Attempt to Optimize Some Properties of Fluorescent Chimeras of Human Small Heat Shock Protein HspB1 by Modifying Linker Length and Nature
P. N. Datskevich, L. K. Muranova, and N. B. Gusev*
Lomonosov Moscow State University, Biological Faculty, 119991 Moscow, Russia; fax: +7 (495) 939-2747; E-mail: NBGusev@mail.ru* To whom correspondence should be addressed.
Received October 2, 2014; Revision received October 12, 2014
Chimerical proteins consisting of enhanced yellow fluorescent protein (EYFP) connected by linkers of different length and nature to the N-terminal end of small heat shock protein HspB1 were obtained and characterized. To obtain fluorescent chimeras with properties similar to those of unmodified small heat shock protein, we used either 12-residue-long linkers of different nature (highly flexible Gly-Ser linker (L1), rigid α-helical linker (L2), or rigid Pro-Ala linker (L3)) or highly flexible Gly-Ser linker consisting of 12, 18, or 21 residues. The wild-type HspB1 formed large stable oligomers consisting of more than 20 subunits. Independent of the length or the nature of the linker, all the fluorescent chimeras formed small (5-9 subunits) oligomers tending to dissociate at low protein concentration. Chaperone-like activity of the wild-type HspB1 and its fluorescent chimeras were compared using lysozyme as a model protein substrate. Under the conditions used, all the fluorescent chimeras possessed higher chaperone-like activity than the wild-type HspB1. Chaperone-like activity of fluorescent chimeras with L1 and L3 linkers was less different from that of the wild-type HspB1 compare to the chaperone-like activity of chimeras with rigid L2 linker. Increase in the length of L1 linker from 12 up to 21 residues leads to decrease in the difference in the chaperone-like activity between the wild-type protein and its fluorescent chimeras. Since the N-terminal domain of small heat shock proteins participates in formation of large oligomers, any way of attachment of fluorescent protein to the N-terminal end of HspB1 leads to dramatic changes in its oligomeric structure. Long flexible linkers should be used to obtain fluorescent chimeras with chaperone-like properties similar to those of the wild-type HspB1.
KEY WORDS: small heat shock proteins, HspB1, fluorescent chimeras, quaternary structure, chaperone-like activityDOI: 10.1134/S0006297915010083
Abbreviations: EYFP, enhanced yellow fluorescent protein; sHsp (or HspB), small heat shock proteins.
Small heat shock proteins (sHsp or HspB) belong to a widely distributed
protein family whose members participate in proteostasis as well as
many other key vital processes [1]. For instance,
sHsp affect protein synthesis [2] and degradation
[3] as well as regulate apoptosis [4], proliferation [5],
cytoskeleton dynamics [6, 7],
and many other processes. Since small heat shock proteins participate
in many processes, any defects in their expression or mutations of
genes coding for sHsp correlate with different oncologic and
neurodegenerative diseases, different types of cataracts, and myopathy
[8].
The presence of a conservative α-crystallin domain having the structure of double β-sheet is the most remarkable and distinguishing property of all small heat shock proteins [9]. This domain is flanked by less conservative and less ordered N- and C-terminal domains regulating oligomeric state and functional activity of sHsp [10, 11]. The human genome contains 10 genes coding for 10 different members of the sHsp family. These proteins differ in their properties and tissue distribution [12]. Of them, HspB1 and HspB5 are the best characterized and possess the most diverse activities.
Since sHsp participate in many diverse processes and therefore are related to different diseases, these proteins are now under intense investigation [13, 14]. Detailed investigations of the structure and properties are required for understanding the mechanism of small heat shock proteins functioning. Fluorescent chimeras obtained by fusing different-colored fluorescent proteins to the target proteins can be used for investigation in the living cell and for study of intracellular location and protein–protein interactions. Chimeric proteins composed of different derivatives of green fluorescent protein (GFP) and human small heat shock proteins have been already used for investigation of certain properties of sHsp [15-19]. However, most of these investigations were performed on the cell level without detailed investigation of the physicochemical properties of these chimeras. However, it is worthwhile to mention that the molecular weight of a fluorescent protein is comparable or even larger than that of the small heat shock protein. Moreover, attachment of the fluorescent protein to the N- or C-terminal ends participating in oligomerization and functioning of sHsp [11, 20] can significantly affect their properties.
Investigations performed in our laboratory [21-23] have shown that the earlier-described fluorescent chimeras of small heat shock proteins differ from the wild-type proteins in their oligomeric state, chaperone-like activity, and ability to form heterooligomers with other small heat shock proteins. The largest differences were detected in the case of HspB1 and HspB5, i.e. sHsp tending to form large oligomers. This makes it difficult to use earlier described fluorescent chimeras for investigation of the structure and properties of sHsp. At the same time, use of fluorescent chimeras opens new perspectives in investigation of different proteins and makes desirable obtaining fluorescent chimeras of sHsp having properties similar to those of the wild-type proteins. It is well known that the length and the nature of the linker sequence can strongly affect properties of fluorescent chimeras [24, 25]. Therefore, to obtain fluorescent chimeras with properties most similar to those of the wild-type protein we tried to construct chimeras carrying enhanced yellow fluorescent protein attached to the N-terminus of HspB1 by linkers of different length and nature.
MATERIALS AND METHODS
Obtaining molecular constructs. Chimeras with 12-residue linkers were obtained by PCR amplification of DNA fragments coding for unmodified human HspB1 and enhanced yellow fluorescent protein (EYFP). In this case, we used primers containing part of the future linker and the site recognized by certain restrictases. The fragments were treated by restrictases, ligated, and cloned into pET23b(+) at NdeI and XhoI sites. The linkers sequence, primers, and restrictases are presented in Table 1.
Table 1. Primers used for obtaining
fluorescent chimeras of HspB1 with different types of linkers

Note: The sites recognized by the corresponding restrictases are
underlined. T7-forward and T7-reverse primers were utilized as the
forward and reverse primers for EYFP and HspB1, respectively. Linker L1
is highly flexible and mobile, linker L2 tends to form α-helix,
and linker L3 is rigid and contains Pro-Ala repeats [24].
cDNA of chimeras with L1 linker (highly flexible unordered linker) of different length were obtained by overlap extension PCR. For this purpose, we used primers containing overlapping sites of the future linker. By using these primers, we amplified cDNA of HspB1 and EYFP containing overlapping fragments of the linker sequence. Using cDNA of two parts of the future chimeras and “external” primers, we obtained the full-size cDNA of chimeric protein by PCR. The full-size insert was cloned in pET23b(+) at NdeI and XhoI sites. The linker sequences and primers are presented in Table 2.
Table 2. Primers utilized for obtaining
fluorescent chimeras of HspB1 with L1 linker of different length

Note: Overlapping fragments of cDNA were obtained utilizing reverse
primers for EYFP and forward primers for HspB1. T7 primers were used as
“external” primers.
All constructs were checked by sequencing.
Expression and isolation of chimeras. Earlier described methods [21] were used for expression and isolation of fluorescent chimeras. Escherichia coli strain BL21(DE3) was chemically transformed by the chimeras. One colony from a Petri dish was inoculated into 40 ml of LB media containing 100 µg/ml of ampicillin and grown overnight at 37°C. The night culture was transferred into 800 ml of 3-fold LB media containing 100 µg/ml ampicillin and incubated for 7 h at 37°C followed by 16 h incubation at 30°C. The cells were collected by centrifugation (10 min, 5000g), suspended in 40 ml of lysis buffer (50 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5 mM PMSF, 14 mM β-mercaptoethanol), and frozen.
The bacterial cells were treated with lysozyme (final concentration 0.05 mg/ml) for 30 min, DNase I (final concentration 2.5 µg/ml in the presence of 1.75 mM MgCl2) for another 30 min, followed by sonication using a Branson S250D ultrasonic disintegrator (40 s burst, four times, at 40% power). The resulting mixture was subjected to centrifugation, and the supernatant was fractionated by ammonium sulfate (0-40% saturation). The pellet obtained after centrifugation (20 min, 23,700g) was dissolved in buffer B (20 mM Tris-acetate, pH 7.6, 10 mM NaCl, 0.1 mM EDTA, and 14 mM β-mercaptoethanol) and dialyzed overnight against 2 liters of buffer B. The sample was centrifuged (30 min, 105,000g) and loaded on a 5-ml HiTrapQ anion-exchange column (Amersham Pharmacia Biotech, GB). The column was washed by buffer B, and the fluorescent chimeras were eluted by a linear gradient of NaCl (10-500 mM). Further purification was achieved by size-exclusion chromatography on a Superdex 200 HiLoad 26/60 (Amersham Pharmacia Biotech) column equilibrated by buffer B supplemented with 150 mM NaCl. The fractions enriched in fluorescent chimeras were collected, concentrated by ultrafiltration, and dialyzed against 2 liters of buffer B containing 2 mM dithiothreitol instead of β-mercaptoethanol. The resulting protein samples were aliquoted and stored frozen at –20°C.
Oligomeric structure of fluorescent chimeras. The quaternary structures of the fluorescent chimeras were analyzed by size-exclusion chromatography on a Superdex 200 HR 10/30 (Amersham Pharmacia Biotech) column. The column equilibrated by buffer B supplemented with 150 mM NaCl and 14 mM β-mercaptoethanol was loaded with 150 µl samples containing from 10 up to 280 µg of protein. The column was run at the rate of 0.5 ml/min, and 400 µl fractions were collected. The column was calibrated with a protein standard with apparent molecular weights indicated in parentheses: thyroglobulin (670 kDa), ferritin (440 kDa), bovine serum albumin (68 kDa), ovalbumin (43 kDa), and chymotrypsinogen (25 kDa).
Chaperone-like activity of fluorescent chimeras. The chaperone-like activity of the fluorescent chimeras was determined by their ability to retard or prevent aggregation of a model protein substrate, lysozyme. All measurements were performed in the buffer of aggregation (50 mM KH2PO4, 50 mM NaCl, pH 7.4) at 37°C. The sample with final volume of 300 µl contained 10 µM of lysozyme and 2-5 µM (per monomer) of the analyzed fluorescent chimeras. Aggregation was initiated by addition of dithiothreitol to the final concentration of 20 mM and was followed by increase in the optical density at 360 nm. Each experiment was repeated at least two times.
RESULTS
To obtain fluorescent chimeras with properties most similar to those of the wild-type protein, we used two experimental approaches. First we obtained chimeras where enhanced yellow fluorescent protein was attached to the N-terminus of HspB1 through identical in length (12 residues) but different in nature linkers. The L1 linker having composition GSAGSAATGGSA is highly mobile and flexible, L2 linker having composition AEAAAKEAAAKA tends to form α-helix, and the L3 linker having composition APAPAPGPAPAP forms a rigid bent conformation. In the second series of experiments we obtained fluorescent chimeras where enhanced yellow fluorescent protein was attached to the N-terminus of HspB1 through identical in nature, but different in length linker. In this case, we used mobile flexible linker consisting of 12, 15, 18, or 21 residues. The composition of these linkers was as follows: 12L1 (GSAGSAATGGSA), 15L1 (GSAGSAATGGSAGSA), 18L1 (GSAGSAATGGSAGSAGSA), and 21L1 (GSAGSAATGGSAGSAGSAGSA).
The quaternary structure of fluorescent chimeras was analyzed by means of size-exclusion chromatography. We loaded on the column different quantities of the wild-type HspB1 and its fluorescent chimeras so that the final molar concentration of the wild-type protein and its chimera was identical. Since the molecular weight of the fluorescent chimeras is roughly 2 times that of the wild-type protein, we loaded 10-130 µg of the wild-type protein and 10-250 µg of its chimera (Figs. 1 and 2). In good agreement with the earlier obtained results [21-23], we found that the wild-type HspB1 forms large stable oligomers with apparent molecular weight of 520-535 kDa. The oligomeric state of the wild-type HspB1 does not depend on protein concentration in the sample loaded on the column. Taking into account that molecular weight of the monomer of the wild-type protein is ~23.7 kDa, we assume that the large oligomers of the wild-type HspB1 contain 22-24 subunits.
Fig. 1. Oligomeric state of wild-type HspB1 and fluorescent chimeras of this protein with different types of linkers. Dependence of elution volume on the quantity of protein loaded on the column of Superdex 200. The column was loaded with the wild-type HspB1 (WT) and its fluorescent chimeras with 12-residue linker having flexible mobile structure (L1), tending to form α-helix (L2), or containing Pro-Ala repeats (L3).
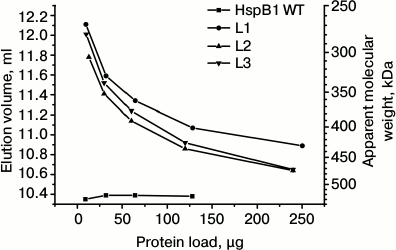
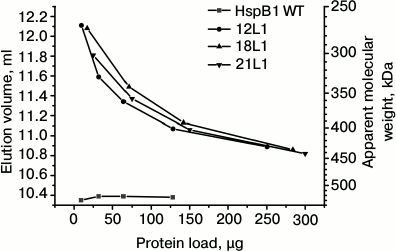
Fig. 2. Oligomeric structure of HspB1 of the wild type and its fluorescent chimeras with different length of flexible mobile linker of L1 type. Dependence of elution volume on the quantity of protein loaded on a Superdex 200 column. The column was loaded by the wild-type HspB1 (WT) and its fluorescent chimeras with L1 linker containing 12 (12L1), 18 (18L1), or 21 (21L1) residues.
The oligomeric state of the chimeras with different linkers is strongly dependent on the concentration of the protein sample loaded on the column (Fig. 1). If small quantities of protein were loaded on the column, the elution volume was equal to 11.8-12.1 ml, thus corresponding to proteins with apparent molecular weight 270-300 kDa. Taking into account that the molecular weight of fluorescent chimeras is equal to ~50 kDa, we assume that under these conditions the fluorescent chimeras form only small oligomers consisting of ~5-6 monomers. Increase in the quantity of protein loaded on the column was accompanied by decreases in elution volume to 10.7-10.9 ml, corresponding to apparent molecular weight 430-470 kDa. Thus, even under these conditions oligomers of fluorescent chimeras contain only ~9-10 monomers, i.e. 2 times less than the corresponding oligomers of the wild-type protein.
It is worthwhile to mention that the change of the linker nature and transition from 12-member flexible to the 12-member rigid Ala-Pro or 12-member α-helical linker had no effect on the dependence of the oligomeric state of the fluorescent chimeras on protein concentration (Fig. 1). Moreover, increase in the linker length from 12 up to 21 residues also does induces no significant change in the oligomeric state of fluorescent chimeras (Fig. 2). Independent on the linker nature or its length, the fluorescent chimeras form unstable oligomers composed of a smaller number of subunits than the wild-type protein.
Chaperone-like activity is one of the most important properties of small heat shock proteins. Therefore, it was desirable to compare chaperone-like activity of the wild-type HspB1 and its fluorescent chimeras. Chaperone-like activity was determined using lysozyme as a model protein substrate. Reduction of disulfide bonds leads to denaturation and aggregation of lysozyme that is accompanied by increase in optical density at 340 nm (Fig. 3, curve 0). Under the conditions used, the wild-type HspB1 retards the process of lysozyme aggregation, but it was unable to prevent aggregation of this model substrate completely (Fig. 3a, curve 1). The fluorescent chimera with flexible 12-member linker (L1) in its turn possessed somewhat higher chaperon-like activity (Fig. 3a, curve 2).
Fig. 3. Chaperone-like activity of wild-type HspB1 and its chimeras with linkers of different nature. a) Comparison of chaperone-like activity of the wild-type HspB1 and its fluorescent chimeras determined with lysozyme as a model protein substrate. Aggregation of reduced lysozyme was recorded in samples containing: 0) isolated lysozyme (10 µM); 1) lysozyme in the presence of wild-type HspB1 (5 µM); 2) lysozyme in the presence of fluorescent chimeras with 12-residue linker having flexible mobile structure L1 (5 µM). b) Chaperone-like activity of chimeras with 12-residue linker of different nature. Aggregation of isolated lysozyme (10 µM) (curve 0) or lysozyme in the presence of 5 µM fluorescent chimeras with flexible mobile linker of L1 type (curve L1), α-helical linker (curve L2), or Ala-Pro linker (curve L3).
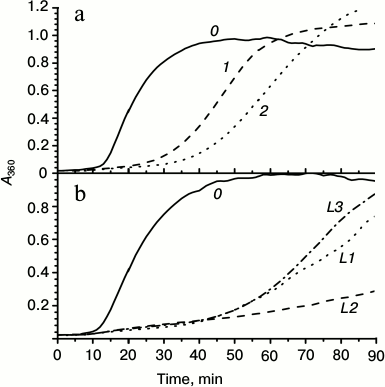
We analyzed the effect of linker nature on the chaperone-like activity of HspB1 (Fig. 3b). All the studied chimeras possessed higher chaperone-like activity than the wild-type protein. However, the chaperone-like activity of chimeras with flexible (L1) or Pro-Ala (L3) linkers was similar, and although higher was nevertheless comparable with the chaperone-like activity of the wild-type protein. Fluorescent chimeras with α-helical linker (L2) possessed the highest chaperone-like activity, which was significantly higher than that of the wild-type protein (Fig. 3b).
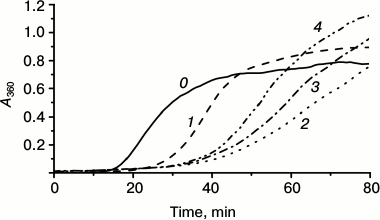
We also analyzed the effect of the linker length on the chaperone-like activity of the fluorescent chimeras. As already mentioned, the chaperone-like activity of chimeras with flexible linker was less different from the corresponding activity of the wild-type protein. Therefore constructing chimeras, we used the flexible L1 linker and changed its length from 12 up to 21 amino acid residues (Fig. 4). All of the studied chimeras possessed higher chaperone-like activity than the wild-type protein (Fig. 4). However, increase in the linker length decreased the difference in chaperone-like of the wild-type protein and its fluorescent chimeras.
Fig. 4. Chaperone-like activity of fluorescent chimeras with flexible mobile linker of L1 type composed of 12, 18, or 21 residues. Aggregation of isolated lysozyme (10 µM) (curve 0) or lysozyme in the presence of 5 µM of HspB1 of the wild-type (curve 1) or fluorescent chimeras of L1 type composed of 12 (curve 2), 18 (curve 3), or 21 (curve 4) residues.
DISCUSSION
Constructing of fluorescent chimeras seems to be an interesting and promising direction for investigation of different proteins including sHsp. However, in the last case constructing fluorescent chimeras is complicated by the fact that the size of the fluorescent protein is comparable or even larger than that of sHsp. Moreover, sHsp tend to form different homo- and heterooligomers [1, 26], and both N- and C-terminal sequences of sHsp are involved in formation of these oligomers [20, 27]. Therefore, a priori attachment of a fluorescent protein to either N- or C-terminus of sHsp will inevitably affect packing of monomers in homo- or heterooligomers.
To overcome these difficulties and obtain fluorescent chimeras with properties similar to those of the wild-type protein, we varied the length and the nature of peptide linker connecting the fluorescent protein to the N-terminus of HspB1.
Varying length or the nature of the peptide linker, we were unable to obtain fluorescent chimeras with oligomeric state similar to that of the wild-type protein. In practically all cases, the fluorescent chimeras formed unstable oligomers containing according to our calculations 5-10 monomers, which is roughly 2-4 times smaller than the number of subunits in the case of large oligomers formed by the wild-type protein (Figs. 1 and 2). As mentioned, the oligomers formed by fluorescent chimeras were unstable and their dilution was accompanied by dissociation leading to accumulation of oligomers of smaller size. In this respect, fluorescent chimeras are significantly different from the wild-type protein forming stable oligomers that do not dissociate under dilution. It is probable that attachment of a bulky fluorescent protein to the N-terminus of the small heat shock proteins makes impossible proper association of sHsp monomers and formation of stable large oligomers. The N-terminal domain is directly involved in formation of large αB-crystallin oligomers [28] and probably of other small heat shock proteins tending to oligomerization. Therefore, modification of the N-terminal domain prevents formation of large oligomers of fluorescent chimeras. Thus, at equal molar concentration, the fluorescent chimeras will form a larger number of small oligomers than the wild-type protein forming only a restricted number of large stable oligomers. According to one hypothesis presented in the literature [29, 30], large oligomers are not very effective in binding denatured proteins. It is supposed that effective chaperone-like activity is achieved by dissociation of large sHsp oligomers followed by binding of denatured protein substrates and reassembling of large oligomers carrying bound unfolding substrates. If this hypothesis is correct, we expect that the fluorescent chimeras tending to dissociation will possess higher chaperone-like activity that the wild-type protein.
Indeed, it was found that the chaperone-like activity of the fluorescent chimeras with lysozyme as a model protein substrate was higher than that of the wild-type protein (Figs. 3 and 4). The fluorescent chimeras with flexible Gly-Ser or Pro-Ala linker possessed similar chaperone-like activity that was comparable with that of the wild-type protein, whereas chimeras with rigid α-helical linker possessed significantly higher chaperone-like activity (Fig. 3b, curve L2). At the same time, all the studied chimeras have similar quaternary structure (see Fig. 1). It is well known that certain substrate-binding sites are located in the flexible N-terminal domain of small heat shock proteins [31, 32]. It is probable that the linker nature affects the location of the fluorescent protein at the functionally important N-terminal domain of HspB1 and therefore, depending on the flexibility of the linker, the fluorescent chimeras will possess different chaperone-like activity.
This suggestion correlates well with the data obtained on fluorescent chimeras with different length of flexible linker. Indeed, increase in the linker length makes the chaperone-like activity of fluorescent chimeras similar to that of the wild-type protein (Fig. 4). It is probable that removal of the fluorescent protein from certain sites in the N-terminal domain will eliminate effect of the fluorescent protein on the interaction of sHsp with its target proteins.
In summary, we might conclude that construction of fluorescent chimeras of sHsp tending to form large oligomers is a rather complex problem. Since both the N- and C-terminal domains of sHsp are involved in formation of large oligomers, construction of fluorescent chimeras with quaternary structure identical to that of the wild-type sHsp is in principle impossible. However, it is possible to obtain fluorescent chimeras with properties less different from those of the wild-type HspB1. For this purpose, it is advisable to use long flexible linkers. In any case, obtaining of fluorescent chimeras usable for cellular investigation requires additional investigations.
This investigation was supported by the Russian Science Foundation (RSF) (grant No. 14-35-00026).
REFERENCES
1.Mymrikov, E. V., Seit-Nebi, A. S., and Gusev, N. B.
(2011) Large potentials of small heat shock proteins, Physiol.
Rev., 91, 1123-1159.
2.Carra, S., Rusmini, P., Crippa, V., Giorgetti, E.,
Boncoraglio, A., Cristofani, R., Naujock, M., Meister, M., Minoia, M.,
Kampinga, H. H., and Poletti, A. (2013) Different anti-aggregation and
pro-degradative functions of the members of the mammalian sHSP family
in neurological disorders, Philos. Trans. R. Soc. Lond. B. Biol.
Sci., 368.
3.Boelens, W. C., Croes, Y., and de Jong, W. W.
(2001) Interaction between alphaB-crystallin and the human 20S
proteasomal subunit C8/alpha7, Biochim. Biophys. Acta,
1544, 311-319.
4.Paul, C., Simon, S., Gibert, B., Virot, S., Manero,
F., and Arrigo, A. P. (2010) Dynamic processes that reflect
anti-apoptotic strategies set up by HspB1 (Hsp27), Exp. Cell
Res., 316, 1535-1552.
5.Crowe, J., Aubareda, A., McNamee, K., Przybycien,
P. M., Lu, X., Williams, R. O., Bou-Gharios, G., Saklatvala, J., and
Dean, J. L. (2013) Heat shock protein B1-deficient mice display
impaired wound healing, PLoS ONE, 8, e77383.
6.Wettstein, G., Bellaye, P. S., Micheau, O., and
Bonniaud, P. (2012) Small heat shock proteins and the cytoskeleton: an
essential interplay for cell integrity? Int. J. Biochem. Cell
Biol., 44, 1680-1686.
7.Seit-Nebi, A. S., Datskevich, P., and Gusev, N. B.
(2013) Commentary on paper: “Small heat shock proteins and the
cytoskeleton: an essential interplay for cell integrity?”
(Wettstein et al.), Int. J. Biochem. Cell Biol., 45,
344-346.
8.Datskevich, P. N., Nefedova, V. V., Sudnitsyna, M.
V., and Gusev, N. B. (2012) Mutations of small heat shock proteins and
human congenital diseases, Biochemistry (Moscow), 77,
1500-1514.
9.Bagneris, C., Bateman, O. A., Naylor, C. E.,
Cronin, N., Boelens, W. C., Keep, N. H., and Slingsby, C. (2009)
Crystal structures of alpha-crystallin domain dimers of
alphaB-crystallin and Hsp20, J. Mol. Biol., 392,
1242-1252.
10.Delbecq, S. P., and Klevit, R. E. (2013) One size
does not fit all: the oligomeric states of alphaB crystallin, FEBS
Lett., 587, 1073-1080.
11.Lelj-Garolla, B., and Mauk, A. G. (2012) Roles of
the N- and C-terminal sequences in Hsp27 self-association and chaperone
activity, Protein Sci., 21, 122-133.
12.Kappe, G., Franck, E., Verschuure, P., Boelens,
W. C., Leunissen, J. A., and de Jong, W. W. (2003) The human genome
encodes 10 alpha-crystallin-related small heat shock proteins:
HspB1-10, Cell Stress Chaperones, 8, 53-61.
13.Arrigo, A. P., and Gibert, B. (2012) HspB1
dynamic phospho-oligomeric structure dependent interactome as cancer
therapeutic target, Curr. Mol. Med., 12, 1151-1163.
14.Cox, D., Carver, J. A., and Ecroyd, H. (2014)
Preventing alpha-synuclein aggregation: the role of the small
heat-shock molecular chaperone proteins, Biochim. Biophys. Acta,
1842, 1830-1843.
15.Sun, X., Fontaine, J. M., Rest, J. S., Shelden,
E. A., Welsh, M. J., and Benndorf, R. (2004) Interaction of human HSP22
(HSPB8) with other small heat shock proteins, J. Biol. Chem.,
279, 2394-2402.
16.Fontaine, J. M., Sun, X., Benndorf, R., and
Welsh, M. J. (2005) Interactions of HSP22 (HSPB8) with HSP20,
alphaB-crystallin, and HSPB3, Biochem. Biophys. Res. Commun.,
337, 1006-1011.
17.Fontaine, J. M., Sun, X., Hoppe, A. D., Simon,
S., Vicart, P., Welsh, M. J., and Benndorf, R. (2006) Abnormal small
heat shock protein interactions involving neuropathy-associated HSP22
(HSPB8) mutants, FASEB J., 20, 2168-2170.
18.Borrelli, M. J., Bernock, L. J., Landry, J.,
Spitz, D. R., Weber, L. A., Hickey, E., Freeman, M. L., and Corry, P.
M. (2002) Stress protection by a fluorescent Hsp27 chimera that is
independent of nuclear translocation or multimeric dissociation,
Cell Stress Chaperones, 7, 281-296.
19.Evgrafov, O. V., Mersiyanova, I., Irobi, J., Van
Den Bosch, L., Dierick, I., Leung, C. L., Schagina, O., Verpoorten, N.,
Van Impe, K., Fedotov, V., Dadali, E., et al. (2004) Mutant small
heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and
distal hereditary motor neuropathy, Nat. Genet., 36,
602-606.
20.Delbecq, S. P., Jehle, S., and Klevit, R. (2012)
Binding determinants of the small heat shock protein,
alphaB-crystallin: recognition of the “IxI” motif, EMBO
J., 31, 4587-4594.
21.Datskevich, P. N., Mymrikov, E. V., Sluchanko, N.
N., Shemetov, A. A., Sudnitsyna, M. V., and Gusev, N. B. (2012)
Expression, purification and some properties of fluorescent chimeras of
human small heat shock proteins, Protein Exp. Purif., 82,
45-54.
22.Datskevich, P. N., Mymrikov, E. V., and Gusev, N.
B. (2012) Utilization of fluorescent chimeras for investigation of
heterooligomeric complexes formed by human small heat shock proteins,
Biochimie, 94, 1794-1804.
23.Datskevich, P. N., and Gusev, N. B. (2014)
Structure and properties of chimeric small heat shock proteins
containing yellow fluorescent protein attached to their C-terminal
ends, Cell Stress Chaperones, 19, 507-518.
24.Chen, X., Zaro, J. L., and Shen, W. C. (2013)
Fusion protein linkers: property, design and functionality, Adv.
Drug Deliv. Rev., 65, 1357-1369.
25.Arai, R., Ueda, H., Kitayama, A., Kamiya, N., and
Nagamune, T. (2001) Design of the linkers which effectively separate
domains of a bifunctional fusion protein, Protein Eng.,
14, 529-532.
26.Arrigo, A. P. (2013) Human small heat shock
proteins: protein interactomes of homo- and hetero-oligomeric
complexes: an update, FEBS Lett., 587, 1959-1969.
27.Shcherbo, D., Souslova, E. A., Goedhart, J.,
Chepurnykh, T. V., Gaintzeva, A., Shemiakina, I. I., Gadella, T. W.,
Lukyanov, S., and Chudakov, D. M. (2009) Practical and reliable
FRET/FLIM pair of fluorescent proteins, BMC Biotechnol.,
9, 24.
28.Jehle, S., Vollmar, B. S., Bardiaux, B., Dove, K.
K., Rajagopal, P., Gonen, T., Oschkinat, H., and Klevit, R. E. (2011)
N-terminal domain of alphaB-crystallin provides a conformational switch
for multimerization and structural heterogeneity, Proc. Natl. Acad.
Sci. USA, 108, 6409-6414.
29.Shashidharamurthy, R., Koteiche, H. A., Dong, J.,
and McHaourab, H. S. (2005) Mechanism of chaperone function in small
heat shock proteins: dissociation of the HSP27 oligomer is required for
recognition and binding of destabilized T4 lysozyme, J. Biol.
Chem., 280, 5281-5289.
30.Leder, L., Stark, W., Freuler, F., Marsh, M.,
Meyerhofer, M., Stettler, T., Mayr, L. M., Britanova, O. V., Strukova,
L. A., Chudakov, D. M., and Souslova, E. A. (2010) The structure of
Ca2+ sensor Case16 reveals the mechanism of reaction to low
Ca2+ concentrations, Sensors, 10,
8143-8160.
31.Ghosh, J. G., Estrada, M. R., and Clark, J. I.
(2005) Interactive domains for chaperone activity in the small heat
shock protein, human alphaB crystallin, Biochemistry, 44,
14854-14869.
32.Jaya, N., Garcia, V., and Vierling, E. (2009)
Substrate binding site flexibility of the small heat shock protein
molecular chaperones, Proc. Natl. Acad. Sci. USA, 106,
15604-15609.