REVIEW: Coelenterazine-Dependent Luciferases
S. V. Markova and E. S. Vysotski*
Institute of Biophysics, Siberian Branch of the Russian Academy of Sciences, 660036 Krasnoyarsk, Russia; fax: +7 (391) 243-3400; E-mail: eugene.vysotski@gmail.com* To whom correspondence should be addressed.
Received February 25, 2015; Revision received March 4, 2015
Bioluminescence is a widespread natural phenomenon. Luminous organisms are found among bacteria, fungi, protozoa, coelenterates, worms, molluscs, insects, and fish. Studies on bioluminescent systems of various organisms have revealed an interesting feature – the mechanisms underlying visible light emission are considerably different in representatives of different taxa despite the same final result of this biochemical process. Among the several substrates of bioluminescent reactions identified in marine luminous organisms, the most commonly used are imidazopyrazinone derivatives such as coelenterazine and Cypridina luciferin. Although the substrate used is the same, bioluminescent proteins that catalyze light emitting reactions in taxonomically remote luminous organisms do not show similarity either in amino acid sequences or in spatial structures. In this review, we consider luciferases of various luminous organisms that use coelenterazine or Cypridina luciferin as a substrate, as well as modifications of these proteins that improve their physicochemical and bioluminescent properties and therefore their applicability in bioluminescence imaging in vivo.
KEY WORDS: bioluminescence, luciferase, luciferin, coelenterazineDOI: 10.1134/S0006297915060073
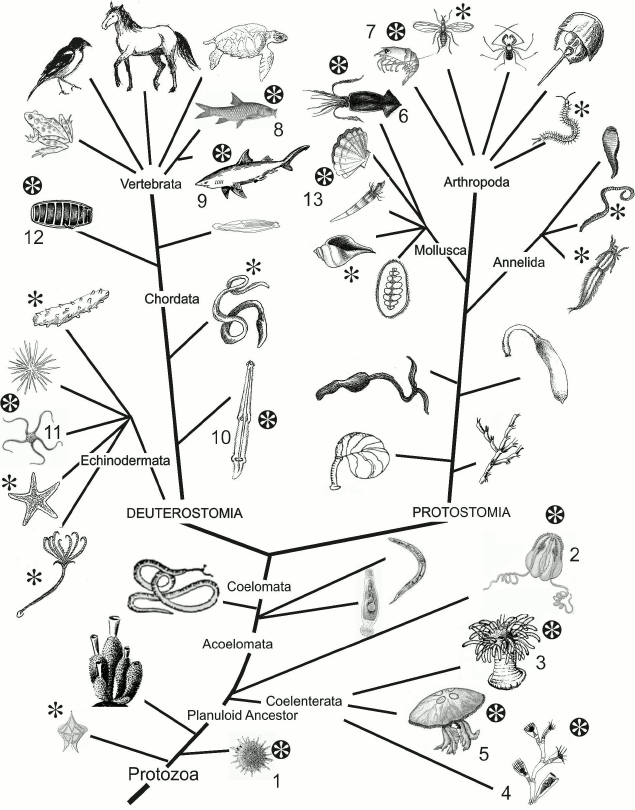
Bioluminescence is a widespread natural phenomenon. Luminous organisms are found among bacteria, fungi, protozoa, coelenterates, worms, molluscs, insects, and fish. There are now known several thousands of bioluminescent species represented by more than 700 genera. Although luminous species can be found among terrestrial organisms (bacteria, insects, worms, and fungi), the ability for bioluminescence is still most widespread in marine dwellers (Fig. 1). At great depths, for example, where light from the surface never penetrates, more than 90% of organisms are luminous [1, 2]. It is assumed that bioluminescence is related to the visual function of organisms and is often used for interspecific and intraspecific communication, for camouflage, for scaring away predators by the use of light flashes, and for attracting prey. However, in many cases the role of bioluminescence for the functioning of certain organisms is still unclear.
Fig. 1. Phylogenetic tree. Taxa including luminous species are marked with black asterisk. The so far identified phylogenetic groups whose representatives use imidazopyrazinone-type luciferin as a bioluminescent reaction substrate are marked with white asterisk: radiolarians (1), ctenophores (2), coelenterates (coral polyps (3), hydroids (4), scyphozoan jellyfish (5)), squids (6), crustaceans (7), some bony fishes (8) and sharks (9), chaetognaths (10), ophiurs (11), tunicates (12), molluscs (13).
Biochemical studies on bioluminescent systems of various organisms have revealed an interesting feature of bioluminescence – the mechanisms underlying visible light emission are considerably different in representatives of different taxa, despite the same final result of this biochemical process. Not only the substrates and cofactors involved in bioluminescent reactions are distinct, but the enzymes that catalyze these reactions differ too [3]. Therefore, the terms “luciferase” and “luciferin” applied to enzymes and substrates of bioluminescent reactions are rather generalizing and functional than structural and chemical concepts.
Among the several substrates of light-emitting reactions identified in luminous marine organisms, the imidazopyrazinone-type luciferins are most frequently found (Fig. 2a). Coelenterazine was identified as a substrate of bioluminescent reactions of such taxonomically distant luminous organisms as the soft coral Renilla [4], the decapods Oplophorus [5, 6], the scyphozoan medusa Periphylla [7, 8], the copepods [9, 10], the ostracods Conchoecia [11], the cephalopods Vampyroteuthis [12], the fish Benthosema pterotum [13], and others (Fig. 1). The hydromedusan and ctenophore Ca2+-regulated photoproteins contain a 2-hydroperoxy-derivative of coelenterazine [3]. The squids Watasenia and Symplectoteuthis, as well as the mollusc Pholas dactylus, use coelenterazine derivatives as the substrates of bioluminescent reactions – coelenterazine disulfate in the case of Watasenia [14], and dehydrocoelenterazine in the cases of Symplectoteuthis [15] and Pholas dactylus [16]. Cypridina (Vargula) luciferin is a substrate of luciferases of ostracods [17], as well as of fish Porichthys notatus, Apogon, and Parapriacanthus [18]. Most probably, many luminous organisms that use imidazopyrazinone-type luciferin for bioluminescence obtain these compounds from their diet, as do the luminous fish Porichthys notatus (Cypridina luciferin) [19, 20] and jellyfish Aequorea (coelenterazine) [21].
Fig. 2. a) Imidazopyrazinone-type luciferins: coelenterazine (1), Cypridina (Vargula) luciferin (2), 3-enol sulfate of coelenterazine (3), 3-enol sulfate of Cypridina luciferin (4), dehydrocoelenterazine (5), and coelenterazine disulfate (6). b) The hypothesized scheme of biosynthesis of coelenterazine and Cypridina luciferin in copepods and ostracods from L-amino acids.
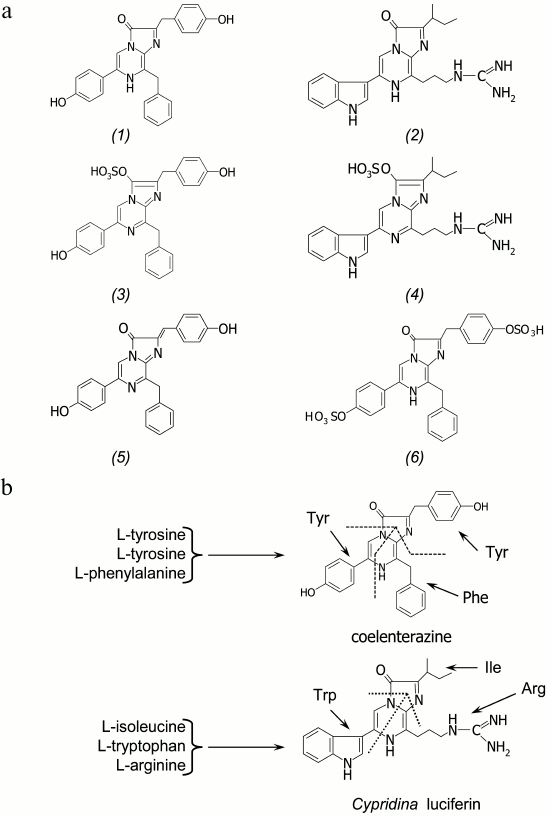
The de novo synthesis of coelenterazine and Cypridina luciferin has been shown only for luminous crustaceans – copepods and ostracods of the family Cypridinidae. With isotopically labeled L-amino acids to feed the crustaceans Metridia pacifica and Cypridina (Vargula) hilgendorfii applied, the copepods were found to synthesize coelenterazine from two tyrosines and one phenylalanine [22], while Cypridina luciferin in ostracods is synthesized from tryptophan, isoleucine, and arginine [23] (Fig. 2b). The imidazopyrazinone ring is apparently formed either by cyclization of a tripeptide (H2N-Phe-Tyr-Tyr-COOH in the case of coelenterazine and H2N-Arg-Trp-Ile-COOH in the case of Cypridina luciferin) that runs like the formation of the GFP chromophore [3] and includes an additional step of decarboxylation, or in a stepwise manner by nonribosomal synthesis [24], which we believe is most likely. It is noteworthy that coelenterazine was also found in rather large amounts in many non-luminous marine animals [3, 25].
A special place among imidazopyrazinone-type luciferins belongs to 3-enol sulfate derivatives (Fig. 2a). These compounds have been found in marine luminous coral Renilla reniformis [26] and ostracods Vargula (Cypridina) hilgendorfii [27]. The 3-enol sulfate derivatives of coelenterazine and Cypridina luciferin are actually preluciferins. These compounds are enzymatically converted into the active substrate by a sulfotransferase catalyzing sulfate group transfer from coelenterazine 3-enol sulfate (or Cypridina luciferin 3-enol sulfate) to an acceptor, adenosine 3′,5′-diphosphate (PAP) [26, 27]. As a result, coelenterazine (or Cypridina luciferin) and 3′-phosphoadenosine 5′-phosphosulfate (PAPS) are formed. This reaction is reversible, i.e. sulfotransferase can catalyze the formation of 3-enol sulfate derivatives of imidazopyrazinones as well [27]. In this case, the PAPS is the donor of a sulfate group. Since 3-enol sulfate derivatives of imidazopyrazinones are much more stable in aqueous solutions, it is assumed that these compounds provide a storage form of a substrate in marine luminous organisms that use either coelenterazine or Cypridina luciferin for bioluminescence.
MAIN TYPES OF COELENTERAZINE-DEPENDENT BIOLUMINESCENT
SYSTEMS
Bioluminescent systems using imidazopyrazinone compounds as the reaction substrates can be divided into photoprotein and luciferase types (Fig. 3). Bioluminescent reactions catalyzed by luciferases are typical enzymatic reactions in which the substrate is oxidized by oxygen with generation of a product (oxyluciferin) in the excited state, whose relaxation to the ground state is accompanied by light emission. Luciferase as the usual enzyme turns over several times, completely utilizing the substrate. The bioluminescent systems of the marine copepods, shrimp Oplophorus, ostracods, scyphozoan medusa, and coral Renilla are of this type [3]. In the case of bioluminescent systems of photoprotein type, the protein forms a stable enzyme–substrate complex existing for a long time. The most studied hydromedusan and ctenophore Ca2+-regulated photoproteins [3, 28-30], for example, are a complex of apoprotein and tightly but noncovalently bound 2-hydroperoxycoelenterazine, coelenterazine activated by oxygen that arises on the formation of a photoprotein from apoprotein, coelenterazine, and molecular oxygen.
Fig. 3. General scheme of bioluminescent reactions of luciferase and photoprotein types. The reaction of Ca2+-regulated photoprotein, which is a stable complex of the protein with coelenterazine activated by oxygen, 2-hydroperoxycoelenterazine, is shown as an example of photoprotein type. Bioluminescence is initiated by binding of calcium ions with protein Ca2+-binding sites.
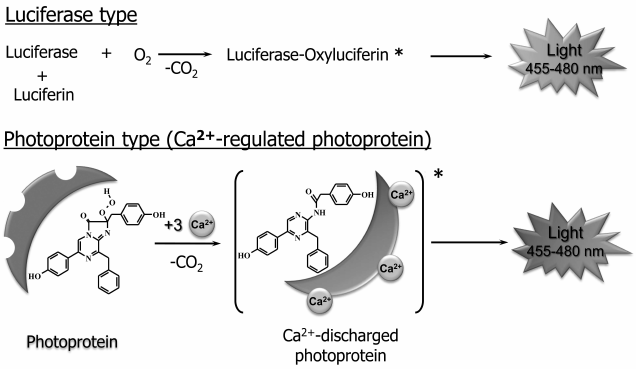
A bioluminescent reaction is initiated in response to binding of calcium ions with Ca2+-binding sites on the surface of the protein molecule. Binding of Ca2+ causes small conformational changes in a substrate-binding cavity of the protein, which disturb the hydrogen bond network that stabilizes 2-hydroperoxycoelenterazine, thereby triggering the reaction of oxidative decarboxylation leading to the formation of the product in the excited state [31]. Unlike Ca2+-regulated photoproteins, the photoproteins of squid Symplectoteuthis oualaniensis [15] and mollusc Pholas dactylus [16], simplectin and pholasin, are a complex of a protein with a covalently bound coelenterazine, which is formed upon binding of dehydrocoelenterazine with the apoprotein. The bioluminescent reaction of the photoproteins of squid and mollusc is initiated by O2 and K+ [15] and the reactive oxygen species [16], respectively. Since a substrate whose oxidation is necessary for light emission is already bound in photoproteins, these, unlike luciferases, can react only once, i.e. cannot turn over several times like a usual enzyme. It should be noted that since the reaction involves one molecule, the amount of the emitted light is always in proportion to the amount of the protein. This feature is very suitable for the use of photoproteins as labels in various types of bioluminescent assays [29].
Diversity OF COELENTERAZINE-DEPENDENT LUCIFERASES
Despite the wide variety of luminous marine organisms that use coelenterazine as a substrate in bioluminescent reaction, genes encoding only four types of coelenterazine-dependent luciferases have been cloned thus far (Table 1). In addition, some more luciferases from different taxa have been partially characterized (Table 2). All cloned coelenterazine-dependent luciferases (Table 1) belong to cofactor-independent monooxygenases as they catalyze a simple reaction (Fig. 3) involving only luciferase, substrate (coelenterazine or Cypridina luciferin), and molecular oxygen. Of these, only luciferase of the soft coral of the Renilla genus is an intracellular protein; the other luciferases cloned from phylogenetically distant groups of marine crustaceans are secreted luciferases. Luciferase and luciferin accumulated separately in a bioluminescent secretory gland of the crustacean are secreted via special pores into seawater [32] where the luciferase oxidizes substrate by the dissolved oxygen with the emission of light.
Table 1. Cloned luciferases that use
coelenterazine or Cypridina luciferin as a bioluminescent
reaction substrate
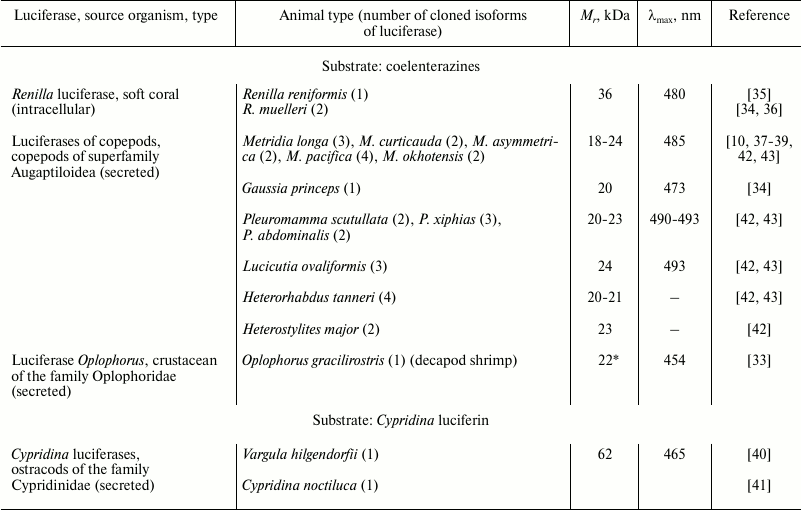
* For the luciferase of Oplophorus, the characteristics of only
the catalytic subunit are given.
Table 2. Partially characterized luciferases
that use coelenterazine as a bioluminescent reaction substrate

Natural luciferases of Renilla and ostracods of the Cypridinidae family were isolated as monomeric proteins from the corresponding luminous organisms. In contrast to these, the natural secreted luciferase of decapod shrimp Oplophorus gracilirostris was isolated as a tetramer consisting of two types of proteins [33], with only one revealing catalytic activity (Table 1). The natural form of secreted luciferases of marine planktonic copepods is unknown, since they have not been isolated. The first luciferases of copepods Metridia longa [10] and Gaussia princeps [34] were cloned by functional screening of cDNA gene libraries, while the cDNA genes encoding the other homologous luciferases of copepods (Table 1) were isolated using degenerate primers constructed from the known sequences [42, 43].
The sequence analysis of the cloned coelenterazine-dependent luciferases clearly shows that luciferases of all four types belonging to phylogenetically distant groups of organisms reveal no homology and are totally different proteins with different molecular masses (Table 1) and properties, although they catalyze the oxidation of the same substrate with the emission of light.
Two other coelenterazine-dependent luciferases from different taxa were isolated and partially characterized (Table 2). Cloning of the luciferase of fish Benthosema pterotum should be forthcoming [13], as the pure protein that can be used to determine its sequence and consequently the following isolation of the luciferase gene has already been achieved. Taking into account the diversity of coelenterazine-dependent luciferases, it will most likely be a completely new luciferase sequence.
Luciferase of the coral Renilla. Luciferase of corals of genus Renilla (class Anthozoa) (RLuc) was one of the first cloned [35] and thus far the only cloned intracellular coelenterazine-dependent luciferase. RLuc is a single-chain protein with a molecular mass of ~36 kDa consisting of 311 amino acids. Apart from luciferase, the bioluminescence of Renilla in vivo involves at least two more proteins – a Ca2+-regulated coelenterazine-binding protein (CBP) [44], which stabilizes the substrate, and the green fluorescent protein (GFP) [45], which is a secondary emitter (Fig. 4). The bioluminescent system of Renilla is well studied – all three proteins have been cloned [34-36] and characterized, and their three-dimensional structures have been determined [46-48].
Fig. 4. Schematic representation of bioluminescent reaction of Renilla luciferase. After binding of three calcium ions, the CBP conformation changes so that coelenterazine becomes available for luciferase. The reaction in the presence of GFP is accompanied by emission with a maximum at ~509 nm; in the absence of GFP, the emission is at ~480 nm.
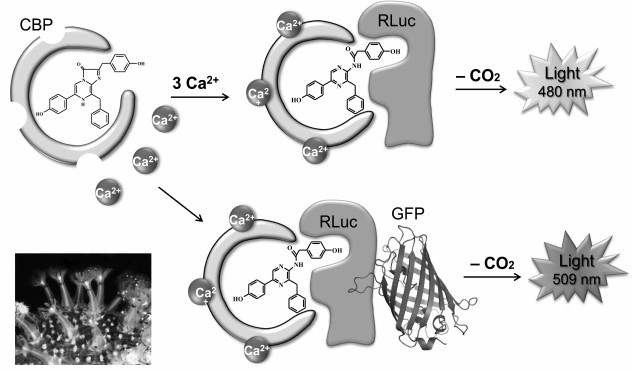
Unlike Ca2+-regulated photoproteins containing 2-hydroperoxycoelenterazine, the coelenterazine-binding protein contains a tightly but noncovalently bound molecule of the native coelenterazine in the inner hydrophobic cavity [47]. The binding of calcium ions by Ca2+-binding sites of CBP initiates small conformational changes in the protein that are enough to make the coelenterazine molecule available for oxidation by RLuc. In the absence of GFP, the oxidation of coelenterazine is accompanied by blue light emission with a maximum at λmax ~ 480 nm (Fig. 4). In the presence of GFP, a bioluminescent reaction, both in vivo and in vitro, is accompanied by light emission with λmax ~ 510 nm, which is due to Forster resonance energy transfer (FRET) from a bioluminescent donor (luciferase) to a fluorescent acceptor (GFP) as a result of the formation of a protein–protein complex. The formation of an RLuc—GFP complex was shown in experiments in vitro [45]. It should be noted that GFP not only shifts the emission spectrum to longer wavelengths, but also significantly increases the quantum yield of the bioluminescent reaction. The efficiency of the luciferase reaction in vitro is also increased several-fold when CBP is used as a substrate instead of free coelenterazine [36], which may be due to the formation of CBP—RLuc complex during the reaction [48]. Hence, it can be assumed that the in vivo bioluminescent reaction of Renilla occurs with the formation of a triple short-lived protein–protein complex CBP—RLuc—GFP, where coelenterazine is delivered into the luciferase active site and the efficient non-radiative energy transfer from the excited product to the chromophore of GFP with following emission of green light takes place.
Proteins involved in the bioluminescent reaction of the coral Renilla are packaged in specialized cells called photocytes. Luminescence is under the control of the nervous system and appears on increase in concentration of calcium ions in photocytes in response to various stimuli [49]. Thus, CBP not only protects coelenterazine from spontaneous oxidation [50], but also performs the function of connecting the nervous and bioluminescent systems, thereby providing a quick light response of the animal to stimuli.
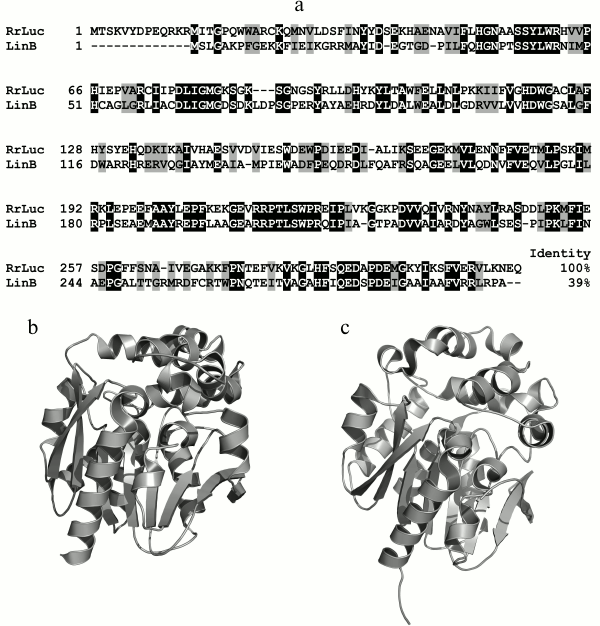
The amino acid sequence of RLuc has no homology with those of other coelenterazine-dependent luciferases, including photoproteins, but reveals similarities with the primary sequences of α/β-hydrolases [51]. RLuc reveals the greatest homology with the amino acid sequences of bacterial dehalogenases LinB of the α/β-hydrolases family. The degree of similarity with haloalkane dehalogenase LinB from Sphingomonas paucimobilis, for example, is 64% (identity – 39%) (Fig. 5a), which clearly implies evolution from a common ancestral sequence. The spatial structure of RLuc also reveals the typical α/β-hydrolase fold (Cα RMSD ~ 1.5 Å) (Fig. 5, b and c) [46]. In this case, the difference in structures of luciferase and α/β-hydrolases is even less than that in structures of the related hydromedusan photoproteins aequorin and obelin (Cα RMSD ~2.27 Å) [52] and functionally identical photoproteins of jellyfish and ctenophores (Cα RMSD ~ 2.19 Å) [53].
Fig. 5. a) Comparison of amino acid sequence of luciferase R. reniformis (RrLuc – AAA29804) with that of the homologous cellular protein haloalkane dehalogenase LinB of S. paucimobilis (LinB – 34810153). Black color indicates identical amino acid residues, gray – with similar properties. b, c) Structures of luciferase from R. reniformis (PDB file 2PSF) and haloalkane dehalogenase LinB (PDB file 1MJ5) from S. paucimobilis, respectively.
RLuc is now one of the most popular bioluminescent reporters in cellular and biomedical research as it can easily be expressed in the native state in almost all cell types. The temperature optimum for the bioluminescence reaction in vitro catalyzed by the wild-type RLuc is 18-37°C, and the pH optimum is 6-7. However, the quantum yield of bioluminescence is quite low, i.e. ~0.07 at 23°C [54]. Being probably optimal for functioning together with CBP and GFP in photocytes of animals, these properties of RLuc cannot always meet the requirements for reporter proteins, e.g. for their use in mammalian cells and tissues. RLuc-based reporters with improved properties were constructed in a standard way – by random mutagenesis with the following selection of suitable variants and by site-directed mutagenesis. To select amino acids for site-directed mutagenesis, the spatial structure of RLuc was used. RLuc mutants with higher specific bioluminescence activity and increased resistance to inactivation by blood serum were obtained [51, 55]. One of the optimized variants with eight mutations (RLuc8) revealed 4-fold increase in specific activity and the 200-fold increase in resistance to inactivation by blood serum.
Animal tissues are known to significantly absorb visible light. The lowest absorption is observed in the range of 600-900 nm, the so-called “transparency window” of biological tissues [56]. Thus, luciferases as reporters emitting the largest number of photons in this spectral region will provide higher sensitivity when used in intact animal tissues. The mutants RLuc-535 and RLuc-547 constructed for these purposes have the increased specific bioluminescent activity and emission spectrum shifted to longer wavelengths by up to 66 nm [57]. Another way to change the emission spectrum is the use of modified coelenterazine as a substrate. For instance, the use of coelenterazine ν shifted the bioluminescence spectrum to longer wavelengths for RLuc and its mutants by almost 40 nm [50, 57]. The spectral maximum of light emission of the mutant RM-Y with a natural coelenterazine is at 535 nm, while that with coelenterazine ν is at 574 nm.
Luciferases of copepods. These small, secreted proteins of ~18.4-24.3 kDa, including a signal peptide for secretion, are responsible for the bioluminescence of some marine copepods. The first cloned luciferases GpLuc and MLuc of the copepods G. princeps and M. longa, respectively, were immediately and successfully applied as bioluminescent reporters in vivo [10, 58] and in vitro [37, 59], and since then the scope of their application has been expanding.
Although bioluminescent species have been found among many copepods [32], so far the genes encoding luciferases have been cloned only from copepods belonging to the superfamily Augaptiloidea. The sequences of 28 full-length genes of luciferases of 12 species of luminous copepods encoding amino acid sequences with a relatively high degree of similarity have been determined (Table 1). Of the several types of luminous copepods, a few genes encoding 2-3 luciferase isoforms from one species have been isolated; for these, differences between isoforms of one species were comparable to those between the luciferases of taxonomically distant species. The sequence identity of isoforms MLuc164 and MLuc7 of M. longa, for example, is only 68%, which is close to the 57% sequence identity of MLuc164 and the luciferase GpLuc of G. princeps (Fig. 6), the copepod species from another genus of the same family Metridinidae. Such significant differences in amino acid sequences of three isoforms of M. longa suggest that the luciferase of these copepods is encoded by at least three paralogous (non-allelic) genes.
Fig. 6. Comparison of amino acid sequences of isoforms (MLuc164 – AAR17541, MLuc39 – ABW06650, and MLuc7 – AJC98141) of M. longa luciferase and the G. princeps luciferase (GpLuc – AAG54095). The identical amino acid residues are marked in black, the ones with similar properties – in gray. A signal peptide providing secretion of the luciferase and the conservative motifs within the repeats are framed; the conservative Cys residues are marked under the amino acid sequences. Deletion points of the N-terminal variable part in the truncated mutants MLuc164 [60] are labeled as M3, M4, and M5.
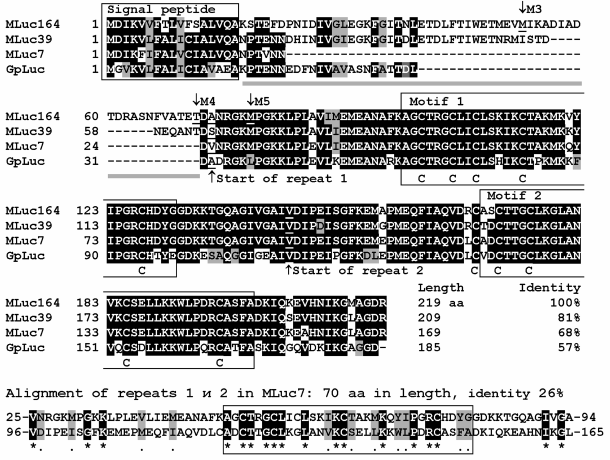
Copepod luciferases can be conditionally divided into three parts: a signal peptide for secretion, a variable N-terminus, and a conservative C-terminus (Fig. 6). The variable part, sometimes constituting up to one third of the length of the amino acid sequence, does not substantially affect the luciferase bioluminescent function – mutants with “truncated” variable part (Fig. 6) reveal even higher activity than the full-length MLuc164 [60]. It should be noted, however, that the deletion of the N-terminal part has an effect on the decay kinetics of the bioluminescent signal, making it faster than that of the wild-type MLuc164. The C-terminal conservative part of the luciferase is formed by two non-identical tandem repeats of length of about 70 amino acid residues, each containing a highly conserved motif of 32 amino acids [10, 61]. Each motif contains five conservative Cys residues (Fig. 6), suggesting the existence of up to five S–S bonds per luciferase molecule. The currently available data on the bioluminescent activity of individual tandem repeats are very contradictory. As reported in [61], each of the tandem repeats of GpLuc reveals bioluminescence activity under expression in E. coli cells. Hence, the authors concluded that the luciferases of copepods have two catalytic domains. Another study, however, reports on the absence of any activity of the same separately synthesized tandem repeats of GpLuc under expression in mammalian cells [62].
Despite widespread and successful application of copepod luciferases as reporters, their biochemical and bioluminescent properties are still insufficiently studied. The natural luciferase of copepods has not yet been isolated and characterized, even partially. Production of recombinant protein in native conformation using simple and inexpensive bacterial expression systems was ineffective due to the presence of numerous disulfide bonds in copepod luciferases. Thus, luciferase of M. longa obtained in E. coli cells is prone to aggregation, which leads to significant heterogeneity of the sample; the luciferase portion in a monomeric form is only a few percent of the total protein amount [37, 63]. In addition, certain properties of even monomeric highly purified recombinant luciferase from bacterial cells are very different from those of the luciferase produced in eukaryotic expression systems. For example, the specific bioluminescent activity of isoforms MLuc164 and MLuc7 obtained with a baculovirus expression system in insect cells is several times higher than the activity of these isoforms produced by bacterial expression [38, 63]. It should be noted that in addition to high bioluminescent activity, the luciferases of copepods have extremely high thermostability [39]; the isoform MLuc7 retains 50% of its activity even after boiling for 1 h [38]. The high thermostability of luciferases, similar to other secreted proteins, is apparently accounted for by the large amount of S–S bonds that stabilize their spatial structure.
The kinetics of luminescence of copepod luciferases under optimal conditions is of “flash”-type with fast decay within a few minutes [37, 58, 60]. However, some applications involving copepod luciferase as a reporter, e.g. high-throughput screening, require a bioluminescent signal that would be stable for tens of minutes. Such a signal can be obtained by changing the conditions of measurement of luciferase activity, as the kinetics are rather dependent on the buffer composition. The addition of 0.1% Triton X-100 to the buffer for measuring activity of GpLuc, for example, significantly slows the decay rate of the bioluminescent signal, but at the same time reduces the specific activity of the luciferase, thus significantly decreasing its detection limit and therefore the sensitivity of the method [64]. To overcome these shortcomings, a number of mutants of GpLuc possessing both high specific activity and slow decay of bioluminescent signal were obtained by mutagenesis [64-66]. The decay of bioluminescence of a double mutant of GpLuc (M60L-M127L), for example, was approximately 14 times slower, and the specific activity was like that of the wild-type GpLuc [65].
Since the redox potential in the cytoplasm of all cells does not facilitate the formation of disulfide bonds, the cytoplasmic expression of copepod luciferases potentially containing up to five S–S bonds does not yield cells with high bioluminescent activity, presumably due to incorrect folding of the luciferase. However, the bioluminescent intensity of cells can be significantly increased by expression of the secreted variant of luciferase with a signal peptide at the N-terminus and the KDEL sequence at the C-terminus, which retains the protein in the endoplasmic reticulum [58]. The secretory signal peptide directs the luciferase into the endoplasmic reticulum, where the protein properly maturates, but its following secretion will be stopped due to the “retention” peptide KDEL. As a result, a significant increase in the bioluminescence of cells will occur.
Luciferases of copepods are natural secreted proteins and therefore are most effective when used as secreted reporters, since this allows real-time monitoring of the intracellular events in vivo repeatedly without disruption of the object under study both in case of cells and in case of small laboratory animals. For GpLuc [58, 67] and MLuc [68], it was shown that the bioluminescence in cell culture medium or in the blood of the animals correlates linearly with the number of cells secreting the reporter in the range of about five orders of magnitude. This, for instance, allows the assessment of the functional state of malignant tumor, the rate of its growth and metastasis, as well as response to therapy by the level of bioluminescent activity in a small blood sample (5 µl) regardless of the localization of the cancer cells in the body [67, 68].
The main hindrance that limits the use of copepod luciferases as reporters in imaging in vivo directly in tissues and intact small laboratory animals is the blue emission considerably absorbed by mammalian tissues. This attenuates the signal and consequently reduces the sensitivity. For copepod luciferases, as in the case of Renilla luciferase, highly active mutants with a red-shifted bioluminescence spectrum are being sought. So far, one of the best variants is the mutant GpLuc (F89W/I90L/H95E/Y97W) called “Monsta”, which is almost five times higher in activity than the wild-type GpLuc when expressed in COS-7 cells, and it has bioluminescent maximum at 503 nm [66]. Although the mutant with red-shifted emission was constructed by mutagenesis, its spectrum is still far from the “transparency window” (>600 nm) of biological tissues.
Recently, several “artificial” luciferases (ALuc) have been constructed. They demonstrate higher stability and increased bioluminescent activity, which for some of these luciferases many times exceeded that of the wild-type GpLuc, the most popular and commonly used secreted bioluminescent reporter [69, 70]. In addition, some of the artificial luciferases revealed a significant red shift of the emission spectra (in the case of ALuc25, up to λmax = 530 nm) [69]. This property along with high bioluminescent activity makes these luciferases the most attractive reporters among the copepod luciferases for use in in vivo imaging in tissues and small laboratory animals. On construction of artificial luciferases, an approach was used based on the assumption that for a stable protein structure the amino acids frequently occurring in the same position are thermodynamically more favorable than the residues rarely occurring in the same position. Thus, the amino acid sequences of artificial luciferases are variants of consensus sequences derived from a comparison of the amino acid sequences of copepod luciferases [69].
Luciferase of shrimp Oplophorus gracilirostris. Natural secreted luciferase of the decapod shrimp O. gracilirostris is a tetramer with molecular mass of ~106 kDa [71] consisting of two monomers each of molecular mass 19 kDa, and the two monomers of 35 kDa. However, it is only the protein with Mr = 19 kDa that reveals luciferase activity; the function of the other protein that is part of the tetramer is not yet reliably established. The quantum yield of the bioluminescent reaction catalyzed by the natural luciferase Oplophorus is one of the highest (0.34 at 22°C) among luciferases using luciferins of imidazopyrazinone type. The temperature optimum of bioluminescent reaction is 40°C, and the pH optimum is ~9.0. Cloning of cDNA genes encoding the luciferase subunits showed that the 19- and 35-kDa proteins are composed of 196 and 359 amino acids, respectively, including putative signal peptides for secretion [33]. The recombinant protein with Mr = 19 kDa produced both in mammalian and E. coli cells was monomeric, thermolabile, prone to aggregation, and had substantially less specific bioluminescent activity compared to the natural luciferase [72]. Based on these observations, it was suggested that the function of the protein with Mr = 35 kDa in the natural luciferase is stabilization of the catalytic subunit. Unlike copepod luciferases containing 10 Cys residues and presumably five S–S bonds that stabilize the structure of the molecule and are critical for activity, the catalytic subunit of luciferase Oplophorus of approximately the same length contains only one cysteine residue that is not essential for the activity of the luciferase [72].
As with the case of luciferases of copepods, no significant similarity is found between the amino acid sequences of the Oplophorus luciferase and both known and hypothetical proteins. However, when using fold-recognition programs that identify the similarity of proteins by the elements of spatial structure, a vague resemblance of the catalytic 19-kDa subunit of Oplophorus luciferase to the well-characterized family of intracellular lipid-binding proteins (iLBPs) was found. Introducing the N166R substitution into the 19-kDa protein, which in accord with a spatial model must enhance the stability of the structure and increase the structural similarity to iLBP, led to increase in its bioluminescent activity ~3-fold and stability by ~50% [73]. This mutant was then used as a template for three rounds of random mutagenesis with intent to produce mutant luciferases with properties (high specific activity, slow decay of bioluminescent signal, increased solubility, and stability) optimized for applications as reporter proteins. The best mutations of each round were introduced in a template for the next round of random mutagenesis. In addition, the selected mutants were also tested with 24 coelenterazine analogs to find a substrate that would provide the highest bioluminescent activity and the lowest background signal compared to coelenterazine. As a result, a thermostable mutant named “NanoLuc” (NLuc) carrying 16 amino acid substitutions and showing several orders greater bioluminescent activity and stability than the wild-type catalytic subunit of the luciferase Oplophorus was produced [73]. As the best substrate for NanoLuc, the coelenterazine analog Furimazine was identified, which in contrast to coelenterazine provides a higher bioluminescent activity and stability in biological media (signal half-life exceeds 4 h) and, consequently, a lower background signal. In lysates of HEK293 cells, NanoLuc with Furimazine was reported to produce bioluminescent signals with slow decay kinetics (signal half-life > 2 h) and intensity ~2.5 million times higher than that of the 19-kDa protein with wild-type coelenterazine under similar conditions, and ~150 times higher than those of Renilla and firefly luciferases [73]. It should be noted, however, that the activity of NanoLuc, as shown in [74], exceeded that of 19-kDa only ~10-fold on soluble expression in E. coli cells, and only ~80-fold on expression in CHO-K1 cells. Also, the specific activity of NanoLuc with coelenterazines h was ~1.5 times greater than with Furimazine. A significant limitation to the use of a new artificial luciferase as a reporter for in vivo imaging in tissues of animals is the emission spectrum (λmax = 460 nm) corresponding to the region of strong absorption of light by mammalian tissues.
Cypridina luciferases. A natural secreted luciferase of ostracod Cypridina (Vargula) hilgendorfii was the first luciferase utilizing the luciferin of imidazopyrazinone type in the bioluminescent reaction that was isolated by chromatography as a highly purified protein and thoroughly characterized at the beginning of the 1960s [3]. This luciferase catalyzes the oxidative decarboxylation of Cypridina luciferin (Fig. 2) in a simple reaction involving only the enzyme, luciferin, and O2. As a result of the reaction, a product in an excited state is formed, and its relaxation to the ground state is accompanied by light emission with a maximum at 448-463 nm. The light emission wavelength depends on the ionic strength of the buffer and is practically independent of pH. The temperature optimum of the bioluminescent reaction is 30°C. The quantum yield of the bioluminescent reaction catalyzed by Cypridina luciferase is 0.3, one of the highest among bioluminescent reactions catalyzed by coelenterazine-dependent bioluminescent proteins [75].
Luciferase of the ostracod Cypridina (Vargula) hilgendorfii was one of the first luciferases using luciferin of imidazopyrazinone type for which the cDNA gene was cloned (Fig. 7) [40]. Later, the highly homologous luciferase of Cypridina noctiluca (identity of amino acid sequences is ~84%) was cloned [41]. Luciferase Cypridina is a one-subunit protein with calculated molecular mass of 62.2 kDa composed of 552-555 amino acid residues. This luciferase comprises a signal peptide for secretion and 32 conservative Cys residues within the mature protein that form 16 S–S bonds (in the natural luciferase, free SH-groups were not found [3]), and two putative N-glycosylation sites (Fig. 7). It is the largest known coelenterazine-dependent bioluminescent protein. The bioluminescent reaction catalyzed by Cypridina luciferase is inhibited by EDTA and EGTA. This suggests the influence of Ca2+ or other divalent cations on the enzymatic activity. However, none of the amino acid sequences characteristic of Ca2+-binding sites was revealed in this luciferase. Active recombinant luciferase can be obtained in secreted form only when expressed in eukaryotic cells (yeast, mammalian cells), which is apparently due to the presence of the large number of S–S bonds in the luciferase that are formed in the endoplasmic reticulum, as well as by the need for posttranslational glycosylation of the protein. It is worthy of note that despite high identity of amino acid sequences, the luciferase of Cypridina noctiluca reveals a much higher activity than that of Cypridina (Vargula) hilgendorfii under secretory expression in eukaryotic cell culture [41].
Fig. 7. Comparison of amino acid sequences of luciferases C. noctiluca (CnLuc – BAD08210) and V. hilgendorfii (VhLuc – AAB86460). Signal peptides providing secretion of the luciferases identified by the program Signal P 4.0 [76] and potential sites of N-glycosylation are framed. The conservative Cys residues are marked under the amino acid sequences. The domains of the D factor type of Willebrand (vWF-D) identified by the BLAST program are underlined with gray lines.

The analysis of amino acid sequences of these luciferases revealed no significant homology with any known protein, including coelenterazine-dependent proteins. However, in the sequences of the luciferases the BLAST program identifies two domains (Fig. 7) similar to domain D of the von Willebrand factor (vWF-D), the secreted multidomain glycoprotein that plays a pivotal role in blood coagulation. Domains of the vWF-D family have been found in many proteins, mostly in secreted glycoproteins such as integrins, growth factors, fibrillar collagens, proteins of the complement system, and IGF-binding proteins (sequence database on the proteins Pfam and UniProt). These domains are supposed to be responsible for multimerization of proteins and their interaction with other proteins [77]. It is not known if the vWF-D domains are relevant to the functioning of the luciferases or bioluminescent systems of ostracods.
It is interesting that luminous ostracods of the family Halocypridoidae, Conchoecia pseudodiscophora in particular, use coelenterazine but not Cypridina luciferin as a substrate for the bioluminescent reaction. It is also likely that their bioluminescence is of non-secreted type, which is accounted for by functioning of intracellular luciferase since the emission is observed strictly within the carapace (dorsal section) of the ostracods [11].
Luciferases of scyphozoan medusa P. periphylla and fish B. pterotum. The scyphozoan medusa P. periphylla contains soluble (L-form) and insoluble forms of luciferases, the latter being isolated as particles [7, 8]. The soluble form of luciferase with molecular mass of 32 kDa is responsible for bioluminescence and was isolated from dome mesoglea and radial lappets of the medusa. The insoluble form is accumulated as aggregates in the ovary, and its content, based on estimates of activity, exceeds 100-fold the amount of the soluble form [8]. In the presence of high concentrations of β-mercaptoethanol, the insoluble aggregates of luciferase form a mixture of active oligomers with molecular masses of 20, 40, and 80 kDa (luciferases A, B, and C, respectively). Based on electrophoresis data, it was concluded that the 40- and 80-kDa luciferases are dimer and tetramer of the luciferase A [8]. The soluble luciferase L is also a complex; the addition of β-mercaptoethanol results in the formation of two proteins with molecular masses of 20 and ~12 kDa. Of these two proteins, only the one with molecular mass of 20 kDa showed luciferase activity. All the luciferases were identical in bioluminescence spectra, with λmax at 465 nm, but differed in biochemical properties (temperature optimum, thermostability, and Km). Luciferases A, B, and C obtained from insoluble particles revealed higher specific activity than the soluble luciferase L. The specific bioluminescent activity of these luciferases, according to the authors, was the highest among the luciferases using coelenterazine as a substrate. Luciferases A, B, and C are extremely resistant to denaturation; their bioluminescent activity drops only slightly at pH 1 and pH 11, and it is even enhanced in 1-2 M guanidine hydrochloride. These luciferases, however, are less stable to heating than luciferase L (32 kDa), which retains activity practically unchanged after boiling for several minutes. These luciferases differ in temperature optimum as well. While bioluminescent activity of luciferase L is maximal at 0°C and decreases almost linearly as the temperature increases, approaching zero at 60°C, luciferases A, B, and C have optimum at ~30°C. In addition, the value of the Michaelis constant (Km ~ 0.2 µM) for luciferases A, B, and C is about six times lower than that for luciferase L. The reason for the differences in properties of the insoluble and soluble forms of luciferases of medusa P. periphylla is not clear. Despite such features as thermostability and high specific activity making this luciferase attractive for use as a reporter protein and the availability of biological material (scyphozoan medusa P. periphylla occurs in abundance in the fjords of Norway), the gene (or genes) encoding this luciferase has not yet been cloned.
Recently, a new coelenterazine-dependent luciferase responsible for bioluminescence of Benthosema pterotum was isolated from photophores of this fish, purified by chromatography, and partially characterized [13]. The luciferase has molecular mass of ~27 kDa, and its Michaelis constant is 0.4 µM, which is close to values for other coelenterazine-dependent luciferases. The bioluminescence spectrum has a maximum at 475 nm, and the temperature optimum for the bioluminescent reaction is 40°C. The luciferase of B. pterotum is rather thermostable (retains more than 50% activity for 1 h at 65°C) and resistant to extreme pH values. An interesting feature of this luciferase is that calcium and especially magnesium ions significantly increase its activity [13]. Considering the fact it was possible to obtain enough highly purified B. pterotum luciferase, as well as the current state of development of protein sequencing and molecular biology, it is reasonable to expect that the gene encoding this luciferase will be cloned soon.
Thus, the known coelenterazine-dependent luciferases, despite the use of the same substrate for bioluminescence and the same chemical mechanism of its oxidative decarboxylation [3], are nonhomologous proteins having no similarity with each other or other known proteins. The only exception is the luciferase Renilla, belonging to the group of α/β-hydrolases.
Independent evolution OF bioluminescent systems IS SOURCE OF DIVERSITY OF bioluminescent proteins
Common cellular functions of all organisms living on the Earth are in the main provided by biochemical processes involving homologous proteins, which are almost the same or sometimes greatly similar, namely, up to the full match of amino acid sequences. An unusual property of bioluminescence as a function of living organisms is that the luminescence of organisms belonging to different taxa involves various proteins catalyzing different reactions. This has become especially clear after cloning of genes and determination of the primary sequences of a number of proteins, as well as elucidation of mechanisms of the bioluminescent reactions of various organisms. The presently cloned bioluminescent enzymes from taxonomically distant organisms show no similarities in their amino acid sequences even when they use the same substrate for the bioluminescent reaction. Such proteins also revealed no similarity between their spatial structures despite the fact that those are more conservative than primary sequences. For some bioluminescent proteins, it was unexpected to find close homologs among ordinary cellular enzymes that provide basic cellular functions [51, 78]. Considering these differences in bioluminescent systems and their components in various organisms, it should be assumed that the ability for bioluminescence in representatives of different taxa arose independently and repeatedly in the course of evolution, involving suitable ancestral proteins.
There are more than 40 known variants of bioluminescent systems [1] that have arisen in evolution independently and in some cases apparently very recently. The supposition on the recent origin of certain variants is supported by the “spotted” distribution of bioluminescence among taxa (Fig. 1), i.e. the presence of organisms with different biochemistry of luminescence in the same taxon, as well as the presence of luminous and non-luminous species, sometimes even closely related, in one taxon. For example, among the four species of the colonial hydroid of Obelia genus dwelling the White Sea, only two are luminous despite similar living conditions of all the four species. In some taxa, such as siphonophores and ctenophores, the non-luminous representatives are rare, and in some taxa, on the contrary, luminous organisms are rare, e.g. among chaetognatha (type Chaetognatha) [1].
Even close relatives sometimes differ significantly in the nature of their luminescence, like in the case of different species of squids or planktonic ostracods that use various modifications of coelenterazine for luminescence and also different types of bioluminescence – secreted and intracellular.
Since the ability to produce light occurred repeatedly during evolution (Fig. 1), it can be reasonably assumed that first, bioluminescence is very important for organisms and second, it can appear rather easily. As noted above, luciferins of imidazopyrazinone type are found in many luminous and non-luminous marine organisms and can be transferred from one organism to another through food chains [3, 25], i.e. a bioluminescent substrate is more conservative than the other components of bioluminescent systems. Perhaps this is also true for other luciferins. Thus, having one available organic molecule capable of providing a light-emitting function, the organism only needs to “adapt” a suitable protein for bioluminescence to appear. It seems that various cellular proteins were often recruited to play the role of luciferase; mutations in these proteins led to the ability to catalyze the available bioluminescent substrate with the emission of light, thus forming all the observed diversity of bioluminescent systems.
Such a variety of organisms (Fig. 1) that use luciferins as substrates of imidazopyrazinone type in bioluminescent reactions and variety of proteins that catalyze these reactions suggest the existence of other numerous luminous organisms that have not yet been investigated and that also use these luciferins as substrates, but the function of luciferases is performed by completely different proteins. Why the imidazopyrazinone-type luciferins are the substrates of bioluminescent reactions in such a variety of luminous organisms remains a mystery.
CLONING OF NOVEL BIOLUMINESCENT PROTEINS
There are two approaches that can be used for cloning of genes encoding bioluminescent proteins from novel luminous organisms. The traditional method involves determination of components required for bioluminescence, primarily substrate and potential cofactors. If the substrate of a bioluminescent reaction is known and available, it is usually not difficult to reproduce the bioluminescent reaction in vitro. (As of today, these are apparently only bioluminescent proteins using luciferins of imidazopyrazinone type in bioluminescent reaction that fully meet this requirement.) In the next step, the bioluminescent protein is isolated from luminous organisms and its amount should be sufficient to determine at least a small part of the amino acid sequence. Then degenerate primers to the known protein sequence are constructed, which are then used for screening of cDNA library by hybridization or PCR to find clones with new genes. With this approach, the cDNA genes encoding several coelenterazine-dependent bioluminescent proteins were cloned: Ca2+-regulated photoprotein aequorin from hydromedusa Aequorea victoria [79, 80], luciferase from ostracod Cypridina (Vargula) hilgendorfii [40], luciferases from soft corals Renilla reniformis [35] and Renilla muelleri [36], luciferase from decapod shrimp Oplophorus gracilirostris [33], and Ca2+-dependent coelenterazine-binding protein from Renilla muelleri [36]. It is evident that this approach can be applied in the case if substantial quantities of the luminous organisms can be collected.
However, most luminous marine organisms are rare and can be caught as single specimens. Furthermore, many bioluminescent animals inhabit great depths, and therefore their gathering is rather complicated and labor consuming. These circumstances make the production of a sufficient amount of natural protein for its characterization and determination of amino acid sequence quite difficult, and therefore the traditional approach of cDNA gene cloning cannot be followed. The method of functional screening of clones of cDNA expression library helps get around this problem (Fig. 8). Modern PCR-based methods allow the construction of a cDNA gene expression library from a very small piece (only a few mg) of tissue. For functional screening, a bioluminescent reaction in vitro must only be reproduced and a sufficient amount of a substrate for screening must be available. High sensitivity of detection of the emitted light allows registration of positive bioluminescent signal from an individual clone among a large group of negative clones. With functional screening applied, the cDNA genes encoding Ca2+-regulated photoproteins of hydroids Obelia longissima [81] and Obelia geniculata [82], jellyfish Clytia gregaria [83] and Mitrocoma cellularia [84], ctenophore Beroe abyssicola [30], as well as luciferases of copepods Metridia longa [10, 37, 38] and Gaussia princeps [34], and GFP of jellyfish C. gregaria [83] were cloned. It should be mentioned that most of the cloned cDNA genes contain a stop codon (or even several stop codons) in the 5′-untranslated region. Nevertheless, this did not hinder isolation of individual colonies by bioluminescent activity, thereby demonstrating the high efficiency of functional screening on cloning of genes encoding bioluminescent and fluorescent proteins.
Fig. 8. Schematic representation of functional screening.
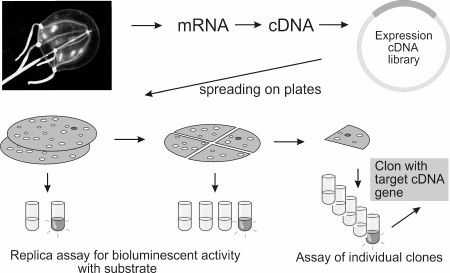
It is obvious that there are some limitations for applying functional screening. Since determination of the structure of the novel bioluminescent substrate is even more difficult than obtaining a highly purified protein for sequencing, this approach can be employed on cloning of novel proteins that use already known and available bioluminescence substrates, such as luciferins of imidazopyrazinone type. In addition, it is desirable that the bioluminescent protein be a one-subunit protein because the presence of another subunit would significantly complicate the search for the corresponding genes, and that the protein does not require posttranslational modifications necessary for its bioluminescent activity. It is likely that the latter of these limitations can be overcome using eukaryotic cells for screening of cDNA expression libraries.
Although the study of bioluminescent systems of various organisms is undoubtedly of fundamental importance, the main driving force that determines interest in the topic is still the usefulness of bioluminescence as an analytical tool. Presently, there is no field of biology, medical science, or pharmacology that does not widely apply bioluminescent methods or has not tested the ones showing suitability. The development of fundamental and applied research in these areas requires the elaboration of effective techniques for noninvasive imaging of molecular processes in vivo in a single living cell and in an intact living organism. Such technologies offer a good opportunity to resolve a wide range of tasks, but are primarily focused on investigation of dynamic processes in vivo in cells and organisms, thus giving a clearer view of the growth and metastatic activity of tumor, the response of tumor cells to therapy, migration of immunocompetent cells in the organism, and protein–protein interactions in a cell. These technologies also allow the study of metabolic activity and its regulation at the level of single cells and the intact organism, the pathways and distribution of proteins in a cell and the intact organism, regulation of gene expression, and a great deal more. In this view, reporters based on luciferases and photoproteins are very useful. Since bioluminescent reactions proceed with high quantum yield and extremely low background, technologies that use luciferases and photoproteins as reporters are capable of providing high-sensitive imaging and a wide linear dynamic range of measurements. Bioluminescent proteins and their substrates are components of bioluminescent systems of luminous organisms and are therefore nontoxic to living cells. Until recently, the use of bioluminescent imaging was limited by the imperfect methods of recording and processing of weak light signals. These problems, however, have been solved with the advent of highly sensitive cooled CCD cameras and microscopes that can provide real-time detection of changes in luminescence in a single cell.
Practically all the bioluminescent proteins available to researchers have already been tested as genetically encoded reporters for monitoring cellular processes. The most popular bioluminescent reporters today are the firefly luciferase, Ca2+-regulated photoproteins, and coelenterazine-dependent luciferases of Renilla, copepods Gaussia and Metridia, as well as their mutant variants with improved characteristics. One of the main applications of luciferases is monitoring of gene expression and tracing of labeled cells in an organism. Luciferase activity is used to estimate cell viability, apoptosis, and various processes associated with cellular metabolism. Luciferases fused with proteins under study are used to monitor metabolism of target proteins and their interaction with other proteins. An effective way to assess protein–protein interactions in vivo is complementation based on the ability of luciferases split into fragments to restore their activity on complementation of these fragments fused with the interacting proteins under study. Another way of assessing protein–protein interactions in vivo by means of luciferase reporters are FRET technologies. This method is based on Forster resonance energy transfer from a bioluminescent donor to a fluorescent acceptor protein, which occurs only with spatial approximation of interacting target proteins fused with the reporters and is accompanied by a change in the emission spectrum.
The availability of bioluminescent proteins with different properties, including mutants with improved characteristics, provides more opportunities for optimal design of experiments. Moreover, differences in substrates, kinetics of bioluminescent reactions, or bioluminescence spectra allow the use of up to three bioluminescent reporters for simultaneous registration of several different processes. Taking into account the diversity of bioluminescent proteins using luciferin of imidazopyrazinone type and continuing studies of new luminous organisms, the appearance of novel luciferases and photoproteins suited for the development of new in vivo imaging technologies should be expected.
This work was supported by the state budget allocated to fundamental research at the Russian Academy of Sciences (project No. 01201351504) and the Russian Academy of Sciences Presidium Program “Molecular and Cell Biology”.
REFERENCES
1.Haddock, S. H., Moline, M. A., and Case, J. F.
(2010) Bioluminescence in the sea, Ann. Rev. Mar. Sci.,
2, 443-493.
2.Widder, E. A. (2010) Bioluminescence in the ocean:
origins of biological, chemical, and ecological diversity,
Science, 328, 704-708.
3.Shimomura, O. (2006) Bioluminescence: Chemical
Principles and Methods, World Scientific Publishing, Singapore.
4.Hori, K., Charbonneau, H., Hart, R. C., and
Cormier, M. J. (1977) Structure of native Renilla reniformis
luciferin, Proc. Natl. Acad. Sci. USA, 74, 4285-4287.
5.Shimomura, O., Masugi, T., Johnson, F. H., and
Haneda, Y. (1978) Properties and reaction mechanism of the
bioluminescence system of the deep-sea shrimp Oplophorus
gracilirostris, Biochemistry, 17, 994-998.
6.Inouye, S., and Shimomura, O. (1997) The use of
Renilla luciferase, Oplophorus luciferase, and
apoaequorin as bioluminescent reporter protein in the presence of
coelenterazine analogues as substrate, Biochem. Biophys. Res.
Commun., 233, 349-353.
7.Shimomura, O., and Flood, P. R. (1998) Luciferase
of the scyphozoan medusa Periphylla periphylla, Biol.
Bull., 194, 244-252.
8.Shimomura, O., Flood, P. R., Inouye, S., Bryan, B.,
and Shimomura, A. (2001) Isolation and properties of the luciferase
stored in the ovary of the scyphozoan medusa Periphylla
periphylla, Biol. Bull., 201, 339-347.
9.Campbell, A. K., and Herring, P. J. (1990)
Imidazolopyrazine bioluminescence in copepods and other marine
organisms, Mar. Biol., 104, 219-225.
10.Markova, S. V., Golz, S., Frank, L. A., Kalthof,
B., and Vysotski, E. S. (2004) Cloning and expression of cDNA for a
luciferase from the marine copepod Metridia longa. A novel
secreted bioluminescent reporter enzyme, J. Biol. Chem.,
279, 3212-3217.
11.Oba, Y., Tsuduki, H., Kato, S., Ojika, M., and
Inouye, S. (2004) Identification of the luciferin–luciferase
system and quantification of coelenterazine by mass spectrometry in the
deep-sea luminous ostracod Conchoecia pseudodiscophora,
Chembiochem, 5, 1495-1499.
12.Robison, B. H., Reisenbichler, K. R., Hunt, J.
C., and Haddock, S. H. (2003) Light production by the arm tips of the
deep-sea cephalopod Vampyroteuthis infernalis, Biol.
Bull., 205, 102-109.
13.Homaei, A. A., Mymandi, A. B., Sariri, R.,
Kamrani, E., Stevanato, R., Etezad, S. M., and Khajeh, K. (2013)
Purification and characterization of a novel thermostable luciferase
from Benthosema pterotum, J. Photochem. Photobiol. B,
125, 131-136.
14.Tsuji, F. I. (2002) Bioluminescence reaction
catalyzed by membrane-bound luciferase in the “firefly
squid” Watasenia scintillans, Biochim. Biophys.
Acta, 1564, 189-197.
15.Chou, C. M., Tung, Y. W., and Isobe, M. (2014)
Molecular mechanism of Symplectoteuthis bioluminescence –
Part 4: Chromophore exchange and oxidation of the cysteine residue,
Bioorg. Med. Chem., 22, 4177-4188.
16.Tanaka, E., Kuse, M., and Nishikawa, T. (2009)
Dehydrocoelenterazine is the organic substance constituting the
prosthetic group of pholasin, Chembiochem, 10,
2725-2729.
17.Kishi, Y., Goto, T., Hirata, Y., Shimomura, O.,
and Johnson, F. H. (1966) Cypridina bioluminescence. I.
Structure of Cypridina luciferin, Tetrahedron Lett.,
29, 3427-3436.
18.Cormier, M. J. (1978) Comparative biochemistry of
animal systems, in Bioluminescence in Action (Herring, P. J.,
ed.) Academic Press, N. Y., pp. 75-108.
19.Tsuji, F. I., Barnes, A. T., and Case, J. F.
(1972) Bioluminescence in the marine teleost, Porichthys
notatus, and its induction in a non-luminous form by
Cypridina (ostracod) luciferin, Nature, 237,
515-516.
20.Tsuji, F. I., Nafpaktitis, B. G., Goto, T.,
Cormier, M. J., Wampler, J. E., and Anderson, J. M. (1975) Spectral
characteristics of the bioluminescence induced in the marine fish
Porichthys notatus by Cypridina (ostracod) luciferin,
Mol. Cell. Biochem., 9, 3-8.
21.Haddock, S. H., Rivers, T. J., and Robison, B. H.
(2001) Can coelenterates make coelenterazine? Dietary requirement for
luciferin in cnidarian bioluminescence, Proc. Natl. Acad. Sci.
USA, 98, 11148-11151.
22.Oba, Y., Kato, S., Ojika, M., and Inouye, S.
(2009) Biosynthesis of coelenterazine in the deep-sea copepod
Metridia pacifica, Biochem. Biophys. Res. Commun.,
390, 684-688.
23.Kato, S., Oba, Y., Ojika, M., and Inouye, S.
(2007) Biosynthesis of Cypridina luciferin in Cypridina
noctiluca, Heterocycles, 72, 673-676.
24.Hur, G. H., Vickery, C. R., and Burkart, M. D.
(2012) Explorations of catalytic domains in non-ribosomal peptide
synthetase enzymology, Nat. Prod. Rep., 29,
1074-1098.
25.Thomson, C. M., Herring, P. J., and Campbell, A.
K. (1997) The widespread occurrence and tissue distribution of the
imidazolopyrazine luciferins, J. Biolum. Chemilum., 12,
87-91.
26.Anderson, J. M., Hori, K., and Cormier, M. J.
(1978) A bioluminescence assay for PAP
(3′,5′-diphosphoadenosine) and PAPS
(3′-phosphoadenylyl sulfate), Methods Enzymol., 57,
244-257.
27.Nakamura, M., Suzuki, T., Ishizaka, N., Sato, J.,
and Inouye, S. (2014) Identification of 3-enol sulfate of
Cypridina luciferin, Cypridina luciferyl sulfate, in the
sea-firefly Cypridina (Vargula) hilgendorfii,
Tetrahedron, 70, 2161-2168.
28.Vysotski, E. S., and Lee, J. (2004)
Ca2+-regulated photoproteins: structural insight into the
bioluminescence mechanism, Acc. Chem. Res., 37,
405-415.
29.Vysotski, E. S., Markova, S. V., and Frank, L. A.
(2006) Calcium-regulated photoproteins of marine coelenterates, Mol.
Biol., 40, 355-367.
30.Markova, S. V., Burakova, L. P., Golz, S.,
Malikova, N. P., Frank, L. A., and Vysotski, E. S. (2012) The
light-sensitive photoprotein berovin from the bioluminescent ctenophore
Beroe abyssicola: a novel type of Ca2+-regulated
photoprotein, FEBS J., 279, 856-870.
31.Liu, Z. J., Stepanyuk, G. A., Vysotski, E. S.,
Lee, J., Markova, S. V., Malikova, N. P., and Wang, B. C. (2006)
Crystal structure of obelin after Ca2+-triggered
bioluminescence suggests neutral coelenteramide as the primary excited
state, Proc. Natl. Acad. Sci. USA, 103, 2570-2575.
32.Evstigneev, P. V., and Bityukov, E. P. (1990)
Bioluminescence of Marine Copepods [in Russian], Naukova Dumka,
Kiev.
33.Inouye, S., Watanabe, K., Nakamura, H., and
Shimomura, O. (2000) Secretional luciferase of the luminous shrimp
Oplophorus gracilirostris: cDNA cloning of a novel
imidazopyrazinone luciferase, FEBS Lett., 481, 19-25.
34.Bryan, B., and Szent-Gyorgyi, C. (1999)
Luciferases, fluorescent proteins, nucleic acids encoding the
luciferases and fluorescent proteins and the use thereof in
diagnostics, WO9949019A2.
35.Lorenz, W. W., McCann, R. O., Longiaru, M., and
Cormier, M. J. (1991) Isolation and expression of a cDNA encoding
Renilla reniformis luciferase, Proc. Natl. Acad. Sci.
USA, 88, 4438-4442.
36.Titushin, M. S., Markova, S. V., Frank, L. A.,
Malikova, N. P., Stepanyuk, G. A., Lee, J., and Vysotski, E. S. (2008)
Coelenterazine-binding protein of Renilla muelleri: cDNA
cloning, overexpression, and characterization as a substrate of
luciferase, Photochem. Photobiol. Sci., 7, 189-196.
37.Borisova, V. V., Frank, L. A., Markova, S. V.,
Burakova, L. P., and Vysotski, E. S. (2008) Recombinant Metridia
luciferase isoforms: expression, refolding and applicability for in
vitro assay, Photochem. Photobiol. Sci., 7,
1025-1031.
38.Markova, S. V., Larionova, M. D., Burakova, L.
P., and Vysotski, E. S. (2015) The smallest natural high-active
luciferase: cloning and characterization of novel 16.5-kDa luciferase
from copepod Metridia longa, Biochem. Biophys. Res.
Commun., 457, 77-82.
39.Takenaka, Y., Masuda, H., Yamaguchi, A.,
Nishikawa, S., Shigeri, Y., Yoshida, Y., and Mizuno, H. (2008) Two
forms of secreted and thermostable luciferases from the marine copepod
crustacean Metridia pacifica, Gene, 425,
28-35.
40.Thompson, E. M., Nagata, S., and Tsuji, F. I.
(1989) Cloning and expression of cDNA for the luciferase from the
marine ostracod Vargula hilgendorfii, Proc. Natl. Acad. Sci.
USA, 86, 6567-6571.
41.Nakajima, Y., Kobayashi, K., Yamagishi, K.,
Enomoto, T., and Ohmiya, Y. (2004) cDNA cloning and characterization of
a secreted luciferase from the luminous Japanese ostracod Cypridina
noctiluca, Biosci. Biotechnol. Biochem., 68,
565-570.
42.Takenaka, Y., Yamaguchi, A., Tsuruoka, N.,
Torimura, M., Gojobori, T., and Shigeri, Y. (2012) Evolution of
bioluminescence in marine planktonic copepods, Mol. Biol. Evol.,
29, 1669-1681.
43.Takenaka, Y., Noda-Ogura, A., Imanishi, T.,
Yamaguchi, A., Gojobori, T., and Shigeri, Y. (2013) Computational
analysis and functional expression of ancestral copepod luciferase,
Gene, 528, 201-205.
44.Charbonneau, H., and Cormier, M. J. (1979)
Ca2+-induced bioluminescence in Renilla reniformis.
Purification and characterization of calcium triggered
luciferin-binding protein, J. Biol. Chem., 254,
769-780.
45.Ward, W. W., and Cormier, M. J. (1979) An energy
transfer protein in coelenterate bioluminescence. Characterization of
the Renilla green fluorescent protein, J. Biol. Chem.,
254, 781-788.
46.Loening, A. M., Fenn, T. D., and Gambhir, S. S.
(2007) Crystal structures of the luciferase and green fluorescent
protein from Renilla reniformis, J. Mol. Biol.,
374, 1017-1028.
47.Stepanyuk, G. A., Liu, Z. J., Markova, S. V.,
Frank, L. A., Lee, J., Vysotski, E. S., and Wang, B. C. (2008) Crystal
structure of coelenterazine-binding protein from Renilla
muelleri at 1.7 Å: why it is not a calcium-regulated
photoprotein, Photochem. Photobiol. Sci., 7, 442-447.
48.Stepanyuk, G. A., Liu, Z. J., Vysotski, E. S.,
Lee, J., Rose, J. P., and Wang, B. C. (2009) Structure based mechanism
of the Ca2+-induced release of coelenterazine from the
Renilla binding protein, Proteins, 74,
583-593.
49.Cormier, M. J. (1978) Application of
Renilla bioluminescence: an introduction, Methods
Enzymol., 57, 237-244.
50.Stepanyuk, G. A., Unch, J., Malikova, N. P.,
Markova, S. V., Lee, J., and Vysotski, E. S. (2010) Coelenterazine-v
ligated to Ca2+-triggered coelenterazine-binding protein is
a stable and efficient substrate of the red-shifted mutant of
Renilla muelleri luciferase, Anal. Bioanal. Chem.,
398, 1809-1817.
51.Loening, A. M., Fenn, T. D., Wu, A. M., and
Gambhir, S. S. (2006) Consensus guided mutagenesis of Renilla
luciferase yields enhanced stability and light output, Protein Eng.
Des. Sel., 19, 391-400.
52.Deng, L., Vysotski, E. S., Markova, S. V., Liu,
Z. J., Lee, J., Rose, J., and Wang, B. C. (2005) All three
Ca2+-binding loops of photoproteins bind calcium ions: the
crystal structures of calcium-loaded apo-aequorin and apo-obelin,
Protein Sci., 14, 663-675.
53.Stepanyuk, G. A., Liu, Z. J., Burakova, L. P.,
Lee, J., Rose, J., Vysotski, E. S., and Wang, B. C. (2013) Spatial
structure of the novel light-sensitive photoprotein berovin from the
ctenophore Beroe abyssicola in the Ca2+-loaded
apoprotein conformation state, Biochim. Biophys. Acta,
1834, 2139-2146.
54.Matthews, J. C., Hori, K., and Cormier, M. J.
(1977). Purification and properties of Renilla reniformis
luciferase, Biochemistry, 16, 85-91.
55.Woo, J., and von Arnim, A. G. (2008) Mutational
optimization of the coelenterazine-dependent luciferase from
Renilla, Plant Methods, 4, 23.
56.Weissleder, R. (2001) A clearer vision for in
vivo imaging, Nat. Biotechnol., 19, 316-317.
57.Loening, A. M., Wu, A. M., and Gambhir, S. S.
(2007) Red-shifted Renilla reniformis luciferase variants for
imaging in living subjects, Nat. Methods, 4, 641-643.
58.Tannous, B. A., Kim, D. E., Fernandez, J. L.,
Weissleder, R., and Breakefield, X. O. (2005) Codon-optimized
Gaussia luciferase cDNA for mammalian gene expression in culture
and in vivo, Mol. Ther., 11, 435-443.
59.Verhaegent, M., and Christopoulos, T. K. (2002)
Recombinant Gaussia luciferase. Overexpression, purification,
and analytical application of a bioluminescent reporter for DNA
hybridization, Anal. Chem., 74, 4378-4385.
60.Markova, S. V., Burakova, L. P., and Vysotski, E.
S. (2012) High-active truncated luciferase of copepod Metridia
longa, Biochem. Biophys. Res. Commun., 417,
98-103.
61.Inouye, S., and Sahara, Y. (2008) Identification
of two catalytic domains in a luciferase secreted by the copepod
Gaussia princeps, Biochem. Biophys. Res. Commun.,
365, 96-101.
62.Remy, I., and Michnick, S. W. (2006) A highly
sensitive protein–protein interaction assay based on
Gaussia luciferase, Nat. Methods, 3, 977-979.
63.Stepanyuk, G. A., Xu, H., Wu, C. K., Markova, S.
V., Lee, J., Vysotski, E. S., and Wang, B. C. (2008) Expression,
purification and characterization of the secreted luciferase of the
copepod Metridia longa from Sf9 insect cells, Protein Expr.
Purif., 61, 142-148.
64.Degeling, M. H., Bovenberg, M. S., Lewandrowski,
G. K., de Gooijer, M. C., Vleggeert-Lankamp, C. L., Tannous, M.,
Maguire, C. A., and Tannous, B. A. (2013) Directed molecular evolution
reveals Gaussia luciferase variants with enhanced light output
stability, Anal. Chem., 85, 3006-3012.
65.Welsh, J. P., Patel, K. G., Manthiram, K., and
Swartz, J. R. (2009) Multiply mutated Gaussia luciferases
provide prolonged and intense bioluminescence, Biochem. Biophys.
Res. Commun., 389, 563-568.
66.Kim, S. B., Suzuki, H., Sato, M., and Tao, H.
(2011) Superluminescent variants of marine luciferases for bioassays,
Anal. Chem., 83, 8732-8740.
67.Chung, E., Yamashita, H., Au, P., Tannous, B. A.,
Fukumura, D., and Jain, R. K. (2009) Secreted Gaussia luciferase
as a biomarker for monitoring tumor progression and treatment response
of systemic metastases, PLoS One, 4, e8316.
68.Lupold, S. E., Johnson, T., Chowdhury, W. H., and
Rodriguez, R. (2012) A real time Metridia luciferase based
non-invasive reporter assay of mammalian cell viability and
cytotoxicity via the β-actin promoter and enhancer, PLoS
One, 7, e36535.
69.Kim, S. B., Torimura, M., and Tao, H. (2013)
Creation of artificial luciferases for bioassays, Bioconjug.
Chem., 24, 2067-2075.
70.Kim, S. B., and Izumi, H. (2014) Functional
artificial luciferases as an optical readout for bioassays, Biochem.
Biophys. Res. Commun., 448, 418-423.
71.Shimomura, O., Masugi, T., Johnson, F. H., and
Haneda, Y. (1978) Properties and reaction mechanism of the
bioluminescence system of the deep-sea shrimp Oplophorus
gracilorostris, Biochemistry, 17, 994-998.
72.Inouye, S., and Sasaki, S. (2007) Overexpression,
purification and characterization of the catalytic component of
Oplophorus luciferase in the deep-sea shrimp Oplophorus
gracilirostris, Protein Expr. Purif., 56,
261-268.
73.Hall, M. P., Unch, J., Binkowski, B. F., Valley,
M. P., Butler, B. L., Wood, M. G., Otto, P., Zimmerman, K., Vidugiris,
G., Machleidt, T., Robers, M. B., Benink, H. A., Eggers, C. T., Slater,
M. R., Meisenheimer, P. L., Klaubert, D. H., Fan, F., Encell, L. P.,
and Wood, K. V. (2012) Engineered luciferase reporter from a deep sea
shrimp utilizing a novel imidazopyrazinone substrate, ACS Chem.
Biol., 7, 1848-1857.
74.Inouye, S., Sato, J., Sahara-Miura, Y., Yoshida,
S., and Hosoya, T. (2014) Luminescence enhancement of the catalytic
19 kDa protein (KAZ) of Oplophorus luciferase by three
amino acid substitutions, Biochem. Biophys. Res. Commun.,
445, 157-162.
75.Shimomura, O., and Johnson, F. H. (1970)
Mechanisms in the quantum yield of Cypridina bioluminescence,
Photochem. Photobiol., 12, 291-295.
76.Petersen, T. N., Brunak, S., von Heijne, G., and
Nielsen, H. (2011) Signal P 4.0: discriminating signal peptides from
transmembrane regions, Nat. Methods, 8, 785-786.
77.Zhou, Y. F., Eng, E. T., Zhu, J., Lu, C., Walz,
T., and Springer, T. A. (2012) Sequence and structure relationships
within von Willebrand factor, Blood, 120, 449-458.
78.Viviani, V. R. (2002) The origin, diversity, and
structure function relationships of insect luciferases, Cell. Mol.
Life Sci., 59, 1833-1850.
79.Prasher, D., McCann, R. O., and Cormier, M. J.
(1985) Cloning and expression of the cDNA coding for aequorin, a
bioluminescent calcium-binding protein, Biochem. Biophys. Res.
Commun., 126, 1259-1268.
80.Inouye, S., Noguchi, M., Sakaki, Y., Takagi, Y.,
Miyata, T., Iwanaga, S., Miyata, T., and Tsuji, F. I. (1985) Cloning
and sequence analysis of cDNA for the luminescent protein aequorin,
Proc. Natl. Acad. Sci. USA, 82, 3154-3158.
81.Illarionov, B. A., Markova, S. V., Bondar, V. S.,
Vysotski, E. S., and Gitelson, J. I. (1992) Cloning and expression of
cDNA for the Ca2+-activated photoprotein obelin from the
hydroid polyp Obelia longissima, Dokl. Akad. Nauk,
326, 911-913.
82.Markova, S. V., Vysotski, E. S., Blinks, J. R.,
Burakova, L. P., Wang, B. C., and Lee, J. (2002) Obelin from the
bioluminescent marine hydroid Obelia geniculata: cloning,
expression, and comparison of some properties with those of other
Ca2+-regulated photoproteins, Biochemistry,
41, 2227-2236.
83.Markova, S. V., Burakova, L. P., Frank, L. A.,
Golz, S., Korostileva, K. A., and Vysotski, E. S. (2010)
Green-fluorescent protein from the bioluminescent jellyfish Clytia
gregaria: cDNA cloning, expression, and characterization of novel
recombinant protein, Photochem. Photobiol. Sci., 9,
757-765.
84.Burakova, L., Natashin, P., Markova, S.,
Eremeeva, E., and Vysotsky, E. (2014) The C-terminal tyrosine deletion
in mitrocomin increases its bioluminescent activity,
Luminescence, 29, 84.