containing highly degraded genomic DNA.
Fig. 1. Capillary electrophoresis by Agilent Bioanalyzer 2100. Sample numbers correspond to the numbers of herbarium specimens (Table 1). DNA was extracted using Method 1 (curves 1), Method 2 (curves 2), and Method 3 (curves 3). Y-axis, fluorescence intensity; X-axis, fragment length (bp).
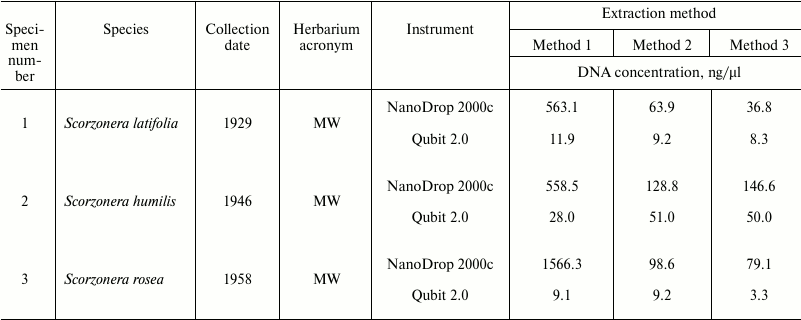
Spectrophotometric analysis of extracted nucleic acids demonstrated that the basic method (Method 1) yields samples with high concentration (94.7-1566 ng/µl). DNA purification using both magnetic beads methods result in 12-fold (Method 2) and 18-fold (Method 3) decreasing of DNA concentrations (to 5.3-133.4 and 5.9-146.6 ng/µl, respectively) (Table 1). At the same time, measurement of DNA concentration by fluorescence measurements using dyes interacting with DNA by intercalation (Qubit 2.0) showed that samples obtained by all three methods contain approximately equal amounts of DNA (Table 3). Measurement of DNA concentration by the Agilent 2100 bioanalyzer confirmed the fluorimeter results (data not shown).
Table 3. DNA concentrations measured by
NanoDrop 2000c spectrophotometer and Qubit 2.0 fluorometer
The values of 260/280 and 260/230 nm absorbance ratios demonstrated that application of the basic method (Method 1) yields low-purity DNA, since OD260/280 values of most samples do not fall into the optimal limit of 1.7-2.0 of pure DNA. At the same time, the CTAB/magnetic beads methods yield samples with noticeably better quality but lower DNA concentration (OD260/280 between 1.7 and 2.0 in 10 out of 12 samples in Method 2, and in 9 out of 12 in Method 3) (Table 1).
As previously reported by several authors, nucleic acid extraction by CTAB accompanied with a chloroform/isoamyl alcohol stage purification yields high quality DNA samples. However, our experiments show that DNA concentration value measured by absorbance at 260 nm distinctly differs from the visible electrophoresis data. We suppose that samples obtained by Method 1 contain some components of lysis buffer, including CTAB, which may interfere with spectrophotometric measurements [11].
The usability of DNA samples for phylogenetically informative molecular markers analysis was tested by amplification of marker nuclear and chloroplast genome gene fragments commonly used for phylogenetic analysis: 5S rRNA (400-750 bp) and rbcL (~600 bp) [12]. The results demonstrated that some samples, produced by Method 1 and used as a template, did not yield PCR amplification products of 5S rRNA genes, while the samples extracted from the same specimens by both magnetic bead-based methods resulted in successful amplification. Products of expected size (400-750 bp) were not detected by electrophoresis for samples 3, 6, and 8 (Method 1), but they were visible on the gel for samples 3 and 6 extracted by Method 2 and for samples 3, 6, and 8 extracted by Method 3 (Fig. 2).
Fig. 2. Agarose electrophoresis of PCR-amplified fragments of 5S rRNA gene. Numbers of lanes coincide with those of herbarium specimens (see Table 1). M is DNA marker GeneRuler, 1000 bp (Thermo Scientific, USA).
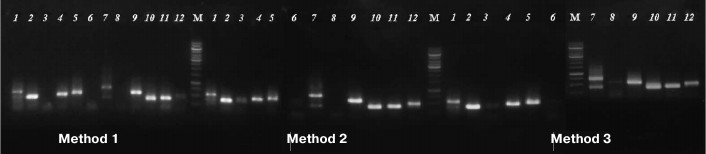
PCR-amplification of the rbcL gene fragment (~600 bp) was successful in 8 of 12 samples isolated by Method 1 and in 11 of 12 samples obtained by Methods 2 and 3 (experimental data not shown).
Plants belonging to the Scorzonera genus commonly contain a large variety of secondary metabolites [13]. Increase in PCR efficiency for templates extracted by magnetic bead-based methods indicates that magnetic purification stage is strictly necessary for pure DNA obtaining because it removes compounds that inhibit the PCR enzymes [14]. Therefore, we suppose the CTAB-lysis/magnetic beads method is a good approach to isolation of DNA from secondary metabolite-rich plant species amenable for PCR and profiling experiments.
Amplification of the chloroplast DNA fragment, which covers most of the full-length rbcL gene (~1200 bp), was successful for only one sample (No. 7) if the extraction Method 1 (without magnetic beads) was used. For the other two methods, the amplification product was detected for samples obtained from specimens Nos. 4 and 7 (experimental data not shown). The fact that there were no full-length rbcL amplification products for most of the samples is a result of the discrepancy of desired amplification product sizes and means of average DNA fragment sizes of the samples. It should be noted that DNA fragmentation does not correlate with the age of specimens. For example, full-length rbcL amplification was obtained by PCR of DNA extracted from specimens collected in 1920 and 1934, but do not from those collected 25 years later. The same results were obtained for herbarium specimens of other plant taxonomy groups [6, 15].
Method of DNA extraction using magnetic beads without chloroform yields DNA samples suitable for PCR amplification of nuclear and chloroplast markers 400-750 bp long. Amplification of fragments longer than 1000 bp is not effective enough if template is derived from old plant material due to DNA degradation through storage of the specimens. The proposed method for plant DNA extraction is rapid, cost-effective, and does not required special toxic and expensive reagents and special facilities for investigator safety and therefore can be widely used for effective DNA extraction from various dried plant samples, e.g. during DNA-barcoding of herbarium specimens, e.g. outside of high-technology molecular biology laboratories.
This study was supported by the Russian Science Foundation (project No. 14-50-00029).
REFERENCES
1.Sukhorukov, A. P., Mavrodiev, E. V., Struwig, M.,
Nilova, M. V., Dzhalilova, Kh. Kh., Balandin, S. A., Erst, A., and
Krinitsyna, A. A. (2015) One-seeded fruits in the core Caryophyllales:
their origin and structural diversity, PLoS One, 10, 1-38.
2.Hajibabaei, M., Singer, G. A. C., Hebert, P. D. N.,
and Hickey, D. A. (2007) DNA barcoding: how it complements taxonomy,
molecular phylogenetics and population genetics, Trends Genet., 23,
167-172.
3.Kress, W. J., and Erickson, D. L. (2007) A
two-locus global DNA barcode for land plants: the coding rbcL gene
complements the non-coding trnH–psbA spacer region, PLoS One, 2,
e508.
4.Kress, W. J., Erickson, D. L., Jones, F. A.,
Swenson, N. G., Perez, R., Sanjur, O., and Bermingham, E. (2009) Plant
DNA barcodes and a community phylogeny of a tropical forest dynamics
plot in Panama, PNAS, 106, 18621-18626.
5.Srinivansan, M., Sedmak, D., and Jewell, S. (2002)
Effect of fixatives and tissue processing on the content and integrity
of nucleic acids, Am. J. Pathol., 161, 1961-1971.
6.Doyle, J. J., and Dickson, E. E. (1987)
Preservation of plant species for DNA restriction endonuclease
analysis, Taxon, 36, 715-722.
7.Doyle, J. J., and Doyle, J. L. (1987) A rapid DNA
isolation procedure for small quantities of fresh leaf tissue, Phyt.
Bull., 19, 11-15.
8.Stewart, C. N., Jr., and Via, L. E. (1993) A rapid
CTAB DNA isolation technique useful for RAPD fingerprinting and other
PCR applications, BioTechniques, 14, 748-749.
9.Porebski, S., Bailey L. G., and Baum, B. R (1997)
Modification of a CTAB DNA extraction protocol for plants containing
high polysaccharide and polyphenol components, Plant Mol. Biol. Rep.,
15, 8-15.
10.Sarkinen, T., Staats, M., Richardson, J. E.,
Cowan, R. S., and Bakker, F. T. (2012) How to open the treasure chest?
Optimising DNA extraction from herbarium specimens, PLoS One, 7,
e43808.
11.Drabkova, L., Kirschner, J., and Vlcek, C. (2002)
Comparison of seven DNA extraction and amplification protocols in
historical herbarium specimens of Juncaceae, Plant Mol. Biol. Rep., 20,
161-175.
12.Levin, R. A., Wagner, W. L., Hoch, P. C.,
Nepokroeff, M., Piers, J. C., Zimmer, E. A., and Sytsma, K. J. (2003)
Family-level relationships of Onagraceae based on chloroplast rbcL and
ndhF data, Am. J. Bot., 90, 107-115.
13.Sar, A., Zidorn, C., Ellmerer, Ernst P., Ozgokce,
F., Ongania, K.-H., and Stuppner, H. (2007) Phenolic compounds from
Scorzonera tomentosa L., HCA, 90, 311-317.
14.Sharma, A. D., Gill, P. K., and Singh, P. (2002)
DNA isolation from dry and fresh samples of polysaccharide-rich plants,
Plant Mol. Biol. Rep., 20, 415-415.
15.Erkens, R. H. J., Cross, H., Maas, J. W.,
Hoenselaar, K., and Chatrou, L. W. (2008) Assessment of age and
greenness of herbarium specimens as predictors for successful
extraction and amplification of DNA, BLUMEA, 53, 407-428.