StructAlign, a Program for Alignment of Structures of DNA–Protein Complexes
Ya. V. Popov1, A. A. Galitsyna1, A. V. Alexeevski1,2,3, A. S. Karyagina2,4,5, and S. A. Spirin1,2,3*
1Lomonosov Moscow State University, Faculty of Bioengineering and Bioinformatics, 119991 Moscow, Russia; fax: +7 (495) 939-41952Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; fax: +7 (495) 939-3181; E-mail: sas@belozersky.msu.ru
3Institute of System Studies, Russian Academy of Sciences, 117218 Moscow, Russia
4Gamaleya Center of Epidemiology and Microbiology, 123098 Moscow, Russia
5Institute of Agricultural Biotechnology, 127550 Moscow, Russia
* To whom correspondence should be addressed.
Received July 3, 2015
Comparative analysis of structures of complexes of homologous proteins with DNA is important in the analysis of DNA–protein recognition. Alignment is a necessary stage of the analysis. An alignment is a matching of amino acid residues and nucleotides of one complex to residues and nucleotides of the other. Currently, there are no programs available for aligning structures of DNA–protein complexes. We present the program StructAlign, which should fill this gap. The program inputs a pair of complexes of DNA double helix with proteins and outputs an alignment of DNA chains corresponding to the best spatial fit of the protein chains.
KEY WORDS: structural bioinformatics, DNA–protein complexes, alignment, web interfaceDOI: 10.1134/S0006297915110073
Spatial structures of homologous proteins are usually highly similar. Comparative analysis of similar structures often assists to reveal functionally important features of proteins. DNA-binding proteins form families, within which not only the fold of the protein molecule is conserved, but also its position on double-stranded DNA helix. In this case, comparative analysis of DNA–protein complexes might be meaningful.
An essential stage of comparative analysis of structures is alignment of these structures. We regard an alignment as establishing a correspondence between residues from two structures (see “Materials and Methods” for detailed definition). There are several programs for alignment of single protein chains (e.g. see [1-4]). Unfortunately, there is no available program for alignment of macromolecular complexes, particularly DNA–protein complexes. Our program, StructAlign, is dedicated to filling this gap. The program inputs two structures of double-stranded DNA helix complexed with proteins and outputs the alignment of nucleotide sequences that corresponds to the optimal superimposition of the complexes. A numerical measure of alignment quality is also provided in the output. The program algorithm uses the fact that two double-stranded DNA helices could always be superimposed according to any gap-free alignment of nucleotides. Thus, the problem of alignment of DNA–protein complexes is solved once the best shift of DNA helices is found. The better shift could be judged by the quality of protein chain superimposition.
MATERIALS AND METHODS
The StructAlign input comprises two structures of proteins complexed with double-stranded DNA helices in PDB format (see http://www.wwpdb.org/documentation/file-format-content/format33/v3.3.html). The program stages are: 1) associate a coordinate system with each nucleotide in both structures (see algorithm below); 2) for each pair of nucleotides (one nucleotide per structure), calculate the value S, which is the measure of similarity of protein positions in relation to the given nucleotides (see algorithm below); 3) select the DNA fragments from both structures so that they have equal lengths and the sum of measures S for corresponding nucleotides is the maximum among all possible pairs of fragments; this pair of fragments gives the alignment of nucleotides of two structures; 4) form output files, including a PDB file with input structures superimposed over pairs of nucleotides with the highest measure S and a text file with the alignment of DNA chains.
The algorithm of construction of the coordinate system for a nucleotide structure is as follows. The center of the phosphorus atom is the origin of the coordinate system. The X-axis matches the direction from the center of the phosphorus atom to the middle of the segment between centers of two oxygen atoms (PDB atom designations OP1 and OP2) covalently bound with the phosphorus atom and not bound with the ribose residue. The Y-axis matches the direction orthogonal to the X-axis and parallel to the plane containing centers of P and C1′ atoms and parallel to the X-axis; from two possible directions, the one with smaller angle towards the direction from P to C1′ is chosen. The Z-axis matches the direction orthogonal to the X-axis and the Y-axis so that XYZ is a right-handed coordinate system (i.e. rotation from X to Y is clockwise when looking in the Z-axis direction).
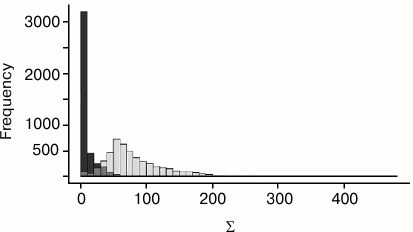
Algorithm of calculation of the measure S. Consider two nucleotides, one from each structure. Construct the coordinate system for each nucleotide and match the coordinate spaces of structures over these coordinate systems. For each protein Cα-atom from the first structure, consider the closest Cα-atom from the second structure after matching. Conversely, for each Cα-atom from the second structure, consider the closest Cα-atom from the first structure. If two Cα-atoms, a from the first structure and b from the second, are mutually closest (i.e. a is the closest to b among all Cα-atoms from the first structure and, conversely, b is the closest to a among all Cα-atoms from the second structure) and the distance d(a,b) between them is less than 4.5 Å, then mark these atoms as corresponding (for the given pair of nucleotides). The constant 4.5 was chosen after testing different values. The similarity measure for the pair of nucleotides is obtained from the sum Σ of values 4.5 Å – d(a,b) over all pairs of corresponding Cα-atoms a and b. The more similar is the location of the proteins in relation to two given nucleotides, the larger is the sum Σ. The value of the measure S is obtained from the specified sum Σ by subtraction of the constant 31 Å. This constant was chosen after testing for several families of DNA–protein complexes. Namely, the nucleotides with equal location relative to protein were determined for several tens of related complexes. For pairs of corresponding (i.e. equally located relative to protein) and non-corresponding nucleotides, the mutually closest Cα-atoms were found. Then two distributions of sums Σ were calculated: for equally located and for other pairs of nucleotides. These distributions are shown in Fig. 1. The 31 Å constant was chosen so that the value S = Σ – 31 Å would be, as a rule, positive for equally located nucleotides and negative for others.
The StructAlign program is implemented in the C programming language. The web interface for the program is written in Python using CGI.
Fig. 1. Σ value distribution for pairs of corresponding (light bars) and non-corresponding (dark bars) nucleotides.
RESULTS AND DISCUSSION
Web interface for the StructAlign program. The web interface is available at http://mouse.genebee.msu.ru/tools/StructAlign.html. It offers the user to enter two PDB IDs and, optionally, protein chain identifiers for corresponding PDB entries. If the chain identifier is not specified, the first chain from the PDB entry is taken. The program aligns two structures, each consisting of one (user specified) protein chain and all existing DNA chains from the corresponding PDB entry.
The program output contains: 1) the alignment score (a value describing the quality of the superimposition of structures); 2) a file in PDB format with aligned structures; 3) a DNA sequences alignment as a preformatted text; 4) an interactive representation of superimposed structures in Jmol.
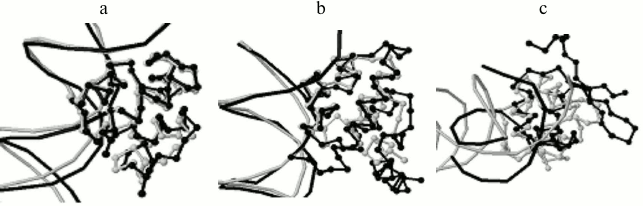
Program output examples: comparison of structures of complexes of phage repressors with DNA. Thirteen structures of DNA–protein complexes were taken where the protein belongs to the SCOP [5] family “Phage Repressors”; namely, 1LMB (protein chains 3 and 4, C1 repressor of λ phage), 1LLI (chains A and B, mutant C1 repressor of λ phage), 1RIO (chains A and B, C1 repressor of λ phage), 1PER (chains L and R, C1 repressor of 434 phage), 1RPE (chains L and R, C1 repressor of 434 phage), 3CRO (chains L and R, Cro protein of 434 phage), 6CRO (chain A, Cro protein from λ phage). All pairs of structures were analyzed with the StructAlign program. 1PER, 1RPE, 3CRO structures turned out to be well pairwise aligned by the program. 1LMB, 1LLI, 1RIO structures are well aligned too. Alignment scores range from 1300 to 5500 Å within these two groups, and the superimposition of structures is visually very good (Fig. 2a). Alignments between structures from different groups are worse, scores range from 400 to 1000 Å and the superimposition is worse (Fig. 2b). The worst alignments were obtained for 6CRO structure with other structures, scores range from 110 to 260 Å and only DNA-recognizing helices of proteins are superimposed (Fig. 2c). This result is not surprising taking into account that Cro protein from λ phage has a fold with a β-hairpin and all other regarded proteins consist of four α-helices. In this case, including these proteins into one SCOP family is not in accordance with the low similarity of their tertiary structures.
Fig. 2. Program results for pairs of phage repressors with DNA. a) 1PER, protein chain L (gray), and 3CRO, protein chain L (black), alignment score 1149 Å; b) 1PER, protein chain L (gray), and 1LMB, protein chain 3 (black), alignment score is 855 Å; c) 1PER, protein chain L (gray), and 6CRO, protein chain A (black), alignment score is 116 Å.
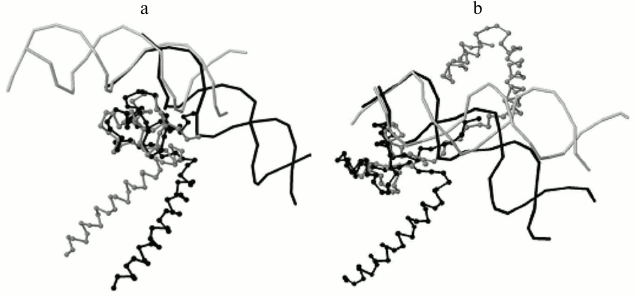
Program output examples: comparison of structures of DNA–protein complexes with Zn2/Cys2 and leucine zipper motifs in proteins. The proteins GAL4 (PDB ID 3COQ) and HAP1 (PDB ID 1HWT) from baker’s yeast (Saccharomyces cerevisiae) contain the DNA-recognizing motif Zn2/Cys2 and the dimerization motif called leucine zipper. Each of these proteins binds DNA as a homodimer, GAL4 as a symmetrical dimer, and HAP1 as an asymmetrical dimer (Fig. 3) [6]. Each monomer structure has a long α-helix, which forms the leucine zipper with the same helix from the other monomer. The pairwise alignment of monomers complexed with DNA results in good match of DNA-recognizing motifs Zn2/Cys2 (Fig. 4). The alignment score for different GAL4 monomers is 2789 Å due to matching of two motifs: Zn2/Cys2 and the α-helix of the leucine zipper. Meanwhile, the alignment score for two complexes with different HAP1 monomers is 246 Å due to matching of only Zn2/Cys2 motifs and the short linker segment between motifs; α-helices diverge far in space (Fig. 4a). The alignments of complexes with any monomer GAL4 and one monomer HAP1 have approximately the same score (250 Å for chain D from 1HWT; see Fig. 4b, and 170 Å for chain C).
Fig. 3. DNA–protein complexes for homodimers with the DNA-recognizing motif Zn2/Cys2 and the dimerization motif leucine zipper. a) Symmetrical dimer of yeast protein GAL4 (PDB 3COQ); b) asymmetrical dimer of yeast protein HAP1 (PDB 1HWT). DNA and one chain of each protein (chain A for 3COQ and chain C for 1HWT) are shown in gray; another chain (chain B for 3COQ and chain D for 1HWT) is shown in black.
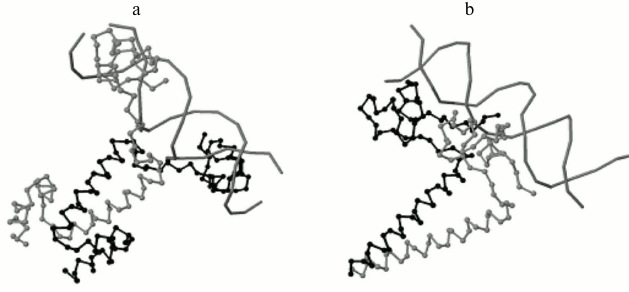
Fig. 4. a) The program result for two monomers of protein HAP1 from the 1HWT structure. The chain C and one copy of DNA chains A and B are shown in gray; the chain D and another DNA copy are shown in black. The DNA-recognizing domains Zn2/Cys2 match after the superimposition of the most similarly located nucleotides relative to protein, and the α-helices of the leucine zipper motif spatially diverge. The alignment score is 246 Å. b) The program result for structures of DNA complexes with proteins GAL4, PDB 3COQ, protein chain A (gray) and HAP1, PDB 1HWT, protein chain D (black). Again, Zn2/Cys2 domains match and α-helices of leucine zipper diverge. The alignment score is 250 Å.
Potential usage of the program. The program could ease the comparative analysis of DNA–protein complexes is several aspects. First, it automatically finds corresponding nucleotides in two similar complexes. Manual search of such nucleotides using structure visualizers might be laborious. Second, it allows visualization of DNA-oriented superimposition of complexes revealing similarly located, in relation to DNA, parts of a protein molecule. Third, the alignment score might have certain significance.
This research was supported by the Russian Science Foundation (project No. 14-50-00029, algorithm development, program testing) and by the Russian Foundation for Basic Research (project No. 13-07-00969, programming).
REFERENCES
1.Taylor, W. R., and Orengo, C. A. (1989) Protein
structure alignment, J. Mol. Biol., 208, 1-22.
2.Holm, L., and Sander, C. (1993) Protein structure
comparison by alignment of distant matrices, J. Mol. Biol.,
233, 123-138.
3.Shindiyalov, I. N., and Bourne, P. E. (1998)
Protein structure alignment by incremental combinatorial extension (CE)
of the optimal path, Protein Eng., 9, 739-747.
4.Krissinel, E., and Henrick, K. (2004)
Secondary-structure matching (SSM), a new tool for fast protein
structure alignment in three dimensions, Acta Crystallogr.,
60, 2256-2268.
5.Murzin, A. G., Brenner, S. E., Hubbard, T., and
Chothia, C. (1995) SCOP: a structural classification of proteins
database for the investigation of sequences and structures, J. Mol.
Biol., 247, 536-540.
6.King, D. A., Zhang, L., Guarente, L., and
Marmorstein, R. (1999) Structure of a HAP1–DNA complex reveals
dramatically asymmetric DNA binding by a homodimeric protein, Nat.
Struct. Biol., 6, 64-71.