Stages of Cell Cannibalism – Entosis – in Normal Human Keratinocyte Culture
A. S. Garanina*, L. A. Khashba, and G. E. Onishchenko
Lomonosov Moscow State University, Biological Faculty, 119991 Moscow, Russia; fax: +7 (495) 939-4309; E-mail: anastasiacit@gmail.com* To whom correspondence should be addressed.
Received July 6, 2015
Entosis is a type of cell cannibalism during which one cell penetrates into another cell and usually dies inside it. Researchers mainly pay attention to initial and final stages of entosis. Besides, tumor cells in suspension are the primary object of studies. In the present study, we investigated morphological changes of both cells-participants of entosis during this process. The substrate-dependent culture of human normal keratinocytes HaCaT was chosen for the work. A combination of light microscopy and scanning electron microscopy was used to prove that one cell was completely surrounded by the plasma membrane of another cell. We investigated such “cell-in-cell” structures and described the structural and functional changes of both cells during entosis. The outer cell nucleus localization and shape were changed. Gradual degradation of the inner cell nucleus and of the junctions between the inner and the outer cells was revealed. Moreover, repeated redistribution of the outer cell membrane organelles (Golgi apparatus, lysosomes, mitochondria, and autophagosomes), rearrangement of its cytoskeleton, and change in the lysosomal, autophagosomal, and mitochondrial state in both entotic cells were observed during entosis. On the basis of these data, we divided entosis into five stages that make it possible to systematize description of this type of cell death.
KEY WORDS: entosis, stages of entosis, cell cannibalism, cell organellesDOI: 10.1134/S0006297915110085
Abbreviations: PCD, programmed cell death; SEM, scanning electron microscopy.
Two basic forms of cell death are currently recognized: necrosis and
programmed cell death (PCD) [1]. Fifteen types of
PCD exist [2]. In 2009, the Nomenclature Committee
on Cell Death included entosis in the list of cell death types for the
first time [3]. Entosis is a type of cell
cannibalism [4] during which one cell invades the
other [5]. It was suggested initially that
detachment of a cell from substrate initiated its internalization [6-10]. However, studies during
recent years indicated that the “cell-in-cell” structures
could be formed without detachment of cells from a matrix [11]. It was shown that entosis is characteristic
predominantly for tumor cells, but also can occur in some normal cell
populations [12].
Despite the fact that numerous researchers are investigating entosis actively, the exact mechanisms of this process remain unknown. The initial and final stages of entosis are, as a rule, investigated in studies, namely, the process of penetration and the fate of the internalized cell. It was shown that E-cadherin molecules in the composition of adhesive junctions, which are formed between entotic cells, play an important role in the first process [13]. Active cell invasion occurs with participation of its actomyosin complex. The fate of this cell can be different: it can leave the outer cell, divide within it, or be subjected to lysosome-mediated degradation [5, 12]. Entosis is a lengthy process (from 20 h and more) [5]. To understand what determines the fate of an internalized cell, it is necessary to identify the sequence of events in both cells-participants during entosis, and to determine mechanisms involved in each stage of this process.
The aim of our work was to identify stages of entosis based on analysis of the sequence of structural changes of cells themselves, as well as localization and functional state of a number of membrane organelles and cytoskeleton components of the cells. This will allow in future to investigate mechanisms of the processes involved in realization of each stage. The culture of HaCaT cells with strong adhesion to substrate was selected as the object of our study. This cell line is a model of normal human keratinocytes, for which the entosis phenomenon has not been yet reported.
MATERIALS AND METHODS
Cell culture. A HaCaT cell culture (normal human keratinocytes) kindly provided by E. A. Vorotelyak (Koltzov Institute of Developmental Biology, Russian Academy of Sciences) was used in this work. Cells were cultivated in DMEM medium (PanEco, Russia) supplemented with 10% fetal bovine serum (FBS) (PAA Laboratories, Austria), 2 mM L-glutamine (PanEco), and antibiotic–antimycotic solution (Sigma, USA) with final concentrations of agents: 100 U/ml of penicillin, 0.1 mg/ml streptomycin, and 0.25 µg/ml of fungizone. Cells were incubated at 37°C in 5% CO2.
Morphological analysis of cells. Cells were fixed in Bouin’s solution (15 ml of picric acid, 5 ml of formalin, and 1 ml of glacial acetic acid) for 30 min and washed with PBS, pH 7.4 (Sigma). Samples were stained with hematoxylin and eosin.
Intravital observations. Intravital observations were conducted during analysis of autophagosomes and mitochondria in cells. A monodansylcadaverine stain (85.5 mM stock solution in dimethyl sulfoxide (DMSO); Sigma) was used for visualization of autophagosomes. Rhodamine 123 (stock solution 1 mg/ml; Sigma) – a potential-dependent stain – was used for visualization of mitochondria. Both stains were introduced for 30 min using 1 µl of stock solution per 1 ml of medium. The cells were washed with medium following staining and mounted onto a camera for in vivo monitoring.
Cytochemical staining. The LysoTracker Red DND-99 cell stain (Invitrogen, USA) designed for labeling live cells was used to visualize vesicles of the acidic compartment. Stock solution was diluted at rate 1 : 5 in DMSO and added to cells using 1 µl of this solution per 1 ml of medium. Then the cells were kept in a CO2-incubator for 30 min. Prior to fixation, cells were washed with DMEM medium without serum. A 4% solution of formaldehyde in PBS was used as a fixative agent for 20 min. To visualize polymerized F-actin, TRITC-labeled phalloidin (Sigma) was used: stock solution 1 mg/ml in methanol, working solution 0.005 mg/ml in PBS. Incubation time with the stain was 45 min. The cell nuclei were additionally stained with DAPI (0.1 µg/ml) (Sigma) for 10 min, and then the preparation was transferred into glycerol–PBS (1 : 1) mixture.
Immunocytochemical staining. Cells were fixed with a 4% formaldehyde solution. To permeabilize the cell membrane, cells were transferred into a 0.2% Triton X-100 solution for 7 min, washed with PBS, and placed into a solution of a first antibody for 1 h at room temperature in a moist chamber. Rabbit polyclonal anti-mannosidase II antibodies (1 : 200; Abcam, USA) were used to visualize Golgi apparatus, mouse monoclonal anti-α-tubulin antibodies (1 : 1000; Sigma) – to visualize microtubule system, and rabbit anti-β-catenin antibodies (1 : 1000; Sigma) – to visualize β-catenin. Then the cells were washed with PBS and placed in solution of secondary antibodies for 1 h at room temperature in a moist chamber. Monoclonal goat anti-mouse IgG antibodies conjugated with Alexa Fluor 488 (1 : 800; Invitrogen, USA) and monoclonal donkey anti-rabbit IgG antibodies conjugated with Alexa Fluor 568 (1 : 800; Invitrogen) were used as secondary antibodies. Samples were analyzed with a Leica light microscope (Leica, Germany) using an N Plan 100×/1.25 oil immersion objective and an Axiovert 200M luminescence microscope (Carl Zeiss Inc., Germany) using PlanApo 63×/1.4 oil immersion objective. The samples were imaged with Leica Application Suite and AxioVision program, respectively. Further image processing was performed using the ImageJ program. Construction of histograms and calculations of standard deviation were performed using Microsoft Office Excel 2007.
Scanning electron microscopy (SEM). Cells were fixed using a 2.5% glutaraldehyde with 2% formalin in 0.1 M PBS and dehydrated according to the standard technique. Sample was subjected to critical point drying (for CO2t = 31°C, P = 73.8 bar). Glass slides with cells were mounted onto metal stages. Samples were sputter coated with Au-Pd (gold–palladium) mixture and analyzed in a SCAN JSM scanning electron microscope (JEOL, Japan).
Correlative light-electron microscopy (CLEM). The following procedure was performed to identify certain cells in SEM: entotic cells were located on the sample fixed with 2.5% glutaraldehyde using phase contrast microscopy (Axiovert 200M, ×63) and marked with a marker. The cell layer around the mark was removed under a differential interference microscope (DIC) (Leica) (HI Plan10×/0.22 and 20×/0.30 objectives) using a thin needle. A standard protocol for SEM preparations was then conducted.
RESULTS
Frequency of entotic cells in the investigated culture. The structures in which one cell was located inside another (“cell-in-cell” phenomenon) were found in the HaCaT cell culture. Characteristic changes in morphology of both cells were observed in the process. The nucleus of the outer cell (cannibal cell) assumed a crescent-like shape and shifted toward the periphery. Usually, no features of the outer cell differed from the neighboring cells: it was well-flattened, showed clearly defined lamella, and had size comparable with the sizes of neighboring cells. The inner cell (internalized cell) usually had rounded shape and was located inside a large non-stainable vacuole structure – entotic vacuole (Fig. 1a). These morphological features were consistent with indicators of entosis described in the literature [5].
Fig. 1. Entosis in HaCaT cell culture. a) Changes in morphology of entotic cells, light microscopy, staining with hematoxylin-eosin. b) Proof of one cell location inside another, CLEM: 1, 4) phase-contrast microscopy; 2, 5) fluorescence microscopy, staining with DAPI; 3, 6) SEM. c) Histogram of frequency of entotic cells in HaCaT cell culture. d) Stages of degradation of an internalized cell: 1) first stage; 2) second stage; 3) third stage; 7) fourth stage; 8, 9) fifth stage; 1-3, 7-9) phase contrast microscopy; 4-6, 10-12) fluorescence microscopy, staining with DAPI.
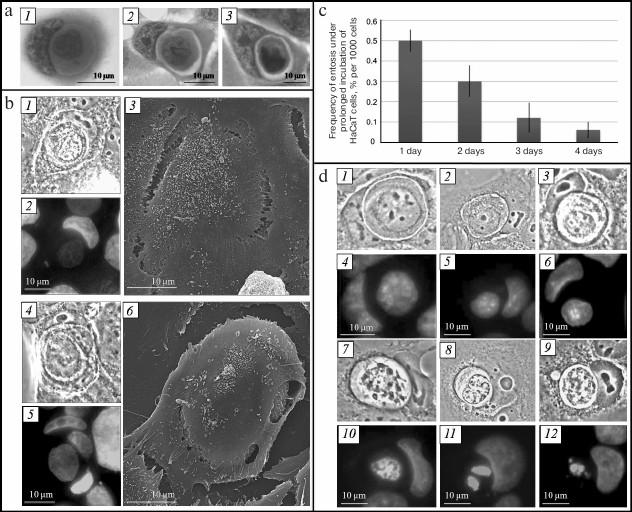
To confirm that one cell was located inside another, we analyzed entotic cells using correlative light-electron microscopy (CLEM). It can be seen in Fig. 1b that the plasma membrane of the outer cell completely covers the inner cell. It must be mentioned that there can be several internalized cells, in particular several cells in one cell or cell-in-cell-in-cell.
The fraction of entotic cells in the culture is on average 0.5% on the first day of cultivation, which decreases to 0.06% by the fourth day (Fig. 1c).
Morphological changes of inner and outer cells during different stages of entosis. By analyzing “cell-in-cell” structures, we revealed that the morphology of the inner cell as well as the state and location of the outer cell organelles can be different. This is related to the fact that the observed entotic cells are at different stages of the process. We divided entosis into five stages according to the following features: morphology of the internalized cell nucleus and cytoplasm, availability of junctions between entotic cells, state and location of organelles in both cells.
Shape and size of inner cell. The inner cell displays rounded shape at the early stage of entosis (immediately after invasion of one cell into another). Its size corresponds to the size of cells of this cell line in suspension (Fig. 1d, 1). Then small collaterals in the form of spines are formed on the surface of the inner cell (second stage of entosis) (Fig. 1d, 2). The cell outlines later become rough, and its size decreases (third and fourth stages of entosis) (Fig. 1d, 3 and 7). At the fifth stage of the process, the inner cell displayes irregular shape, and its size is significantly reduced in comparison with the initial size (Fig. 1d, 8 and 9).
Availability of space between plasma membrane of the inner cell and entotic vacuole membrane. At the early stage of entosis, the plasma membrane of the inner cell is in tight contact with the entotic vacuole membrane and, hence, space between them is not observed. At the second stage of entosis, when only a few contact points between the two membranes are retained, the boundaries of the inner cell and of the entotic vacuole become clearly visible (Fig. 1d, 2). At the third and fourth stages of entosis, the distance between these two membranes reaches its maximum value due to reduction of the inner cell size (Fig. 1d, 3 and 7). At the last stage of entosis, the space between the plasma membrane of the inner cell and entotic vacuole membrane decreases again (Fig. 1d, 9).
State of inner cell nucleus and cytoplasm. In majority of cases, the inner cell is subjected to degradation during entosis. As a consequence, the morphology of its nucleus changes. At the first stage of entosis it has rounded shape, is enriched with euchromatin, and several nucleoli are clearly visible (Fig. 1d, 4). This state of the inner cell nucleus is retained at the second stage of entosis (Fig. 1d, 5). However, the inner cell nucleus becomes slightly deformed at the third stage, assuming irregular shape (Fig. 1d, 6). At the fourth stage it shrinks significantly, chromatin undergoes gradual condensation, and nucleoli are not revealed (Fig. 1d, 10). At the final stage the nucleus of the inner cell is subjected to fragmentation (Fig. 1d, 11 and 12). Simultaneously with the morphology changes of the inner cell nucleus, enhancement of cytoplasmic vacuolation occurs.
Adhesive junctions between inner and outer cells. It is known that development of adhesive junctions between outer and inner cells (E-cadherin mediated) is required for the “cell-in-cell” structure formation. We conducted stage-by-stage analysis of adhesive junctions between entotic cells. For this purpose, cells were stained with anti-β-catenin antibodies binding to the intracellular domain of E-cadherin [14]. At early stages of entosis, the antibodies stained a thick circle between the membranes of the internalized cell and of the entotic vacuole (Fig. 2a, 3). At the second stage of entosis, β-catenin was observed along the periphery of the inner cell and along the periphery of the entotic valuole (Fig. 2a, 6). At the third stage, only few junction points between the internalized cell and entotic valuole were observed that contained β-catenin. However, this pattern was rare. As a rule, the anti-β-catenin antibody staining between internalized cell and entotic vacuole was not observed commencing from the third stage (Fig. 2a, 9). Hence, we showed that the molecules mediating adhesive junctions, such as β-catenin, undergo gradual degradation during entosis.
Fig. 2. Entosis in HaCaT cell culture. a) Visualization of adhesive junctions between entotic cells: 1-3) first stage; 4-6) second stage; 7-9) fourth stage; 1, 4, 7) phase-contrast microscopy; 2, 5, 8) fluorescence microscopy, DAPI staining; 3, 6, 9) fluorescence microscopy, staining with anti-β-catenin antibodies. b) Visualization of Golgi apparatus: 1-3) mononuclear cell; 4-6) second stage of entosis; 7-9) fourth stage; 1, 4, 7) phase-contrast microscopy; 2, 5, 8) fluorescence microscopy, DAPI staining; 3, 6, 9) fluorescence microscopy, staining with anti-mannosidase II antibodies. c) Visualization of lysosomes: 1-3) first stage; 4-6) second stage; 7-9) fourth stage; 1, 4, 7) phase-contrast microscopy; 2, 5, 8) fluorescence microscopy, DAPI staining; 3, 6, 9) fluorescence microscopy, staining with LysoTracker Red. d) Visualization of autophagosomes: 1, 4) first stage; 2, 5) second stage; 3, 6) fifth stage; 1-3) phase-contrast microscopy; 4-6) fluorescence microscopy, staining with monodansylcadaverine. e) Visualization of mitochondria: 1, 4) mononuclear cell; 2, 5) second stage of entosis; 3, 6) fourth stage; 1-3) phase-contrast microscopy; 4-6) fluorescence microscopy, staining with rhodamine 123.
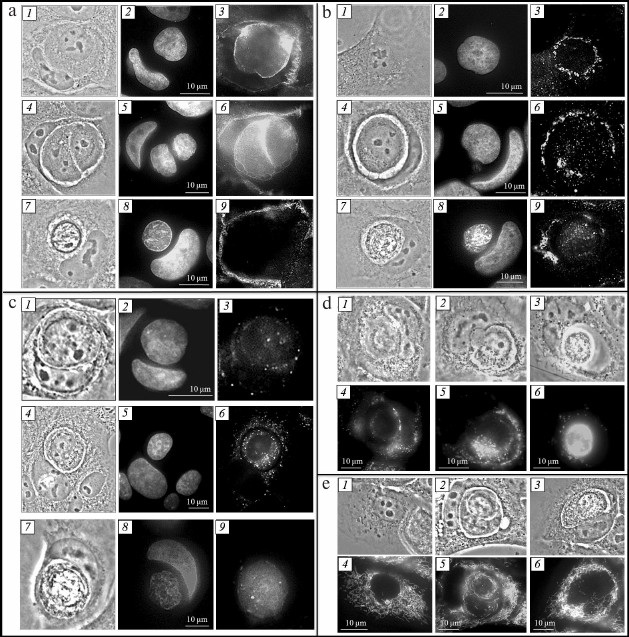
Golgi apparatus of inner and outer cells. Dictyosomes of the Golgi apparatus in mononuclear cells were usually located along the periphery of the cell nucleus (Fig. 2b, 3). Localization of the Golgi apparatus changed in the outer cell during entosis. At the early stages, the dictyosomes of the cannibal cell Golgi apparatus moved to the area of the entotic vacuole, surrounding it completely (Fig. 2b, 6). This location of the outer cell Golgi apparatus was retained up to the fourth stage. In some cases, repeated redistribution of dictyosomes in the cannibal cell occurred – they returned to the area close to the nucleus. The Golgi apparatus of the internalized cell was observed in the perinuclear space during the first two stages of entosis. Starting with the third stage, the dictyosomes of the the inner cell Golgi apparatus usually broke down into separate cisterns and vesicles (Fig. 2b, 9). The Golgi apparatus was not observed in the inner cell at the fifth stage. Thus, we demonstrated that redistribution of the outer cell Golgi apparatus dictyosomes occurred during entosis, and prolonged integrity of the Golgi apparatus was observed in the internalized cell.
Lysosomes and autophagosomes of inner and outer cells. It was shown that lysosomes in mononuclear cells were located predominantly in the area close to nucleus. At the first stage of entosis, the lysosomes of the cannibal cell started to redistribute and localized along the periphery of the entotic vacuole. However, their numbers were low (Fig. 2c, 3). At the second stage, the number of lysosomes in the outer cell increased significantly (Fig. 2c, 6). From this point on, lysosomes chaotically located in the cytoplasm were observed in the cannibal cell. Their number decreased in the process. The lysosomes of the internalized cell were assembled close to the nucleus, and their numbers were low at the first stage of entosis (Fig. 2c, 3). At the second and third stages, the number of lysosomes was likely increasing and they became redistributed – they were found throughout the entire cytoplasm of the cell (Fig. 2c, 6). Next, the lysosomes of the inner cell started merging, its cytoplasm acidified, and by the fifth stage, the entire entotic vacuole was stained with LysoTracker Red (Fig. 2c, 9).
Autophagosomes located throughout the outer cell cytoplasm were observed at the initial stage of entosis (Fig. 2d, 4). At the second stage, the autophagosomes of the cannibal cell were usually localized along the periphery of the entotic vacuole (Fig. 2d, 5). At the final stages of entosis, larger autophagosomes were observed close to the entotic vacuole in the outer cell (Fig. 2d, 6). Their number was significantly lower than at the first stages of entosis. Single autophagosomes were found in the cytoplasm of the inner cell during the initial stage of entosis. Thereafter, the number of autophagosomes in the inner cell increased gradually (second and third stages), they started merging (fourth stage), and at the late stage of entosis diffuse monodansylcadaverine staining of the entire internalized cell was observed.
So, it can be suggested that both the acidic vesicular compartment and autophagosomes of outer and inner cells participate in degradation of the internalized cell.
Mitochondria of the inner and outer cells. The mitochondria of HaCaT cell line are thread-like, although smaller bead-like mitochondria can also be found. The mitochondria are usually distributed uniformly throughout the entire cytoplasm of the cell (Fig. 2e, 4). The mitochondria of the outer cell during entosis (all stages) were mainly assembled around the entotic vacuole (Fig. 2e, 5 and 6). It must be noted that the mitochondria of the internalized cell localized around the cell nucleus were able to sustain membrane potential during early stages of entosis (first and second stages) (Fig. 2e, 5). However, the mitochondrial membrane potential of the inner cell decreased gradually starting with the third stage of entosis, and mitochondria of the inner cell were not observed at the final stages (Fig. 2e, 6). Hence, redistribution of the outer cell mitochondria occurs during entosis, and mitochondria of the internalized cell remain in their functional state up to the third stage.
Cytoskeleton of inner and outer cells.' 'Actin cytoskeleton. Invasion of one cell into another during entosis occurs through the work of actomyosin complex of the invading cell [5, 13]. We studied the location of actin microfilaments in both entotic cells at different stages of entosis. The actin microfilaments were observed along the periphery of cannibal cell, as individual stress fibers in the cell cytoplasm and along the periphery of the entotic vacuole during all stages of entosis. One or two areas with high actin content in the area proximal to the nucleus of the outer cell were observed in the internalized cell during the early stage of entosis (Fig. 3a, 3). Moreover, actin microfilaments were located in the cortex zone of the inner cell. Later, the actin cytoskeleton of the internalized cell was observed along the periphery of the cell and in the cytoplasm as short chaotically located filaments (Fig. 3a, 6 and 9). At the final stage of entosis, only a few short actin filaments were retained in the inner cell cytoplasm.
Fig. 3. Entosis in HaCaT cell culture. a) Visualization of actin cytoskeleton: 1-3) first stage; 4-6) second stage; 7-9) third stage; 10-12) fifth stage; 1, 4, 7, 10) phase-contrast microscopy; 2, 5, 8, 11) fluorescence microscopy, DAPI staining; 3, 6, 9, 12) fluorescence microscopy, staining with phalloidin. b) Visualization of microtubule system: 1-3) mononuclear cell; 4-6) first stage of entosis; 7-9) second stage; 10-12) fifth stage; 1, 4, 7, 10) phase-contrast microscopy; 2, 5, 8, 11) fluorescence microscopy, DAPI staining; 3, 6, 9, 12) fluorescence microscopy, staining with anti-α-tubulin antibodies.
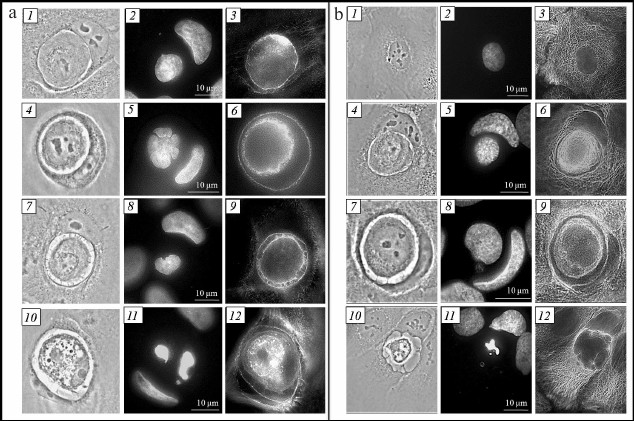
Microtubule system. Repeated redistribution of the cannibal cell membrane organelles occurs during entosis. It is known that movement of cellular organelles is accomplished with the help of microtubules [15, 16]. In this connection, we investigated localization of microtubules during entosis. The microtubule system in the HaCaT cell culture appears as a three-dimensional network (Fig. 3b, 3). Two variants of microtubule distribution in the cannibal cell were observed at early stages of entosis: (i) in the case of a well-flattened cell, the 3D network structure was well conserved (sometimes with slightly more pronounced concentration around entotic vacuole) (Fig. 3b, 6); (ii) in the case of a moderately flattened cell, microtubules were localized predominantly around the entotic vacuole in the form of dense bundles (Fig. 3b, 9). Such localization of microtubules was maintained up to the late stage of entosis. At the final stage, the microtubule cytoskeleton of the cannibal cell always resembled three-dimensional network structure (Fig. 3b, 12). It was remarkable that the mucrotubules in the rounded internalized cell were assembled and located near the nucleus as thick bundles during the first and second stages of entosis. As the inner cell degraded, its microtubules were gradually disassembed (third and fourth stage), and they were not observed at the fifth stage. Thereby, the microtubules of the outer cell are rearanged in some cases surrounding the entotic vacuole at the early stages of entosis; the microtubules of the internalized cell are not disassemblled for a long period of time.
DISCUSSION
In this work, we described entosis in HaCaT culture of normal human keratinocytes for the first time. The highest frequency of entosis of 0.5% in this cell line is observed during the first day of cultivation. According to the literature data, the frequency of entosis in tumor cell lines is significantly higher [5]. It was suggested that entosis is initiated in the majority of cell lines by detachment of cells from the substrate. The cells of HaCaT line, being normal cells, exhibit higher adhesion to the substrate in comparison with tumor cells [17]. It seems likely that precisely the difference in adhesion to substrate could explain the difference in the number of entotic cells in the cultures of normal and tumor cells.
By comparing morphological characteristics of cells and their organelles, we were able for the first time to present entosis as a sequence of events related to changes in organization, functional activity, and integrity of the cannibal cell and the internalized cell organelles. Based on our data, we identified five stages of entosis (after invasion of one cell into another).
We suggest that the changes occuring in both entotic cells reflect the processes required for degradation of the internalized cell. Following the invasion of one cell into another, the former is found surrounded by the plasma membrane of the cannibal cell (Fig. 4a). A large number of adhesive junctions is maintained between the cells in the process.
Fig. 4. Schematic representation of the sequence of events after invasion of one cell into another. a) Localization of cell organelles and composition of entotic vacuole membrane immediately after invasion; b) activation of autophagy-related proteins and first modification of entotic vacuole membrane; c) complete replacement of entotic vacuole plasma membrane with autophagosome membrane; d) activation of lysosomes and second modification of entotic vacuole membrane; e) formed entotic vacuole membrane; f) acidification of entotic vacuole and degradation of internalized cell. Designations: GA, Golgi apparatus; AP, autophagy-related proteins; IC, inner cell; L, lysosome; M, mitochondrion; OC, outer cell; EV, entotic vacuole; N, nucleus.
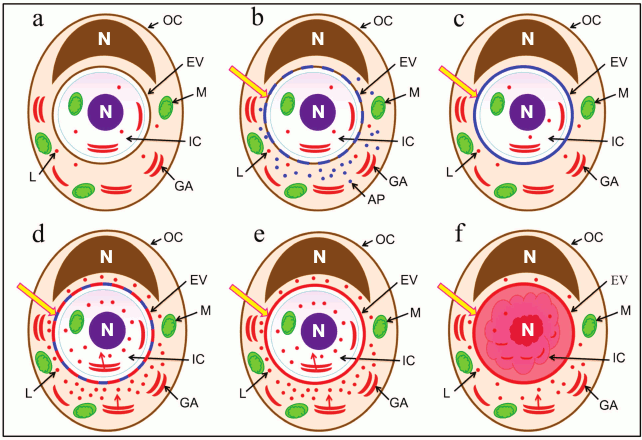
At the early stage of entosis, the first modification of the entotic vacuole membrane starts through the autophagy-related proteins (Fig. 4, b and c). According to the literature, autophagy-related proteins such as Atg5, Atg7, Vps34, and LC3 participate in the entotic vacuole membrane rearrangement [18, 19]. Moreover, integral lysosome-associated membrane proteins, LAMP, are introduced into the entotic vacuole membrane [5]. This explains the gradual disruption of the junctions between the cells. At the same time, the location of the outer cell Golgi apparatus changes. It is known that the Golgi apparatus participates in protein maturation and formation of lysosomes [20, 21]. We suggest that redistribution of the Golgi apparatus at the first stage of entosis, as a result of which it localizes along the periphery of entotic vacuole, is required for formation of additional lysosomes and autophagosomes that is observed at the second stage of entosis, close to the entotic vacuole in particular. This is related to the fact that at the end of the second-beginning of the third stage of entosis, the lysosomes of the cannibal cell are merged with modified membrane of the entotic vacuole (Fig. 4d). It is likely that by these means the lysosomal membrane proteins, which protect the outer cell from hydrolases, are brought into the entotic vacuole membrane (Fig. 4e) [22]. In addition, the release of lysosomal hydrolases of the cannibal cell inside the entotic vacuole takes place (Fig. 4f).
Extended preservation of the Golgi apparatus and microtubule system in the internalized cell is necessary for production of additional lysosomes and autophagosomes and to ensure their transport within the cell. Hence, the lysosomes of both the cannibal cell and the internalized cell itself take part in degradation of the inner cell.
Redistribution of mitochondria in the outer cell as well as the ability of the inner cell mitochondria to maintain membrane potential for an extended period of time during entosis could be related to the necessity for production of a large amount of energy required for functioning of other cellular organelles. Emergence of large autophagosomes in the cannibal cell close to the entotic vacuole at the fifth stage of entosis is due to the fact that either excessive or damaged cell organelles of the outer cell could be there or the fragments of the entotic vacuole with the debris of the internalized cell.
Thus, we have presented a full picture of structural–functional changes occuring in cells during entosis. Nevertheless, the fate of the entotic vacuole and all the matter iside it remains unknown – does complete degradation of the vacuole content occur or its extrusion? The mechanisms involved in different stages of entosis are to be investigated in the future and compared for normal and tumor cells.
This work was financially supported by the Russian Science Foundation (project No. 14-50-00029).
REFERENCES
1.Kerr, J. F. R., Wyllie, A. H., and Curruie, A. R.
(1972) Apoptosis: a basic biological phenomenon with wide-ranging
implications in tissue kinetics, Br. J. Cancer, 26,
239-257.
2.Galluzzi, L., Bravo-San Pedro, J. M., Vitale, I.,
Aaronson, S. A., Abrams, J. M., Adam, D., Alnemri, E. S., Altucci, L.,
Andrews, D., Annicchiarico-Petruzzelli, M., Baehrecke, E. H., Bazan, N.
G., Bertrand, M. J., Bianchi, K., Blagosklonny, M. V., Blomgren, K.,
Borner, C., Bredesen, D. E., Brenner, C., Campanella, M., Candi, E.,
Cecconi, F., Chan, F. K., Chandel, N. S., Cheng, E. H., Chipuk, J. E.,
Cidlowski, J. A., Ciechanover, A., Dawson, T. M., Dawson, V. L., De
Laurenzi, V., De Maria, R., Debatin, K. M., Di Daniele, N., Dixit, V.
M., Dynlacht, B. D., El-Deiry, W. S., Fimia, G. M., Flavell, R. A.,
Fulda, S., Garrido, C., Gougeon, M. L., Green, D. R., Gronemeyer, H.,
Hajnoczky, G., Hardwick, J. M., Hengartner, M. O., Ichijo, H., Joseph,
B., Jost, P. J., Kaufmann, T., Kepp, O., Klionsky, D. J., Knight, R.
A., Kumar, S., Lemasters, J. J., Levine, B., Linkermann, A., Lipton, S.
A., Lockshin, R. A., Lopez-Otin, C., Lugli, E., Madeo, F., Malorni, W.,
Marine, J. C., Martin, S. J., Martinou, J. C., Medema, J. P., Meier,
P., Melino, S., Mizushima, N., Moll, U., Munoz-Pinedo, C., Nunez, G.,
Oberst, A., Panaretakis, T., Penninger, J. M., Peter, M. E.,
Piacentini, M., Pinton, P., Prehn, J. H., Puthalakath, H., Rabinovich,
G. A., Ravichandran, K. S., Rizzuto, R., Rodrigues, C. M., Rubinsztein,
D. C., Rudel, T., Shi, Y., Simon, H. U., Stockwell, B. R., Szabadkai,
G., Tait, S. W., Tang, H. L., Tavernarakis, N., Tsujimoto, Y., Vanden
Berghe, T., Vandenabeele, P., Villunger, A., Wagner, E. F., Walczak,
H., White, E., Wood, W. G., Yuan, J., Zakeri, Z., Zhivotovsky, B.,
Melino, G., and Kroemer, G. (2015) Essential versus accessory aspects
of cell death: recommendations of the NCCD 2015, Cell Death
Differ., 22, 58-73.
3.Kroemer, G., Galluzzi, L., Vandenabeele, P.,
Abrams, J., Alnemri, E. S., Baehrecke, E. H., Blagosklonny, M. V.,
El-Deiry, W. S., Golstein, P., Green, D. R., Hengartner, M., Knight, R.
A., Kumar, S., Lipton, S. A., Malorni, W., Nunez, G., Peter, M. E.,
Tschopp, J., Yuan, J., Piacentini, M., Zhivotovsky, B., and Melino, G.
(2009) Classification of cell death: recommendations of the
Nomenclature Committee on Cell Death, Cell Death Differ.,
12 (Suppl. 2), 1463-1467.
4.Yuan, J., and Kroemer, G. (2010) Alternative cell
death mechanisms in development and beyond, Genes Dev.,
24, 2592-2602.
5.Overholtzer, M., Mailleux, A. A., Mouneimne, G.,
Normand, G., Schnitt, S. J., King, R. W., Cibas, E. S., and Brugge, J.
S. (2007) A nonapoptotic cell death process, entosis, that occurs by
cell-in-cell invasion, Cell, 131, 966-979.
6.Doukoumetzidis, K., and Hengartner, M. O. (2008)
Dying to hold you, Nature, 451, 530-531.
7.Guadamillas, M. C., Cerezo, A., and Del Pozo, M. A.
(2011) Overcoming anoikis – pathways to anchorage-independent
growth in cancer, J. Cell. Sci., 124, 3189-3197.
8.Ishikawa, F., Ushida, K., Mori, K., and Shibanuma,
M. (2015) Loss of anchorage primarily induces non-apoptotic cell death
in a human mammary epithelial cell line under atypical focal adhesion
kinase signaling, Cell Death Dis., 6, 1-12.
9.Krajcovic, M., Johnson, N. B., Sun, Q., Normand,
G., Hoover, N., Yao, E., Richardson, A. L., King, R. W., Cibas, E. S.,
Schnitt, S. J., Brugge, J. S., and Overholtzer, M. (2011) A non-genetic
route to aneuploidy in human cancers, Nat. Cell Biol.,
13, 324-330.
10.Mazzone, M., Selfors, L. M., Albeck, J.,
Overholtzer, M., Sale, S., Carroll, D. L., Pandya, D., Lu, Y., Mills,
G. B., Aster, J. C., Artavanis-Tsakonas, S., and Brugge, J. S. (2010)
Dose-dependent induction of distinct phenotypic responses to Notch
pathway activation in mammary epithelial cells, Proc. Natl. Acad.
Sci. USA, 107, 5012-5017.
11.Wan, Q., Liu, J., Zheng, Z., Zhu, H., Chu, X.,
Dong, Z., Huang, S., and Du, Q. (2012) Regulation of myosin activation
during cell–cell contact formation by Par3-Lgl antagonism:
entosis without matrix detachment, Mol. Biol. Cell, 23,
2076-2091.
12.Overholtzer, M., and Brugge, J. S. (2008) The
cell biology of cell-in-cell structures, Nat. Rev. Mol. Cell
Biol., 9, 796-809.
13.Sun, Q., Cibas, E. S., Huang, H., Hodgson, L.,
and Overholtzer, M. (2014) Induction of entosis by epithelial cadherin
expression, Cell Res., 24, 1288-1298.
14.Gama, A., and Schmitt, F. (2012) Cadherin cell
adhesion system in canine mammary cancer, Vet. Med. Int.,
2012, 1-8.
15.Hirokawa, N. (1996) Organelle transport along
microtubules – the role of KIFs, Trends Cell Biol.,
6, 135-141.
16.Welte, M. A. (2004) Bidirectional transport along
microtubules, Curr. Biol., 14, 525-537.
17.Balzer, E. M., and Konstantopoulos, K. (2012)
Intercellular adhesion: mechanisms for growth and metastasis of
epithelial cancers, Wiley Interdiscip. Rev. Syst. Biol. Med.,
4, 171-181.
18.Florey, O., Gammoh, N., Kim, S. E., Jiang, X.,
and Overholtzer, M. (2014) V-ATPase and osmotic imbalances activate
endolysosomal LC3 lipidation, Autophagy, 11, 88-99.
19.Florey, O., Kim, S. E., Sandoval, C. P., Haynes,
C. M., and Overholtzer, M. (2011) Autophagy machinery mediates
macroendocytic processing and entotic cell death by targeting single
membranes, Nat. Cell Biol., 13, 1335-1343.
20.Green, S. A., Zimmer, K. P., Griffiths, G., and
Mellman, I. (1987) Kinetics of intracellular transport and sorting of
lysosomal membrane and plasma membrane proteins, J. Cell Biol.,
105, 1227-1240.
21.Rohrer, J., and Kornfeld, R. (2001) Lysosomal
hydrolase mannose 6-phosphate uncovering enzyme resides in the
trans-Golgi network, Mol. Biol. Cell, 12,
1623-1631.
22.Fukuda, M. (1991) Lysosomal membrane
glycoproteins. Structure, biosynthesis, and intracellular trafficking,
J. Biol. Chem., 266, 21327-21330.